

Abstract
Cyclooxygenase 2 (COX-2) overexpression has been described in sporadic colonic neoplasia, but its role in ulcerative colitis (UC) neoplastic progression remains unexplored. Although the specific role of cyclooxygenase in colonic neoplasia is uncertain, its inhibition by nonsteroidal anti-inflammatory drugs decreases the risk of sporadic colonic adenocarcinoma and causes regression of adenomas in familial adenomatous polyposis. To investigate the role of COX-2 in UC-associated neoplasia, we assessed COX-2 protein and mRNA expression throughout the spectrum of UC-associated neoplastic lesions in four total colectomy specimens, using immunocytochemistry and a novel TaqMan reverse transcriptase-polymerase chain reaction assay. The findings were correlated with DNA ploidy and inflammatory activity. We found COX-2 overexpression throughout the neoplastic spectrum in UC (P < 0.0001, R2=0.53), even in diploid samples that were negative for dysplasia. Overall, neoplastic change explained 53% of the variation in COX-2 expression, whereas inflammatory activity explained only 11%. COX-2 was overexpressed in all aneuploid samples and in 38% of diploid samples (P = 0.0074). cDNA representational difference analysis was also performed and revealed that COX-2 mRNA was an up-regulated cDNA representational difference analysis difference product. COX-2 overexpression occurs early in UC-associated neoplasia, and the increase cannot be explained by inflammatory activity alone. The data suggest that COX-2-specific inhibitors may have a chemopreventative role in UC but the possibility that they could exacerbate UC inflammatory activity needs to be tested.
Cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2) are cyclooxygenase enzymes that convert arachidonic acid to inflammatory and other physiological mediators, including prostaglandins, prostacyclin, and thromboxane. 1,2 COX-1 is constitutively expressed in most tissues, including the gastrointestinal tract, at a relatively stable level and is thought to help protect the gastrointestinal tract from injury. 1,2 COX-2 is an inducible cyclooxygenase whose production is stimulated by interleukin-1, tumor necrosis factor, and many other mediators. 1,2 COX-2 is thought to play a role in the reparative process after mucosal injury in the gastrointestinal tract. 1,2
Multiple studies suggest that COX-2 plays a role in sporadic colorectal neoplasia, based on its overexpression in colonic adenomas and carcinomas, as shown by both immunohistochemistry and reverse transcriptase-polymerase chain reaction (RT-PCR). 3-5 Cyclooxygenase inhibitors such as nonsteroidal anti-inflammatory drugs (NSAIDs) substantially decrease the risk of colorectal cancer, as well as the number and size of adenomas in familial adenomatous polyposis patients. 6-8 Experimentally, NSAIDs prevent colonic adenocarcinoma in rodents with an familial adenomatous polyposis phenotype (the APC min mouse model). 9,10 Understanding the role of COX-2 in colonic neoplasia is thus particularly important because of these therapeutic implications.
Extensive ulcerative colitis (UC) of >8 years’ duration is an important risk factor for colonic epithelial dysplasia and adenocarcinoma. 11,12 Neoplastic lesions in UC differ from sporadic adenomas and carcinomas in that they generally occur in younger individuals and in flat mucosa within large fields of genetic abnormalities, rather than as isolated and visible polypoid lesions. 12,13 Nonetheless, many of the genetic abnormalities observed in sporadic neoplasms, including alterations in the APC, p53, bcl-2, and K-ras genes, microsatellite instability, and aneuploidy, among others, are also found in UC neoplasia, albeit with different prevalence and timing in many instances. 14-21 Because of these similarities and the significant role COX-2 has been shown to play in sporadic colorectal neoplasia, we sought to investigate its potential role in UC-associated neoplasia. To determine this, we examined COX-2 expression at the protein and mRNA levels on numerous spatially mapped mucosal samples in total colectomy specimens from UC patients who had developed dysplasia or carcinoma. We used immunohistochemistry on fixed tissues and a novel 5′-nuclease or real-time (TaqMan) RT-PCR assay on fresh-frozen epithelium that had been isolated from stromal elements. Finally, COX-2 expression was also studied at the RNA level in one patient using cDNA representational difference analysis. Representational difference analysis is a subtractive hybridization and PCR amplification technique for detecting genetic differences between tissues or cells. In this case, we compared COX-2 cDNA in a sample that was negative for dysplasia to samples that were either indefinite for dysplasia or had low-grade dysplasia. These particular co-expressions were chosen to examine differences in expression in early UC neoplasia, where cancer prevention or therapy is most likely to be effective.
