

Abstract
Astrocytic tumors frequently exhibit defects in the expression or activity of proteins that control cell-cycle progression. Inhibition of kinase activity associated with cyclin/cyclin-dependent kinase co-complexes by cyclin-dependent kinase inhibitors is an important mechanism by which the effects of growth signals are down-regulated. We undertook the present study to determine the role of p57KIP2 (p57) in human astrocytomas. We demonstrate here that whereas p57 is expressed in fetal brain tissue, specimens of astrocytomas of varying grade and permanent astrocytoma cell lines do not express p57, and do not contain mutations of the p57 gene by multiplex-heteroduplex analysis. However, the inducible expression of p57 in three well-characterized human astrocytoma cell lines (U343 MG-A, U87 MG, and U373 MG) using the tetracycline repressor system leads to a potent proliferative block in G1 as determined by growth curve and flow cytometric analyses. After the induction of p57, retinoblastoma protein, p107, and E2F-1 levels diminish, and retinoblastoma protein is shifted to a hypophosphorylated form. Morphologically, p57-induced astrocytoma cells became large and flat with an expanded cytoplasm. The inducible expression of p57 leads to the accumulation of senescence-associated β-galactosidase marker within all astrocytoma cell lines such that ∼75% of cells were positive at 1 week after induction. Induction of p57 in U373 astrocytoma cells generated a small population of cells (∼15%) that were nonviable, contained discrete nuclear fragments on Hoechst 33258 staining, and demonstrated ultrastructural features characteristic of apoptosis. Examination of bax and poly-(ADP ribose) polymerase levels showed no change in bax, but decreased expression of poly-(ADP ribose) polymerase after p57 induction in all astrocytoma cell lines. These data demonstrate that the proliferative block imposed by p57 on human astrocytoma cells results in changes in the expression of a number of cell cycle regulatory factors, cell morphology, and a strong stimulus to cell senescence.
The most common brain tumor is the astrocytoma accounting for ∼65% of all primary brain tumors. The malignant astrocytoma has a very poor prognosis primarily because of its highly proliferative and invasive nature. As with other neoplasms with increased proliferative potential, malignant astrocytomas demonstrate dysregulation of various components of the cell cycle machinery. Altered expression of positive growth regulators such as growth factors, cyclins, and cyclin-dependent kinases (CDKs), or the loss of negative regulators, including cyclin-dependent kinase inhibitors (CKIs) and the retinoblastoma protein (pRB) have all been demonstrated in malignant astrocytomas. 1,2 The CDKs phosphorylate pRB to release cells from cell-cycle arrest. In contrast with CDKs, the CKIs inhibit cyclin-CDK complexes and transduce internal or external growth suppressive signals. Accordingly, all CKIs may be construed as candidate tumor suppressor genes.
The CKIs are divided into two families, the INK4 and the CIP/KIP, which are defined on the basis of their structural homology and mechanism of action. The CIP/KIP family includes three structurally related members, p21CIP1/WAF1, 3,4 p27KIP1, 5,6 and a recently isolated and cloned third member, p57KIP2 (p57). 7-10 These three CKIs share a common N-terminal domain for binding to and inhibiting the kinase activity of CDK-cyclin complexes. Mouse p57 consists of four structurally distinct domains, a CDK inhibitory domain, a proline-rich domain, an acidic-repeat domain, and a carboxy-terminal domain. Human p57 differs from that of mouse by virtue of sequences containing proline-alanine repeats in its internal domain. The human p57 gene is located in 11p15.5, which frequently undergoes maternal allele loss of heterozygosity in several cancers, including Wilms’ tumor, and tumors associated with Beckwith-Wiedemann syndrome. 10-16
p57 has been shown to be a potent inhibitor of several G1 cyclin/CDK complexes, and its overexpression leads to cell-cycle arrest in G1 phase. 10 Recently, Watanabe et al 17 showed that human p57 protein, like p21, contains proliferating cell nuclear antigen-binding domain within its C terminus that, when separated from its N-terminal CDK-cyclin binding domain, can prevent DNA replication. Hashimoto et al 18 showed that the 310 helix region of p57, but not of p21 or p27, was indispensable for the inhibition of cyclin A/CDK2 and cyclin E/CDK2 complexes. Thus, the 310 helix motif may confer a specific regulatory mechanism by which p57 differentially regulates CDK2 and CDK4 activities. p57 mRNA is expressed at high levels in embryonic tissues such as skeletal muscle, heart, lung, and brain. Interestingly, cells expressing p57 have been shown to be terminally differentiated. 10 Using multiple mutant mice, Zhang et al 19,20 showed that the CKIs p57 and p21 function redundantly to control cell-cycle exit and differentiation of lens fiber cells, placental trophoblasts, and myoblasts. As such, p57 is now thought of as a critical terminal effector of signal transduction pathways that control cell differentiation. 8,10,19 Recently, expression of p57 was shown to inhibit the conversion of conditionally immortal human mammary epithelial cells to the fully immortal state, suggesting that p57 may provide an additional barrier against indefinite proliferation. 21
The human brain is a unique organ from a cell kinetic standpoint. Neurons become incapable of cell division in the early postnatal period. On the other hand, astrocytes retain their proliferative potential as is demonstrated in the process of reactive gliosis. Interestingly, astrocytes have the highest propensity to undergo malignant transformation of any cell type in the brain. Because p57 is expressed in cells with a high proliferative potential within embryonic brain tissue, in the present study we sought to determine the role of p57 in well-characterized, permanent human astrocytoma cell lines.
Materials and Methods
Astrocytoma Cell Lines, Culture Conditions, and Tumor Specimens
Three well-characterized malignant astrocytoma cell lines were used in this study: U 343 MG-A (U343), U87 MG (U87), and U373 MG (U373) (generous gifts of Bengt Westermark, Uppsala, Sweden). 22,23 U343 is a subclone of the original malignant astrocytoma that expresses the astrocyte differentiation marker, glial fibrillary acidic protein; it grows anchorage dependently, and is nontumorigenic in athymic mice. 24 U87 is derived from a patient with a glioblastoma multiforme, and is tumorigenic in athymic mice. 25 U373 is derived from an anaplastic astrocytoma and is also tumorigenic in athymic mice. 23 The p53 status of these three cell lines has previously and recently been determined: U87 and U343 are wild type and U373 is mutant for p53 26-28 (David Malkin, The Hospital for Sick Children, Toronto, personal communication). All astrocytoma cell lines were grown in monolayer culture in α-MEM supplemented with 10% fetal bovine serum and penicillin/streptomycin/fungizone (Life Technologies, Inc., Gaithersburg, MD) at 37°C in 5% CO2.
Astrocytoma tumor specimens were taken at the time of craniotomy and tumor excision. A specimen of nonneoplastic human brain from a 14-year-old female was taken after craniotomy for epilepsy surgery. Human fetal brain was obtained after elective second-trimester abortion after informed consent from the parents. 29 Permission to use this material was granted by the Research Ethics Board, The Hospital for Sick Children.
Polymerase Chain Reaction (PCR)/Heteroduplex Analysis and DNA Sequencing
Five overlapping PCR fragments spanning the entire coding region of the human p57 gene were generated using published primer pairs: 7,30 fragment I, primers 2 + 6 (TCTTCTCGCTGTCCTCTCCT + CGCCCCACCTGCACCGTCT); fragment II, primer set 3 (CTTCCAGCAGGACATGCCGCTG + TGGAGCCAGGACCGGGACT); fragment III, primer set 4 (ACTGCCTAGTGTCCCGGTC + GTCAGCGAGAGGCTCCTGG); fragment IV, primers 7 + 9 (TCAAGAGAGCGCCGAGCAG + GCGGGCCCTTTAATGCCAC); fragment V, primers 10 + 12 (TCTCCCGGCCCCCTCTCGG + CAAAACCGAACGCTGCTCTG).
Fragments were amplified from 500 ng of genomic DNA in PCR reactions containing 10% DMSO, 0.6 μmol/L primers, 0.25 mmol/L dNTP, and 2 units Taq polymerase (Life Technologies, Inc.) in the supplied buffer (2 mmol/L MgCl2). Reactions were amplified by touch-down PCR with 35 sequentially linked cycles of 94°C denaturation for 30 seconds, annealing temperature for 30 seconds, and extension at 72°C for 30 seconds. Annealing was initiated at 68°C with a 1°C per 2 cycle step-down to 15 cycles at 58°C, followed by a 7-minute extension. Heteroduplex formation was achieved by denaturation at 94°C for 5 minutes and annealing at 65°C for 5 minutes. PCR products were purified using Qiaquick PCR purification columns (Qiagen Inc., Mississauga, Ontario, Canada), followed by electrophoresis in 0.4-mm thick hydrolink mutation detection enhancement gels (BioWhittaker Molecular Applications, Rockland, Maine) gels containing 15% urea and 6.25% formamide (J. T. Baker, Phillipsburg, NJ). Electrophoresis was performed in 0.6× Tris borate-ethylenediaminetetraacetic acid and run at 500 V for 17 hours before transfer to Hybond-N+ membranes (Amersham, Oakville, Ontario, Canada) and hybridization with internal primers for each PCR fragment: primer 13 (CCTTCCCAGTACTAGTGCGC), primer 6, primer 7, primer 8, and primer 11 (TCAGCAAAGCCGGCGGGGA) for fragments I, II, III, IV, and V, respectively. Samples containing heteroduplex species were directly sequenced in both orientations using the fragment specific-PCR primer pairs by Thermo Sequenase terminator cycle sequencing (Amersham) of gel-purified PCR products.