Materials and Methods
Patients and Histology
Total colectomy specimens from four UC patients (1B, 2J, 3S, and 4R) were evaluated at 64 different sites chosen to represent the entire UC neoplastic spectrum. All patients underwent resection at the University of Washington Medical Center for either high-grade dysplasia or adenocarcinoma, and all had long-standing, pancolonic UC of >10-years’ duration. The University of Washington Human Subjects Division approved this research. The colectomy specimens were mapped histologically in a grid-like manner as previously described. 22 The biopsies were classified independently by two gastrointestinal pathologists (MPB and RCH) as negative for dysplasia, indefinite for dysplasia, low-grade dysplasia, high-grade dysplasia, or invasive carcinoma. Histological criteria used were those defined by the Inflammatory Bowel Disease Dysplasia Morphology Study Group, with the exception that the indefinite category was not subdivided. 23 The degree of active inflammation was assessed semiquantitatively in each sample as either inactive (no intraepithelial granulocytes), cryptitis only present, one to three crypt abscesses present per section, more than three crypt abscesses per section, or as having ulcers/erosions with granulation tissue.
Immunohistochemistry
Deparaffinized sections were immunostained with a monoclonal antibody to COX-2 (Cayman Chemicals, Ann Arbor, MI) at a dilution of 1:1,000. Negative controls consisted of substitutions of mouse ascites fluid for the primary antibody. Sections were subjected to heat-induced epitope retrieval using a microwave oven, as previously described. 24 Antigens were localized using a standard avidin-biotin method with 3,3′-diaminobenzidine as the chromogen and nickel chloride enhancement. The degree of staining was assessed by two pathologists independently and graded semiquantitatively using criteria previously described: 25 0, no overexpression of COX-2 in comparison to the normal colonic epithelium from control patients without UC or other disorders; 1+, mild overexpression; 2+, moderate overexpression; and 3+, marked, uniform overexpression of COX-2. Inflammatory cells within the lamina propria were variably positive and provided an internal positive control in some areas of all sections evaluated.
Flow Cytometry
Samples evaluated by flow cytometry were selected primarily from histologically abnormal areas, although areas with negative histology were also included. The biopsies for flow cytometry were obtained fresh and cryopreserved as previously described. 26 Nuclear isolation, staining, and flow cytometric analysis of DNA-content histograms were also performed as previously described. 26
Epithelial Isolation and mRNA Preparation for TaqMan RT-PCR
Epithelial cells were isolated from biopsies by the following method: the submucosal side of biopsy specimens was affixed to the end of a 2.5-mm diameter wooden stick with cyanoacrylate glue, and then soaked for 5 minutes in Hanks’ buffer with 20 mmol/L dithiothreitol/5 mmol/L ethylenediaminetetraacetic acid at 4°C. Samples were transferred to 2 ml of shaking solution (Hanks’ buffer with 20 mmol/L dithiothreitol/5 mmol/L CaCl2/5 mmol/L MgCl2/10% DMSO at 4°C) and vortexed for 5 seconds, after which the stick with residual stroma was removed. Staining with anti-cytokeratin antibody confirmed that the cells in suspension at the end of this procedure were >90% pure epithelial. RNA isolation was done using TRIzol (Life Technologies, Inc., Rockville, MD).
TaqMan RT-PCR
This assay utilizes the 5′→3′ exonuclease activity of the thermostable T. aquaticus DNA polymerase during PCR to degrade an oligonucleotide probe complementary to an internal (nonprimer) sequence within the PCR template of interest. 27 Fluorogenic probes are used that contain a 5′ fluorescent reporter group and a 3′ quencher group. 27,28 When perfectly matched, the probe is degraded by the Taq exonuclease, resulting in separation of reporter from quencher and increased fluorescent emission from the reaction. Mismatched probes are not degraded by the Taq polymerase, with no corresponding increase in fluorescence. The threshold cycle number is determined on a thermal cycling fluorimeter and is used to quantitate the presence of the sequence of interest. In an optimized PCR system, the threshold cycle decreases by one cycle as the concentration of template doubles.
Exonic primers flanking intron 5 of the human COX-2 gene (PTGS-2; GB No. D28235) were designed for RT-PCR to produce 204- and 918-bp products in cDNA and genomic DNA, respectively. The probe oligonucleotide was labeled at the 5′ end with the fluorescent dye FAM (6-carboxy-fluorescein) and modified at the 3′ end with a quencher-minor groove binder ligand. The minor groove binder stabilizes probe-template annealing and allows use of shorter probes with better sequence specificity and lower fluorescent background. 28 Assessment of β-actin RNA for quality and normalization was done with the TaqMan β-actin Control Reagent kit (Perkin-Elmer-Cetus, Emeryville, CA), which utilizes standard TaqMan probe chemistry. The COX-2 primer and probe sequences and their locations are shown in Table 1 ▶ .
Table 1.
COX-2 TaqMan RT-PCR Primers, Probe Sequence, Location
| Primer/probe | Sequence | Exon | Nucleotides |
|---|---|---|---|
| Sense primer | 5′ GCC TTC TCT AAC CTC TCC | Exon 4 | 3876–3893 |
| α-Sense primer | 5′ CTG ATG CGT GAA GTG C | Exon 5 | 4679–4694 |
| TaqMan probe | 5′ FAM AGC CCT TCC TCC TG-MGB | Exon 4 | 3905–3918 |
Abbreviations: FAM, 6-carboxyfluorescein; MGB, minor groove binder.
One microgram of all RNAs and control genomic DNA was DNase treated before RT-PCR to eliminate DNA contamination. All RT-PCR reactions used the Titan One-Tube RT-PCR System (Boehringer Mannheim, Indianapolis, IN) and were done in triplicate (COX-2) and duplicate (β-actin) sets. Each RT-PCR reaction contained: 100 ng total RNA (or DNA controls), 200 μmol/L dNTPs, 0.4 μmol/L primers, 0.1 μmol/L COX-2 or 0.04 μmol/L β-actin probe, 1.5 mmol/L MgCl2, 5 mmol/L dithiothreitol, 5 U of RNase inhibitor, 1× RT-PCR buffer, and the AMV RT and Taq polymerase enzymes provided with the kit. For RT, reactions were incubated at 55°C for 30 minutes followed by 3 minutes at 95°C, then 40 cycles of 94°C to 62°C for PCR. All reactions were performed and analyzed with an ABI 5700 Sequence Detection System, which monitors fluorescence increase at each cycle of the PCR reaction.
Representational Difference Analysis
Driver and tester pairs of varying histological grades were selected from UC colon 2J and were matched for degree of inflammation. RNA was isolated from colonic epithelium by the guanidine isothiocyanate method (Stratagene, La Jolla, CA) and cDNA was prepared using a cDNA synthesis kit (Roche Biochemicals, Indianapolis, IN). cDNA representational difference analysis was performed as previously described. 29,30 After four rounds of hybridization/amplification, the discrete difference products were isolated for further analysis through subcloning and direct sequencing. Sequences were then compared against a database for homology to known genes with a BLAST search (http://www.ncbi.nlm.nih.gov/blast/).
Statistical Analysis
COX-2 overexpression was compared among the five histological diagnoses by the Kruskal-Wallis rank test. The association of COX-2 overexpression with inflammation was examined via Pearson correlation coefficient and a linear regression model. The association of COX-2 overexpression and aneuploidy was examined via a logistic regression model. The association of COX-2 expression at the RNA versus protein levels, as assessed by TaqMan RT-PCR and immunohistochemistry, respectively, was assessed by the Spearman and Pearson correlation coefficients. Each model was adjusted for the potential effect of patient. All tests were two-sided.