Plasmids and Transfection
The tetracycline-repressor gene expression system was used to induce expression of p57. 31 The pUHD15–1neo plasmid (generous gift of S. Reed, The Scripps Research Institute, La Jolla, CA) contains the Escherichia coli tetracycline repressor element fused to the VP16 transactivation domain of herpesvirus. This fusion protein is driven by a cytomegalovirus promoter and the vector has the neomycin resistance gene for selection. pUHD15–1neo (25 μg) was transfected into U 343 MG-A cells using calcium phosphate. Neomycin-resistant clones were selected in 900 μg/ml geneticin (G418; Life Technologies, Inc.) in α-MEM and stable expression of the fusion protein was determined by Western blot analysis of total cell lysates using a polyclonal antisera to VP16 (kindly provided by C. J. Ingles, Toronto, Canada). Several clones were analyzed for VP16 expression for each cell line, and the majority expressed VP16. Clones that demonstrated high-level expression of VP16 were selected for transfection with pUHD10-3 (generously provided by H. Bujard, Heidelberg, Germany). pUHD10-3 contains a multiple cloning site downstream from tandem tetracycline operator sequences and a CMV promoter. A full-length human p57 cDNA (kind gift of S. J. Elledge, Houston, TX) was inserted into the multiple cloning site of pUHD10-3, and this plasmid (25 μg) was co-transfected with pgk-puro (1 μg) for selection of stable lines. These clones were also maintained in 4 μg/ml tetracycline (Sigma, St. Louis, MO). Puromycin (Sigma) was used for selection at 1 μg/ml and G418 concentration was maintained at 500 μg/ml.
To induce expression of p57, astrocytoma cells were washed three times in phosphate-buffered saline (PBS) before identical medium without tetracycline was added. To screen for p57 expression, total cell lysates were extracted and Western blot analysis was performed. To determine the effect of induction of p57 expression on the growth and morphology of the astrocytoma cell clones, 2 to 5 × 10 5 cells were plated in 10-cm 2 dishes. The following day, fresh medium was added. Cell proliferation assays and flow cytometric analysis was performed as described below.
Cell Proliferation Assay
Cell growth was assayed by counting cells at defined intervals. Briefly, cells were trypsinized and resuspended in media, and an aliquot of cells was counted using a hemocytometer. Each count represented an average of three counts on three separate determinations. Cell proliferation assays were repeated in duplicate. Cell viability was determined on the basis of trypan blue exclusion as described previously. 32,33
Flow Cytometric Analysis
To determine the proportion of cells present in a particular cell cycle phase, flow-assisted cell sorting (FACS) analysis of DNA content was performed. Briefly, 2 to 8 × 10 5 cells were trypsinized, washed in PBS, and resuspended in ice-cold 80% ethanol. Cells were kept at 4°C until propidium iodide (Sigma) DNA staining was performed. For different samples, the concentration of cells was kept equivalent. For staining, fixed cells were resuspended in propidium iodide and DNase-inactivated RNase A (Sigma) (final concentration 1 mg/ml) and were incubated for 30 minutes at room temperature in the dark. Stained cells were filtered through mesh-capped tubes and DNA content was analyzed on a Becton-Dickinson FACScan (San Jose, CA). Percent cell-cycle phase was determined using Cell Fit software (Becton-Dickinson) on three separate runs for each cell clone.
Antibodies
Antibodies to bax (N-20), p107 (SC-318), p130 (SC-317), E2F-1 (SC-193), and E2F-4 (SC-866x) were obtained from Santa Cruz Biotech Inc. (Santa Cruz, CA); antibodies to pRB (14001A), poly(ADP ribose) polymerase (PARP) (C2-10) and p57 (65021A) were obtained from Pharmingen (Richmond, CA).
Western Blot
Total cell lysates (20 μg) prepared in 120 mmol/L NaCl, 0.5% Nonidet P-40, 50 mmol/L TrisCl, pH 8.0, were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene fluoride membranes (Immobilon-P; Millipore, Bedford, MA) by semidry electrotransfer. Blots were rehydrated before immunodetection, and then were blocked in 5% skim milk dissolved in PBS with 0.1% Tween 20 (PBS-T) at room temperature for 1 hour. Primary and secondary antibody incubations were performed in PBS-T at room temperature for 1 hour. Goat anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies were used at 1:5,000 concentrations. Blots were immersed in enhanced chemiluminescence detection reagent (Amersham) for 1 minute; chemiluminescence was detected on Kodak X-OMAT AR film (Eastman Kodak, Rochester, NY).
Western blots for p57, p107, p130, E2F1, E2F2, and pRB were obtained at multiple time points during p57 induction for the different astrocytoma cell clones. Western blots for PARP and Bax levels were obtained 8 days after p57 induction.
Immunocytochemistry
Astrocytoma cells were seeded onto glass coverslips and maintained in medium with or without tetracycline for 1 to 10 days. For senescence-associated β-galactosidase (SA-β-gal) staining, coverslips were harvested, washed in PBS, and fixed with 2% formaldehyde/0.2% glutaraldehyde in PBS for 5 minutes at room temperature. SA-β-gal (pH 6.0) was detected as reported. 34 Coverslips were rinsed in PBS, counterstained with neutral fast red, rinsed with distilled water, and mounted onto microscope slides using mounting media (DAKO, Carpinteria, CA). In some experiments, tetracycline was added back to the medium on day 5 after p57 induction, and the cells were then harvested for SA-β-gal staining on days 5, 7, and 10.
Morphological Analysis
The morphology of control and p57-induced U 343 MG-A astrocytoma cells was observed using a Leitz (Fluovert FS; Leica Microsystems, Heerbrugg, Switzerland) light and immunofluorescence microscope. Induced morphological changes were observed for variable periods of time. In some experiment, p57 expression was repressed after induction at various time points by adding tetracycline to the medium. Cell morphology and culture conditions were then assessed by phase microscopy.
Fluorescence Microscopy
To identify cells undergoing apoptosis, astrocytoma cells were stained with Hoechst 33258 (Sigma) and quantified by fluorescent microscopic analysis. 35 Briefly, uninduced and p57-induced astrocytoma cells were trypsinized, centrifuged, washed with PBS, and resuspended with 1% glutaraldehyde for 30 minutes for fixation. The cells were rinsed again and stained with 1 μmol/L Hoechst 33258 for 10 minutes previous to viewing under the fluorescence microscope.
Electron Microscopy
Uninduced and p57-induced human astrocytoma cells were harvested and lightly pelleted before fixation in 2.5% paraformaldehyde in phosphate buffer for 2 to 4 hours. Pellets were rinsed thoroughly with phosphate buffer before being postfixed with phosphate-buffered osmium tetroxide for 1 hour. The cells were then dehydrated in an ascending series of ethanols and embedded in epon-araldite via propylene oxide. After polymerization, ultrathin sections were cut on a diamond knife using an ultramicrotome and mounted on grids. The grids were then stained with ethanolic uranyl acetate and lead citrate. All specimens were viewed and photographed in a transmission electron microscope (JEOL 1200EXII; JEOL Peabody, MA).
Results
Inducible Expression of p57 Leads to G1 Arrest and Changes in Cell Morphology
p57 was expressed under the control of the tetracycline operator (tetO) in U343, U87, and U373 astrocytoma cell lines constitutively expressing high levels of the tetracycline repressor protein (tetR)-VP16 fusion protein. Of the many clones screened for induction of p57 expression for each cell line, three clones (clone 9, U343C9; clone 2 U87C2; and clone 3, U373C3) showed tightly regulated, tetracycline-dependent expression of p57 (Figure 1) ▶ . Induction of p57 occurred within 24 hours for U343C9 and U373C3 astrocytoma cells, and by the third day for U87C2 cells (Figure 2) ▶ . High expression levels of p57 for all cell lines could be maintained for periods longer than 9 days after induction. The specimen of human fetal brain showed expression of p57, whereas the parental astrocytoma cell lines and specimen of nonneoplastic brain from a 14-year-old patient with epilepsy did not. No mutations in the p57 gene were observed from the heteroduplex DNA detection analysis in the three astrocytoma cell lines examined and in a panel of human astrocytoma specimens (data not shown)
Figure 1.

Western analysis of inducible expression of p57 in human astrocytoma cell lines. Before transfection with a human p57 cDNA, U343, U87, and U373 astrocytoma cell lines did not express p57. With tetracycline in the medium (+), p57-transfected astrocytoma cell clones did not express p57. However, after the removal of tetracycline from the medium (−), clones were identified with strong expression of p57 in the three astrocytoma cell lines. A specimen of nonneoplastic human brain does not express p57.
Figure 2.

Temporal analysis of p57 expression in astrocytoma cell clones in the uninduced and induced states. Without induction, there is no expression of p57 in U343 astrocytoma cells (uninduced). After induction, p57 is strongly expressed in U343C9 and U373C3 astrocytoma cell clones on day 1, and in U87C3 cells on day 3. High expression levels of p57 for all cell clones could be maintained for periods longer than 9 days after induction. A specimen of human fetal brain is shown to express p57.
To investigate whether induction of exogenous p57 can arrest the cell cycle, we performed a proliferation assay and FACS analysis for all three p57-transfected astrocytoma cell clones. Induction of p57 rapidly blocked proliferation of U343C9, U87C2, and U373C3 astrocytoma cells (Figure 3A) ▶ . FACS analysis revealed that p57-induced astrocytoma cells rapidly accumulate in G1 phase of the cell cycle (Figure 3B) ▶ . The differences in growth rates between the induced and uninduced astrocytoma cell clones were statistically significant, and indicate that the overproduction of p57 can functionally arrest the growth of astrocytoma cell lines in G1.
Figure 3.

Growth inhibitory effects of p57 expression on human astrocytoma cell clones. A: Induction of p57 causes a potent proliferative block in all astrocytoma cell clones as demonstrated by growth curve analysis. Open rectangles, uninduced astrocytoma cells; filled circles, p57-induced astrocytoma cells. Error bars show the SD of three separate counts for each data point. B: Flow cytometric analysis demonstrates that induction of p57 causes astrocytoma cells to accumulate in G1 phase of the cell cycle. Open rectangles, uninduced astrocytoma cells; filled circles, p57-induced astrocytoma cells. Error bars show the SD of the results of three separate FACS analyses for each astrocytoma cell clone.
Uninduced U343C9 cells have a tightly packed, cobblestone appearance and resemble the morphology of the parental U343 MG-A astrocytoma cell line. Induction of p57 causes the cells to become large, round, and flat with abundant cytoplasm containing perinuclear vacuoles (Figure 4) ▶ . These morphological changes are apparent by day 3, coincident with the G1 cell cycle arrest, and are very pronounced by day 5. Uninduced U87C2 cells are generally bipolar in configuration, much like the parental cell line from which they are derived. After p57 induction, U87C2 cells became large and triangular with markedly expanded cytoplasms (Figure 4) ▶ . U373C3 astrocytoma cells were round and flat with a ruffled peripheral plasma membrane before p57 induction. After p57 induction, these cells similarly developed an expanded cytoplasm, but maintained their overall round, flat shape (Figure 4) ▶ .
Figure 4.

Morphological alterations in human astrocytoma cell clones after the induction of p57, day 5. Uninduced U343C9 cells are characterized by their tightly packed, cobblestone appearance. The cells are cuboidal with a high nuclear:cytoplasmic ratio. After p57 expression, these cells become large, round, and flat with abundant cytoplasmic-containing perinuclear vacuoles. Uninduced U87C2 cells are bipolar in configuration. After p57 expression, these cells become large and frequently triangular in shape with a markedly expanded cytoplasm. U373C3 cells are characterized by being round and flat with a ruffled peripheral plasma membrane before p57 induction. After p57 expression, they also developed an expanded cytoplasm, but maintained their overall round, flat shape. Phase microscopy for all panels, ×350.
Induction of p57 Alters the Expression of Cell Cycle Regulatory Factors
To determine whether the induction of p57 was associated with alterations in the expression of downstream cell-cycle regulatory proteins, we next performed Western blot analyses of the pRB and E2F family proteins (Figure 5) ▶ . The expression levels of pRB and p107 were sharply reduced by day 3 after p57 induction, whereas p130 expression levels were unchanged. Expression of the E2F family proteins was also determined in p57-induced U343C9 cells. E2F-1 expression levels were repressed whereas no change in the levels of E2F-4 was observed.
Figure 5.

Expression of pRB- and E2F-family proteins in uninduced (right) and p57-induced (left) U87C2 human astrocytoma cells. Without induction of p57, expression levels of pRB- and E2F-family proteins are not significantly altered throughout a 5-day time interval. However, after induction of p57, decreased levels of pRB, p107, and E2F-1 are observed. pRB is shifted to a faster migrating, hypophosphorylated form. Levels of p130 and E2F-4 are unchanged in this analysis throughout the 5-day time interval.
Increased Expression of SA-β-gal in p57-Induced Astrocytoma Cells
Induction of p57 in U343C9, U87C2, and U373C3 astrocytoma cells led to the identification of SA-β-gal-positive cells (Figure 6) ▶ . The generation of SA-β-gal-positive astrocytoma cells began for each cell clone by day 1 after induction and peaked between 5 to 7 days at which time all cell clones had ∼75% SA-β-gal-positive cells (Figure 7) ▶ . The induction of SA-β-gal-positive cells through forced p57 expression was partially reversible as replenishment of the medium with tetracycline on day 5 led to fewer SA-β-gal-positive cells on days 7 and 10 (Table 1) ▶ .
Figure 6.

Induction of SA-β-gal-positive cells after p57 induction. Uninduced astrocytoma cell clones (left) demonstrated rare cells positive for the SA-β-gal marker. After induction of p57 (day 5), the majority of astrocytoma cells are seen to be positive for SA-β-gal (right). Phase microscopy for U87C2 and U373C3 p57-induced cell panels, ×250; phase microscopy for all other panels, ×125.
Figure 7.
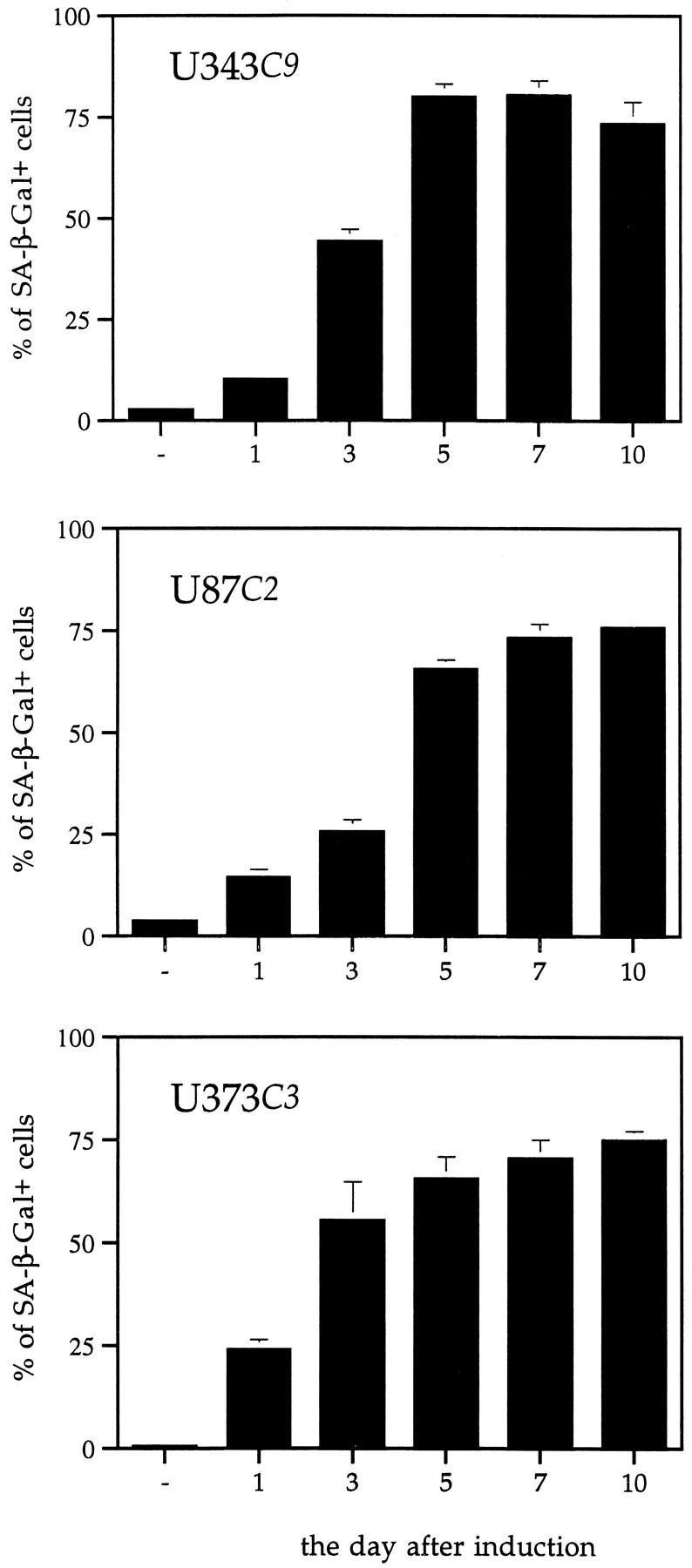
Generation of SA-β-gal-positive cells over time after p57 induction. Uninduced astrocytoma cells have rare SA-β-gal-positive cells. After p57 induction, there is an increase in the number of SA-β-gal-positive astrocytoma cells throughout time such that by day 7 ∼75% of p57-induced cells express the marker.
Table 1.
Percent SA-β-Gal-Positive U343 Astrocytoma Cells
| Day 5 | Day 7 | Day 10 | |
|---|---|---|---|
| p57-uninduced, tetracycline placed in medium on day 1 | 1.5 ± 0.8 | 3.4 ± 1.6 | 3.4 ± 1.2 |
| p57-induced, tetracycline removed from medium on day 1 | 80.0 ± 3.2 | 80.4 ± 3.4 | 73.3 ± 5.4 |
| p57-uninduced, tetracycline added back to medium 5 days after p57 induction | 77 ± 4.2 | 38.7 ± 7.1 | 18.7 ± 2.3 |
The induction of p57 expression in U343 astrocytoma cells led to the generation of a high percentage of cells which stained positively for SA-β-gal when compared to p57-uninduced cells. When tetracycline was added back to the medium on day 5, the percentage of cells which stained positively for SA-β-gal decreased suggesting that the senescent state was at least partially reversible.
Response of Astrocytoma Cells to p57 Induction
After p57 induction, we detected a statistically significant decline in viable cells in U343C9 and U373C3 astrocytoma cells as determined by trypan blue dye exclusion from days 3 to 7 (Figure 8) ▶ . p57 induction had a greater effect on reducing the number of viable cells among U373C3 astrocytoma cells than among U343C9. No such decline in cell viability was apparent for U87C2 astrocytoma cells.
Figure 8.
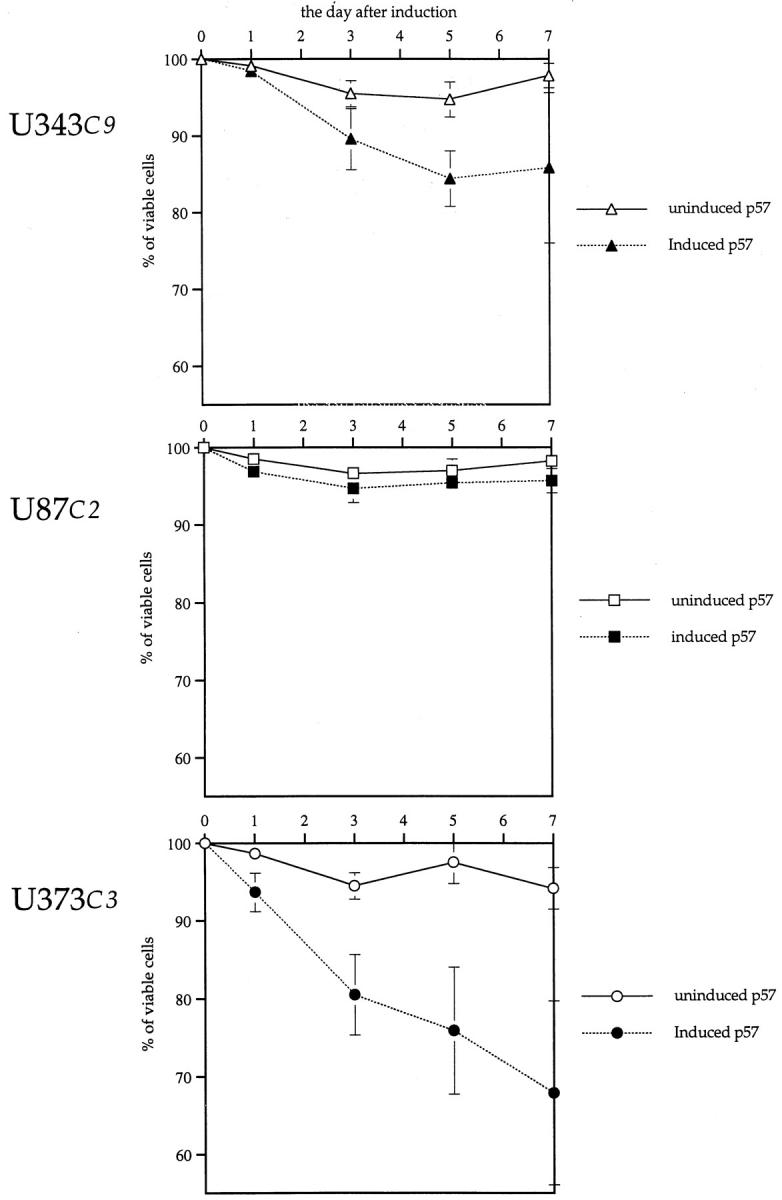
Viability of human astrocytoma cells after p57 induction. After p57 induction, U343C9 and U373C3 astrocytoma cells responded by having diminished numbers of viable cells as determined by trypan blue dye exclusion when compared to uninduced cells. U87C2 astrocytoma cells showed no loss in cell viability after p57 induction.
Hoechst 33258 staining of all three astrocytoma clones revealed rare abnormalities in nuclear configuration among U87C2 and U343C9 cells, but multiple fragmented micronuclei for U373C3 astrocytoma cells induced to express p57 (Figure 9) ▶ . Approximately 15% of U373C3 nuclei were abnormal after Hoechst 33258 staining of p57-induced cells (Figure 10) ▶ . Electron microscopy of p57-induced and uninduced astrocytoma cells confirmed the findings of the Hoechst 33258 staining analysis. Unlike p57-induced U87C2 and U343C9 astrocytoma cells which showed a preponderance of cells with normal ultrastructural features, many p57-induced U373C3 cells were characterized by convolution of nuclear and cytoplasmic outlines, apoptotic bodies, and compaction and margination of nuclear chromatin (Figure 11) ▶ .
Figure 9.
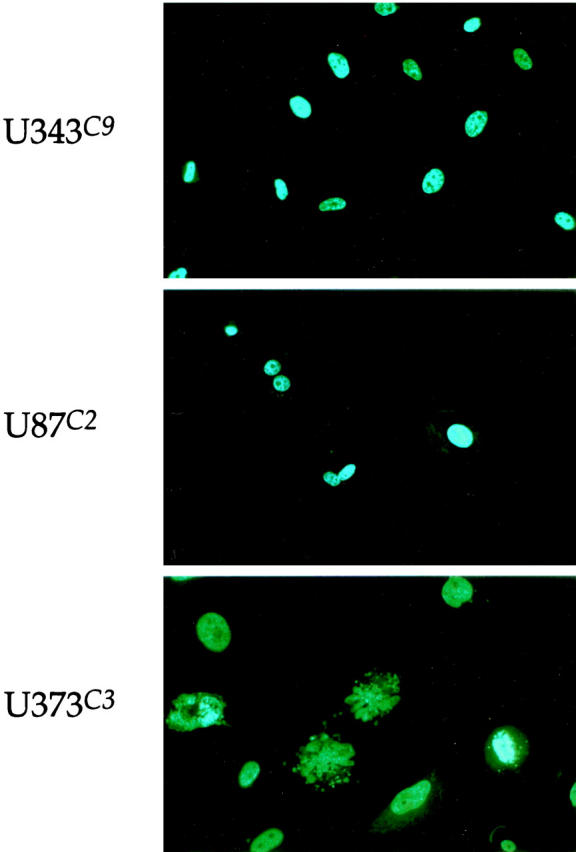
Hoechst 33258 staining of p57-induced human astrocytoma cell clones, day 5. Nuclear integrity was unaltered in U343C9 and U373C2 astrocytoma cells. However, U373C3 cells were characterized by exhibiting fragmentation of the nucleus in ∼15% of cells. Fluorescence microscopy, ×200.
Figure 10.

Number of cells demonstrating micronuclear fragmentation by Hoechst 33258 staining. Unlike U343C9 and U87C2 astrocytoma cell clones which showed no increase in numbers of cells with nuclear fragmentation after p57 induction, ∼15% of U373C3 astrocytoma cells demonstrated micronuclear fragmentation.
Figure 11.
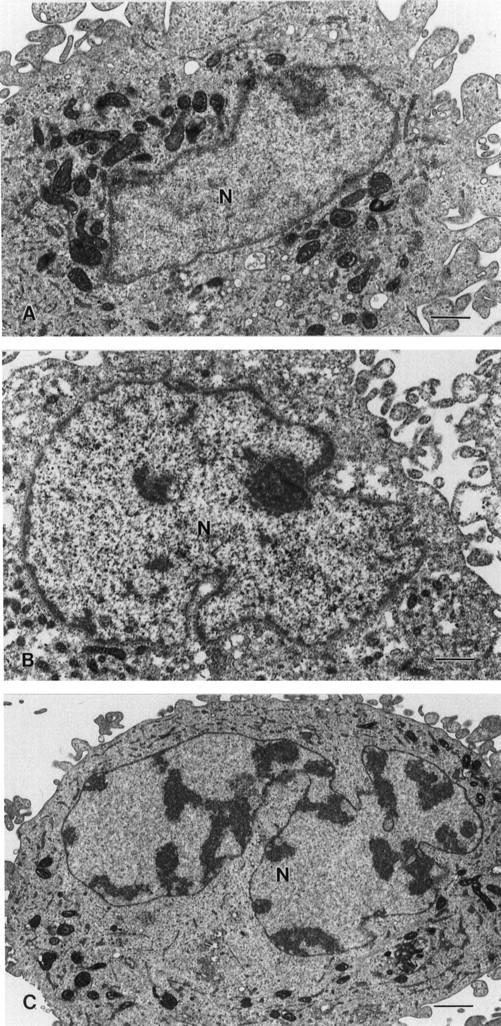
Ultrastructural features of p57-induced human astrocytoma cells. A: Typical nucleus (N) from U343C9 cells. B: Typical nucleus (N) from U87C2 cells. C: Apoptotic nucleus (N) from U373C3 cells. Approximately 15% of these cells were found to contain nuclei similar to this with dense peripheral chromatin and bizarre shapes. Scale bars, 1 μm.
Western analysis of uninduced and p57-induced astrocytoma clones for Bax (Figure 12) ▶ revealed no change in expression levels. However, induction of p57 among astrocytoma cell clones led to diminished PARP expression (Figure 12) ▶ . The reduction in PARP expression was particularly marked for U373C3 cells.
Figure 12.

Western analysis for PARP and Bax in uninduced and p57-induced astrocytoma cell clones. There is no change in Bax expression with p57 induction. After p57 induction, PARP levels are observed to decrease. This is most marked for U373C3 astrocytoma cells. No cleavage products for PARP were observed.
Discussion
We have shown that the inducible expression of p57 in three different permanent astrocytoma cell lines has a profound effect on their proliferation and morphology. Within 3 days after induction of p57, U343C9, U87C2, and U373C3 astrocytoma cells were rapidly growth arrested and accumulated in G1 phase of the cell cycle. After the induction of p57, pRB and the pRB family protein, p107, are diminished; pRB is shifted to a faster migrating, hypophosphorylated form. Likewise, E2F-1 is repressed whereas E2F-4 levels are unchanged after induction of p57. The inducible expression of p57 in all three astrocytoma cell lines led to the identification of SA-β-Gal-positive cells which accumulated during the first week of induction such that ∼75% of all cells in culture were positive. Although equivalent numbers of cells expressed this marker of cell senescence in each astrocytoma cell line, interestingly a population of U373C3 astrocytoma cells responded to p57 induction by following an apoptotic pathway as determined by Hoescht 33258 staining and electron microscopy.
Previous studies have shown that p57 is a gene which undergoes genomic imprinting. Genomic imprinting is a process that results in the expression of only one allele of a gene depending on its parental origin. As such, it is thought to play an important role in embryonal development. Genomic imprinting may also play a role in certain pediatric tumors such as Wilms’ tumor as a highly selective loss of maternal alleles for the Wilm’s tumor gene is found not uncommonly in this tumor type. 36 In fact, a potential role for p57 in tumorigenicity was postulated on the basis of its chromosomal mapping to 11p15, a region which frequently demonstrates loss of heterozygosity in a number of common cancers in adults such as lung, breast, and bladder carcinoma, as well as Wilms’ tumor in children. 10-16,37 Not all attempts to manipulate p57 expression levels have resulted in tumor suppression or formation. For example, p57−/− transgenic mice have delayed differentiation without tumor formation. 38,39 Although a previous report suggested that germline deletions in the proline-alanine-rich (PAPA-repeat) region of p57 are associated with increased risk of a variety of cancers, including breast cancer, a recent report by Li et al 40 failed to substantiate this observation in the context of breast cancer patients. In our study, p57 expression was documented within human fetal brain. However, none of the astrocytoma cell lines expressed p57, and none contained mutations in the p57 gene. Absence of p57 mutations in the human astrocytomas examined here and within a variety of other cancer types 30,40,41 suggest that other mechanisms of transcriptional or posttranslational silencing must be involved in the loss of p57 protein expression in astrocytic tumors and other cancers. Two possible mechanisms of gene inactivation include methylation in the promoter region of the p57 gene and histone deacetylation. Recently, Shin et al 41 demonstrated that formation of inactive chromatin through histone deacetylation is a general mechanism for inactivation of both p21 and p57 genes in gastric cancer cells, and that methylation of the promoter region of the p57 gene occurred in five of eight gastric cancer cell lines as an alternative pathway for inactivation of p57.
It is becoming clear that a common feature of cancer cells is the abrogation of cell-cycle checkpoints, either by aberrant expression of positive regulators such as cyclins and CDKs, or the loss of negative regulators, including CKIs and pRB. It has previously been suggested that transformation of glial cells into malignant astrocytic tumors also involves significant dysfunction of this cell cycle-control machinery. 1,2 Pedram et al 42 have shown that fetal rat diencephalic astrocytes can be stimulated to progress through G1/S phase by the endogenous neuropeptide, endothelin-3. Atrial natriuretic peptide was inhibitory for cell proliferation, and induced the expression of p57 among other CKIs. In fact, multiple CKIs were shown to be necessary to restrain cell-cycle progression in astrocytes, an observation that may have relevance for inhibition of human astrocytoma cells.
In human astrocytic tumors, many reports have demonstrated alterations in the expression and activity of cell-cycle regulatory proteins, especially the loss of p16 expression. 43-48 We have previously shown that induction of p16 leads to a cell-cycle block in the U343 human astrocytoma cell line. 49,50 To our knowledge, however, alterations in the p57-cyclin/CDK complexes-pRB/E2F pathway have not been described previously in malignant astrocytomas. Our data demonstrate for the first time that induction of p57 in p57-negative human astrocytoma cell lines can potently block the proliferation and alter the morphology of three different human astrocytoma cell lines. This p57-induced cell-cycle arrest is accompanied by alterations in the expression and activity of a number of cell-cycle regulatory proteins.
As examples, our data demonstrate the presence of a potential autoregulatory mechanism which may control the levels of expression of members of the pRB/E2F family proteins. It has been shown that both pRB and p107 have E2F binding sites in their promoter regions, and E2F-1 also contains E2F binding sites in its promoter region. pRB phosphorylation is critical for control of cell-cycle progression in G1. 51,52 After pRB phosphorylation, E2F, freed from the repressive effects of pRB family proteins, are then able to activate transcription of genes required for S phase progression. 52-54 It seems reasonable that pRB-E2F-1 complexes repress activated transcription from the pRB and p107 promoters. The repression of these factors suggests a model where initially, pRB and E2F family proteins form complexes that aggressively block transcription of factors required for cell-cycle progression. These same complexes are also responsible for inhibiting expression of the pRB and E2F family proteins, loss of the latter ensuring that E2F-dependent cell-cycle progression is not possible. Other investigators have previously demonstrated that different pRB family proteins show distinct binding specificities for different E2F family proteins. 50,55-61 pRB seems to associate specifically with E2F-1, E2F-2, and E2F-3, 59 whereas p107 binds E2F-4 56,61 and p130 binds E2F-4 and E2F-5. 58,61 In p57-induced, growth-arrested U343 cells, the expression of pRB and E2F-1 is repressed, and pRB becomes quantitatively hypophosphorylated.
We also demonstrate here that p57-induced astrocytoma cells undergo a change in morphology—cells becoming large and flat and having abundant cytoplasm. This morphological change is reminiscent of the phenotype exhibited by human osteosarcoma cell line, SAOS-2, after exogenous expression of pRB. 16,30,62-64 In the presence of pRB, SAOS-2 cells become flat and round with a greatly expanded cytoplasm in their growth-arrested state. These flat cells resemble senescent primary fibroblasts after extended in vitro passage. In a report by Uhrbom et al, 34 ∼40% of U1242 MG astrocytoma cells induced to express p16 developed a senescent cell phenotype at 7 days as determined by SA-β-gal staining. In our study, ∼75% of astrocytoma cells were SA-β-gal-positive 7 days after p57 induction.
Cellular or replicative senescence is a state of permanent growth arrest and altered cell function after a finite number of cell divisions. Cellular senescence is thought to be a tumor suppressive mechanism, and a contributing factor in aging. 65,66 Three features distinguish senescent from presenescent cells: a block-to-cell proliferation, increased resistance to apoptotic death, and changes in differentiated functions. 65 Although our data suggest that inducible p57 expression causes a cell senescent phenotype among astrocytoma cells, the effect of p57 on the generation of SA-β-gal cells was at least partially reversible. Of the several growth regulatory transcriptional modulators known to be repressed in senescent cells, the repression of E2F-1 after p57 induction holds particular significance in our study on human astrocytoma cells.
Although senescent cells are thought to be resistant to apoptotic cell death, one of the astrocytoma cell lines induced to express p57, U373C3, exhibited a population of cells which underwent apoptosis as determined by Hoechst 33258 staining for micronuclear fragmentation and electron microscopy. Why this particular cell line responded in a different manner to p57 induction is an interesting yet unanswered question. Apoptosis is a genetically encoded cell death program defined by typical morphological and biochemical changes. 67 Although ultrastructural characterization of nuclear and plasma membrane alterations remains one of the most important determinants of apoptosis, Hoechst 33258 staining for fragmented nuclei has also been used in a number of studies. 68-74 Apoptosis has been shown to be induced in human astrocytomas after ionizing radiation and treatment with DNA-damaging agents. 75-79 Several apoptosis-related molecules are involved in astrocytoma cell death including bcl-2, interleukin-1-β-converting enzyme, and p53. 80-85
Interestingly, of the three astrocytoma cell lines examined in this study, U87 and U343 express wild-type p53 whereas U373 is mutant for p53. 26,27,80-86 As there have been several cancer cell systems in which apoptosis has been demonstrated in p53-inactivated cells, 87-96 it is conceivable that the apoptosis observed in U373C3 cells after p57 induction occurs in a p53-independent manner. We showed that Bax levels were unchanged but PARP levels were decreased without cleavage formation after p57 induction in all cell clones. In response to DNA damage, PARP activity increases, resulting in poly-(ADP) ribosylation of many nuclear proteins, including PARP itself. In cells which have become activated to undergo apoptosis, the 116-kd PARP protein becomes cleaved by caspase-3 producing 85-kd and 25-kd fragments and resulting in loss of normal PARP function. 97,98 Although we did not observe cleaved fragments of PARP in any of the astrocytoma cell clones examined here, down-regulation of PARP without cleavage product formation was observed for U343C9, U87C2, and U373C3 astrocytoma cells. This phenomenon has been described previously in replicatively senescent fibroblasts. 99 Our inability to detect cleaved PARP fragments in U373C3 in particular may relate to the small number of cells that are undergoing apoptosis compared to the large number of cells that continue to express the senescent cell phenotype.
In summary, we have shown that inducible expression of p57 in three different astrocytoma cell lines is a strong stimulus against cell proliferation and for cell senescence. Ongoing studies in the laboratory are attempting to determine precisely what role p57, in conjunction with other CKIs, may have in inhibiting the growth of human astrocytic tumors in vivo.
Acknowledgments
We thank Dr. S. J. Elledge for the gift of human p57 cDNA.
Footnotes
Address reprint requests to James T. Rutka, M.D., The Division of Neurosurgery, Suite 1502, The Hospital for Sick Children, 555 University Ave., Toronto, Ontario, Canada M5G 1X8. E-mail: rutka@sickkids.on.ca.
Supported by a grant from the National Cancer Institute of Canada (to J. T. R. and P. A. H.), and Brainchild. P. B. D. was a fellow of the Medical Research Council of Canada. J. T. R. is recipient of a Scientist Award from the Medical Research Council of Canada.
References
- 1.Dirks PB, Murakami M, Hubbard SL, Rutka JT: Cyclins and cyclin-dependent kinase expression in human astrocytoma cell lines. J Neuropathol Exp Neurol 1997, 56:291-300 [DOI] [PubMed] [Google Scholar]
- 2.Dirks PB, Rutka JT: Current concepts in neuro-oncology. The cell cycle—a review. Neurosurgery 1997, 40:1000-1015 [DOI] [PubMed] [Google Scholar]
- 3.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogetstein B: WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75:817-825 [DOI] [PubMed] [Google Scholar]
- 4.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D: p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366:701-704 [DOI] [PubMed] [Google Scholar]
- 5.Polyak K, Lee M-H, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagne J: Cloning of p21Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 8:59-66 [DOI] [PubMed] [Google Scholar]
- 6.Toyoshima H, Hunter T: p27, a novel inhibitor of G1 cyclin-cdk protein kinase activity, is related to p21. Cell 1994, 78:67-74 [DOI] [PubMed] [Google Scholar]
- 7.Lee MP, DeBaun M, Randhawa G, Reichard BA, Elledge SJ, Feinberg AP: Low frequency of p57KIP2 mutations in Beckwith-Wiedemann syndrome. Am J Hum Genet 1997, 61:304-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee M-H, Reynisdottir I, Massague J: Cloning of p57kip2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev 1995, 9:639-649 [DOI] [PubMed] [Google Scholar]
- 9.Luo Y, Hurwitz J, Massague J: Cell cycle inhibition by independent CDK and PCNA binding domains in p21cip1. Nature 1995, 375:159-161 [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ: p57kip2, a structurally distinct member of the p21cip1 Cdk inhibitory family, is a candidate tumour suppressor gene. Genes Dev 1995, 9:650-662 [DOI] [PubMed] [Google Scholar]
- 11.Hatada I, Mukai T: Genomic imprinting of p57kip2, a cyclin-dependent kinase inhibitor in mouse. Nat Genet 1995, 11:204-206 [DOI] [PubMed] [Google Scholar]
- 12.Kondo M, Matsuoka S, Uchida K, Osada H, Nagatake M, Takagi K, Harper JW, Takahashi T, Elledge SJ, Takahashi T: Selective maternal-allele loss in human lung cancers of the maternally expressed p57kip2 gene at 11p15.5. Oncogene 1996, 12:1365-1368 [PubMed] [Google Scholar]
- 13.Orlow I, Iavarone A, Crider-Miller SJ, Bonilla F, Latres E, Lee M-H, Gerald WL, Massague J, Weissman BE, Cordon-Cardo C: Cyclin-dependent kinase inhibitor p57kip2 in soft tissue sarcomas and Wilms’ tumor. Cancer Res 1996, 56:1219-1221 [PubMed] [Google Scholar]
- 14.Reid LH, Crider-Miller SJ, West A, Lee M-H, Massague J, Weissmann BE: Genomic organization of the human p57kip2 gene and its analysis in the G401 Wilms’ tumor assay. Cancer Res 1996, 56:1214-1218 [PubMed] [Google Scholar]
- 15.Taniguchi T, Okamoto K, Reeve AE: Human p57kip2 defines a new imprinted domain on chromosome 11p but is not a tumor suppressor gene in Wilms’ tumor. Oncogene 1997, 14:1201-1206 [DOI] [PubMed] [Google Scholar]
- 16.Thompson JS, Reese KJ, DeBaun MR, Perlman EJ, Feinberg AP: Reduced expression of the cyclin-dependent kinase inhibitor gene p57kip2 in Wilms’ tumor. Cancer Res 1996, 56:5723-5727 [PubMed] [Google Scholar]
- 17.Watanabe H, Pan ZQ, Schreiber-Agus N, DePinho RA, Hurwitz J, Xiong Y: Suppression of cell transformation by the cyclin-dependent kinase inhibitor p57KIP2 requires binding to proliferating cell nuclear antigen. Proc Natl Acad Sci USA 1998, 95:1392-1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashimoto Y, Kohri K, Kaneko Y, Morisaki H, Kato T, Ikeda K, Nakanishi M: Critical role for the 310 helix region of p57 (kip2) in cyclin-dependent kinase 2 inhibition and growth suppression. J Biol Chem 1998, 273:16544-16550 [DOI] [PubMed] [Google Scholar]
- 19.Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ: Cooperation between Cdk inhibitors p27 (KIP1) and p57 (KIP2) in the control of tissue growth and development. Genes Dev 1998, 12:3162-3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ: p21 (CIP1) and p57 (KIP2) control muscle differentiation at the myogenin step. Genes Dev 1999, 13:213-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nijjar T, Wiginton D, Garbe JC, Waha A, Stampfer MR, Yaswen P: p57KIP2 expression and loss of heterozygosity during immortal conversion of cultured human mammary epithelial cells. Cancer Res 1999, 59:5112-5118 [PubMed] [Google Scholar]
- 22.Ponten J, Westermark B: Properties of human malignant glioma cells in vitro. Med Biol 1978, 56:184-193 [PubMed] [Google Scholar]
- 23.Westermark B, Ponten J, Hugosson R: Determinants for the establishment of permanent tissue culture lines from human gliomas. Acta Pathol Microbiol Scand 1973, 81:791-805 [DOI] [PubMed] [Google Scholar]
- 24.Carlsson J, Nilsson K, Westermark B: Formation and growth of multicellular spheroids of human origin. Int J Cancer 1983, 31:523-533 [DOI] [PubMed] [Google Scholar]
- 25.Martuza RL, Malick A, Makert JM, Ruffner KL, Coen DM: Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252:854-856 [DOI] [PubMed] [Google Scholar]
- 26.Asai A, Miyagi Y, Sugiyama A, Gamanuma M, Hong SH, Takamoto S, Nomura K, Matsutarri M, Takakura K, Kuchino Y: Negative effects of wild-type p53 and s-Myc on cellular growth and tumorigenicity of glioma cells. Implication of the tumor suppressor genes for gene therapy. J Neurooncol 1994, 19:259-268 [DOI] [PubMed] [Google Scholar]
- 27.Badie B, Goh CS, Herweigjer H, Boothman DA: Combined radiation and p53 gene therapy of malignant glioma cells. Cancer Gene Ther 1999, 6:155-162 [DOI] [PubMed] [Google Scholar]
- 28.Costanzi-Strauss E, Strauss BE, Naviaux RK, Haas M: Restoration of growth arrest by p16INK4, p21WAF1, pRB, and p53 is dependent on the integrity of the endogenous cell-cycle control pathways in human glioblastoma cell lines. Exp Cell Res 1998, 238:51-62 [DOI] [PubMed] [Google Scholar]
- 29.Rutka JT, Giblin JR, Balkissoon R, Wen D, Myatt C, McCulloch JR, Rosenblum ML: Characterization of fetal human brain cultures. Development of a potential model for selectively purifying human glial cells in culture. Dev Neurosci 1987, 9:154-173 [DOI] [PubMed] [Google Scholar]
- 30.O’Keefe D, Dao D, Zhao L, Sanderson R, Warburton D, Weiss L, Anyane-Yeoboa K, Tycko B: Coding mutations in p57KIP2 are present in some cases of Beckwith Wiedemann syndrome but are rare or absent in Wilm’s tumors. Am J Hum Genet 1997, 61:295-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gossen M, Bujard H: Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 1992, 89:5547-5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rutka JT, Giblin JR, Dougherty DV, Liu HC, McCulloch JR, Bell CW, Stern RS, Wilson CB, Rosenblum ML: Establishment and characterization of five cell lines derived from human malignant gliomas. Acta Neuropathol 1987, 75:92-103 [DOI] [PubMed] [Google Scholar]
- 33.Rutka JT, De Armond SJ, Gilbin J, McCulloch JR, Wilson CB, Rosenblum ML: Effect of retinoids on the proliferation, morphology and expression of glial fibrillary acidic protein of an anaplastic astrocytoma cell line. Int J Cancer 1988, 42:419-427 [DOI] [PubMed] [Google Scholar]
- 34.Uhrbom L, Nister M, Westermark B: Induction of senescence in human malignant glioma cells by p16INK4A. Oncogene 1997, 15:505-514 [DOI] [PubMed] [Google Scholar]
- 35.Watanabe-Fukunaga R, Brainnan CI, Copeland NG, Jenkins NA, Nagata F: Lymphoproliferation disorder in mice explained by defects in FAS antigen that mediates apoptosis. Nature 1992, 356:314-317 [DOI] [PubMed] [Google Scholar]
- 36.Reeve AE, Sih SA, Raizis AM, Feinberg AP: Loss of heterozygosity at a second locus on chromosome 11 in sporadic Wilms’ tumor cells. Mol Cell Biol 1989, 9:1799-1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuoka S, Thompson JS, Edwards MC, Barletta JM, Grundy P, Kalikin LM, Harper JW, Elledge SJ, Feinberg AP: Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Natl Acad Sci USA 1996, 93:3026-3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Y, Frisen J, Lee MH, Massague J, Barbacid M: Ablation of the CDK inhibitor p57kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 1997, 11:973-983 [DOI] [PubMed] [Google Scholar]
- 39.Zhang P, Liegeois NJ, Wong C, Finegold M: Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997, 387:151-158 [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Millikan RC, Newman B, Conway K, Tse CK, Liu ET: p57 (KIP2) polymorphisms and breast cancer risk. Hum Genet 1999, 104:83-88 [DOI] [PubMed] [Google Scholar]
- 41.Shin JY, Kim HS, Park J, Park JB, Lee JY: Mechanism for inactivation of the KIP family cyclin-dependent kinase inhibitor genes in gastric cancer cells. Cancer Res 2000, 60:262-265 [PubMed] [Google Scholar]
- 42.Pedram A, Razandi M, Hu RM, Levin ER: Astrocyte progression from G1 to S phase of the cell cycle depends upon multiple protein interaction. J Biol Chem 1998, 273:13966-13972 [DOI] [PubMed] [Google Scholar]
- 43.Giani C, Finocchiaro G: Mutation rate of CDKN2 gene in malignant glioma. Cancer Res 1994, 54:6338-6339 [PubMed] [Google Scholar]
- 44.He J, Allen JR, Collins VP: CDK4 amplification is an alternative mechanism to p16 homozygous deletion in glioma cell lines. Cancer Res 1994, 54:5804-5807 [PubMed] [Google Scholar]
- 45.Jen JJ, Harper JW, Bigner D, Papadopoulis N, Markowitz S, Wilson J, Keyler K, Vogelstein B: Deletion of p16 and p15 in brain tumors. Cancer Res 1994, 54:6353-6358 [PubMed] [Google Scholar]
- 46.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tartigian SV, Stockert E, Day RD, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in the genesis of many tumor types. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 47.Nishikawa R, Furnari FB, Lin H, Arap W, Berger MS, Cavenee WK: Loss of p16 expression is frequent in high grade glioma. Cancer Res 1995, 55:1941-1945 [PubMed] [Google Scholar]
- 48.Sonoda Y, Yoshimoto T, Sekiya T: Homozygous deletion of the MTS1/p16 and MTS2/p15 genes and amplification of the CDK4 gene in glioma. Oncogene 1995, 11:2145-2149 [PubMed] [Google Scholar]
- 49.Dirks PB, Patel K, Hubbard SL, Ackerley C, Hamel PA, Rutka JT: Retinoic acid and cyclin-dependent kinase inhibitors synergistically alter proliferation and morphology of U343 astrocytoma cells. Oncogene 1997, 15:2037-2048 [DOI] [PubMed] [Google Scholar]
- 50.Dirks PB, Rutka JT, Hubbard SL, Mondal S, Hamel PA: The E2F-family proteins induce distinct cell cycle regulatory factors in p16-arrested, U343 astrocytoma cells. Oncogene 1998, 17:867-876 [DOI] [PubMed] [Google Scholar]
- 51.Ewen ME: The cell cycle and the retinoblastoma protein family. Cancer Metastasis Rev 1994, 13:45-66 [DOI] [PubMed] [Google Scholar]
- 52.Weinberg RA: The retinoblastoma protein and cell cycle control. Cell 1995, 81:323-330 [DOI] [PubMed] [Google Scholar]
- 53.Beijersbergen RL, Carlee L, Kerkhoven RM, Bernards R: Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev 1995, 9:1340-1353 [DOI] [PubMed] [Google Scholar]
- 54.Beijersbergen RL, Bernards R: Cell cycle regulation by the retinoblastoma family of growth inhibitor proteins. Biochem Biophys Acta 1996, 1287:103-120 [DOI] [PubMed] [Google Scholar]
- 55.DeGregori J, Kowalik T, Nevins JR: Cellular targets for activation by the E2F1 transcription factor include DNA synthesis and G1/S regulatory genes. Mol Cell Biol 1995, 15:4215-4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ginsberg D, Vairo G, Chittenden T, Yiao Z-X, Xu G, Wydner KL, DeCaprio JA, Lawrence JB, Livingston DM: E2F-4, a new member of E2F transcription factor family, interacts with p107. Genes Dev 1994, 8:2665-2679 [DOI] [PubMed] [Google Scholar]
- 57.Hiebert SW, Chellappan SP, Horowitz JM, Nevins JR: The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev 1992, 6:177-185 [DOI] [PubMed] [Google Scholar]
- 58.Hijmans EM, Voorhoeve PM, Beijersbergen RL, Van’t Veer LJ, Bernards R: E2F-5, a new E2F family member that interacts with p130 in vivo. Mol Cell Biol 1995, 15:3082–3089 [DOI] [PMC free article] [PubMed]
- 59.Lees JA, Saito M, Vidal M, Valentine M, Loo KT, Harlow E, Dyson N, Helin K: The retinoblastoma protein binds to a family of E2F transcription factors. Mol Cell Biol 1993, 13:7813-7825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shirodkar S, Ewen M, DeCaprio JA, Morgan J, Livingston DM, Chittenden T: The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell 1992, 68:157-166 [DOI] [PubMed] [Google Scholar]
- 61.Vairo G, Livingston DM, Ginsberg D: Functional interaction between E2F-4 and p130: evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev 1995, 9:869-881 [DOI] [PubMed] [Google Scholar]
- 62.Huang H-JS, Yee J-K, Shew J-Y, Chen P-L, Chen P-L, Bookstein R, Friedmann T, Lee E Y-HP, Lee WH: Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 1988, 242:1563–1566 [DOI] [PubMed]
- 63.Qian Y, Luckey C, Horton L, Esser M, Templeton DJ: Biological function of the retinoblastoma protein requires distinct domains for hyperphosphorylation and transcription factor binding. Mol Cell Biol 1992, 12:5363-5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qin X-Q, Chittenden T, Livingston DM, Kaelin WGJ: Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev 1992, 6:953-964 [DOI] [PubMed] [Google Scholar]
- 65.Dimri GP, Testori A, Acosta M, Campisi J: Replicative senescence, aging, and growth-regulatory transcription factors. Biol Signals 1996, 5:154-162 [DOI] [PubMed] [Google Scholar]
- 66.Dimri GP, Lee X, Basile G, Acosta M, Scott HG, Roskelley C, Medrano EE, Linskens M, Rubeij I, Pereira-Smith O: A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995, 92:9363-9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kerr JFR, Winterford CM, Harmon BV: Apoptosis: its significance in cancer and cancer therapy. Cancer 1994, 73:2013-2026 [DOI] [PubMed] [Google Scholar]
- 68.Ahlemeyer B, Kreiglstein J: Retinoic acid reduces staurosporine-induced apoptotic damage in chick embryonic neurons by suppressing reactive oxygen species production. Neurosci Lett 1998, 246:93-96 [DOI] [PubMed] [Google Scholar]
- 69.Boix J, Llecha N, Yuste VJ, Comella JX: Characterization of the cell death process induced by staurosporine in human neuroblastoma cell lines. Neuropharmacology 1997, 36:811-821 [DOI] [PubMed] [Google Scholar]
- 70.Iyer S, Chaplin DJ, Rosenthal DS, Boulares AH, Li LY, Smulson ME: Induction of apoptosis in proliferating human endothelial cells by the tumor-specific antiangiogenesis agent coombretastatin A-4. Cancer Res 1998, 58:4510-4514 [PubMed] [Google Scholar]
- 71.Keane RW, Srinivasan A, Foster LM: Activation of CPP32 during apoptosis of neurons and astrocytes. J Neurosci Res 1997, 48:168-180 [DOI] [PubMed] [Google Scholar]
- 72.Simm A, Bertsch G, Frank H, Zimmerman U, Hoppe J: Cell death of AKR-2B fibroblasts after serum removal: a process between apoptosis and necrosis. J Cell Sci 1997, 110:819-828 [DOI] [PubMed] [Google Scholar]
- 73.Taniwaki T, Yamada T, Asahara H, Ohyagi Y, Kira J: Ceramide induces apoptosis in immature cerebellar granule cells in culture. Neurochem Res 1999, 24:685-690 [DOI] [PubMed] [Google Scholar]
- 74.Yan Q, Sage EH: Transforming growth factor-beta1 induces apoptotic cell death in cultured retinal endothelial cells but not pericytes: association with decreased expression of p21waf1/cip1. J Cell Biochem 1998, 70:70-83 [PubMed] [Google Scholar]
- 75.Hueber A, Winter S, Weller M: Chemotherapy primes malignant glioma cells for CD95 ligand-induced apoptosis up-stream of caspase 3 activation. Eur J Pharmacol 1998, 352:111-115 [DOI] [PubMed] [Google Scholar]
- 76.Kondo S, Yin D, Morimura T, Takeuchi J: Combination therapy with cisplatin and nifedipine inducing apoptosis in multidrug-resistant human glioblastoma cells. J Neurosurg 1995, 82:469-474 [DOI] [PubMed] [Google Scholar]
- 77.Kondo S, Barna BP, Morimura T: Interleukin-1 beta-converting enzyme mediates cisplatin-induced apoptosis in malignant glioma cells. Cancer Res 1995, 55:6166-6171 [PubMed] [Google Scholar]
- 78.Raaphorst GP, Mao J, Yang H, Goel R, Niknafs B, Shirazi FH, Yazdi HM, Rippstein P, Ng CE: Evaluation of apoptosis in four human tumour cell lines with differing sensitivities to cisplatin. Anticancer Res 1998, 18:2945-2951 [PubMed] [Google Scholar]
- 79.Yount GL, Haas KDA, Levine KS, Aldape KD, Israel MA: Ionizing radiation inhibits chemotherapy-induced apoptosis in cultured glioma cells: implications for combined modality therapy. Cancer Res 1998, 58:3819-3825 [PubMed] [Google Scholar]
- 80.Alderson LM, Castleberg RL, Harsh GRt, Louis DN: Human gliomas with wild-type p53 express bcl-2. Cancer Res 1995, 55:999–1001 [PubMed]
- 81.Das R, Reddy EP, Chatterjee D, Andrews DW: Identification of a novel Bcl-2 related gene, BRAG-1, in human glioma. Oncogene 1996, 12:947-951 [PubMed] [Google Scholar]
- 82.Krishna M, Smith TW, Recht LD: Expression of bcl-2 in reactive and neoplastic astrocytes: lack of correlation with presence or degree of malignancy. J Neurosurg 1995, 83:1017-1022 [DOI] [PubMed] [Google Scholar]
- 83.Lang FF, Yung WK, Raju U, Libunao F, Terry NH: Enhancement of radiosensitivity of wild-type p53 human glioma cells by adenovirus-mediated delivery of the p53 gene. J Neurosurg 1998, 89:125-132 [DOI] [PubMed] [Google Scholar]
- 84.Schlapbach R, Fontana A: Differential activity of bcl-2 and ICE enzyme family protease inhibitors on Fas and puromycin-induced apoptosis of glioma cells. Biochim Biophys Acta 1997, 1359:174-180 [DOI] [PubMed] [Google Scholar]
- 85.Weller M, Frei K, Groscurth P: Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells. Induction and modulation of sensitivity by cytokines. J Clin Invest 1994, 94:954-964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costanzi-Strauss E, Strauss BE, Naviaux RK, Haas M: Restoration of growth arrest by p16, p21, pRB and p53 is dependent on the integrity of the endogenous cell cycle control pathways in human glioblastoma cell lines. Exp Cell Res 1998, 238:51-62 [DOI] [PubMed] [Google Scholar]
- 87.Burger H, Nooter K, Boersma AW, van Wingerden KE, Loijenga LH, Jochemsen AG, Stoter G: Distinct p53-independent apoptotic cell death signalling pathways in testicular germ cell tumour cell lines. Int J Cancer 1999, 81:620-628 [DOI] [PubMed] [Google Scholar]
- 88.He AW, Cory JG: p53-independent anisomycin induced G1 arrest and apoptosis in L1210 cell lines. Anticancer Res 1999, 19:421-428 [PubMed] [Google Scholar]
- 89.Khalid MH, Yagi N, Hiura T, Shibata S: Immunohistochemical analysis of p53 and p21 in human primary glioblastomas in relation to proliferative potential and apoptosis. Brain Tumor Pathol 1998, 15:89-94 [DOI] [PubMed] [Google Scholar]
- 90.McDonald AC, Brown R: Induction of p53-dependent and p53-independent cellular responses by topoisomerase 1 inhibitors. Br J Cancer 1998, 78:745-751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sipos L, Szegedi Z, Fedorcsak I, Afra D, Szende B: Apoptosis and p53 expression in human gliomas. Pathol Oncol Res 1998, 4:267-270 [DOI] [PubMed] [Google Scholar]
- 92.Stahler F, Roemer K: Mutant p53 can provoke apoptosis in p53-deficient Hep3B cells with delayed kinetics relative to wild-type p53. Oncogene 1998, 17:3507-3512 [DOI] [PubMed] [Google Scholar]
- 93.Strasser-Wojak EM, Hartmann BL, Geley S: Irradiation induces G2/M cell cycle arrest and apoptosis in p53-deficient lymphoblastic leukemia cells without affecting Bcl-2 and Bax expression. Cell Death Differ 1998, 5:687-693 [DOI] [PubMed] [Google Scholar]
- 94.Sun SY, Yue P, Wu GS, El-Deiry WS, Shroot B, Hong WK, Lotan R: Mechanisms of apoptosis induced by the synthetic retinoid CD437 in human non-small cell lung carcinoma cells. Oncogene 1999, 18:2357-2365 [DOI] [PubMed] [Google Scholar]
- 95.Vikhanskaya F, Vignati S, Beccaglia P: Inactivation of p53 in a human ovarian cancer cell line increases the sensitivity to paclitaxel by inducing G2/M arrest and apoptosis. Exp Cell Res 1998, 241:96-101 [DOI] [PubMed] [Google Scholar]
- 96.Zamble DB, Jacks T, Lippard SJ: p53-dependent and -independent responses to cisplatin in mouse testicular teratocarcinoma cells. Proc Natl Acad Sci USA 1998, 95:6163-6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier G: Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 1993, 53:3976-3985 [PubMed] [Google Scholar]
- 98.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit V: Yama/CPP32b, a mammalian homolog of ced-3, is a CrmA-inhibitable protease that cleaves the death substrate Poly(ADP-ribose) polymerase. Cell 1995, 81:801-809 [DOI] [PubMed] [Google Scholar]
- 99.Salminen A, Helenius M, Lahtinen T, Korhonen P, Tapiola T, Soininen H, Solovyan V: Down-regulation of Ku autoantigen, DNA-dependent protein kinase, and poly(ADP-ribose) polymerase during cellular senescence. Biochem Biophys Res Commun 1997, 238:712-716 [DOI] [PubMed] [Google Scholar]