Results
COX-2 Immunohistochemistry
COX-2 immunohistochemistry showed moderate 2+ to marked 3+ diffuse cytoplasmic overexpression in all of the biopsies with dysplasia, regardless of grade (Figure 1) ▶ . The expression of COX-2 in dysplastic lesions (low-grade dysplasia and high-grade dysplasia) was present in most (>90%) of the cells (Figure 2,C and D) ▶ . The expression of COX-2 was not uniform, however, in adenocarcinoma (Figure 2, E–H) ▶ . Although some foci of adenocarcinoma had marked and uniform overexpression of COX-2, others showed staining in only 50% of the cells (Figure 2, E and F) ▶ , and one section of adenocarcinoma had no overexpression (Figure 2, G and H) ▶ . The high-grade dysplastic epithelium overlying this negative carcinoma focus and carcinoma elsewhere in the same colon showed marked overexpression of COX-2 (Figure 1 ▶ , UC colon 3S). The proportion of sites that had at least 1+ COX-2 overexpression was 83%, 100%, 100%, 60%, and 25% for the adenocarcinoma, high-grade dysplasia, low-grade dysplasia, indefinite, and negative for dysplasia categories, respectively (P < 0.0001, R 2 = 0.53). Overall, neoplastic change explained 53% of the variation in COX-2 expression. Specifically, 15 of 25 samples (60%) that were indefinite for dysplasia showed 1+ to 2+ overexpression of COX-2 and five of 20 samples (25%) that were negative for dysplasia also showed 1+ to 2+ overexpression (Figure 2, A and B) ▶ . The distribution within individual colonic crypts of COX-2 staining was variable, and revealed no consistent localization to either the crypt base, mid-crypt, or surface epithelial regions.
Figure 1.

Distribution of COX-2, dysplasia, inflammation, and DNA ploidy in UC colectomy specimens.
Figure 2.

COX-2 immunohistochemistry (right) throughout UC-associated neoplasia (left). A: UC mucosa that is negative for dysplasia and without active inflammation (H&E, ×115). B: COX-2-positive immunostaining (black reaction product against green counterstain) on parallel section to A. Note the focal 2+ positivity at the epithelial surface and scattered positive internal control lymphocytes. Other negative for dysplasia sites demonstrated crypt-only immunopositivity (not shown). (Immunocytochemical DAB/nickel chloride with methyl green counterstain, ×115.) C: UC mucosa with low-grade dysplasia and no active inflammation (H&E, ×60). D: COX-2-positive immunostaining (black reaction product against green counterstain) on parallel section to C. (Immunocytochemical DAB/nickel chloride with methyl green counterstain, ×60.) E: UC adenocarcinoma, poorly differentiated (H&E, ×115). F: COX-2-positive immunostaining (black reaction product against green counterstain) on parallel section to E adenocarcinoma; note that only ∼50% of carcinoma cells are positive. (Immunocytochemical DAB/nickel chloride with methyl green counterstain, ×115.) G: UC adenocarcinoma, poorly differentiated (H&E, ×230). H: COX-2-positive immunostaining (black reaction product against green counterstain) on parallel section to G adenocarcinoma; note that the carcinoma is negative for COX-2 and that internal control lymphocytes (arrow) are positive. (Immunocytochemical DAB/nickel chloride with methyl green counterstain, ×230.)
COX-2 RNA Results
COX-2 RNA analysis by TaqMan RT-PCR was performed on eight UC mucosal samples from three of the UC patients at sites also examined by immunohistochemistry and within three different non-UC controls. The controls consisted of two sporadic colonic adenocarcinomas and their matched distant normal colonic mucosa both from two different patients, along with another normal colonic mucosal sample from a third patient with diverticular disease. All negative control PCR reactions without added RNA, included in all experiments to control for possible reagent contamination by template, showed no fluorescent signal. DNase-treated samples showed significantly less fluorescent signal than nontreated controls, indicating that genomic DNA contamination was present in all starting RNA preparations, and that DNase treatment before TaqMan RT-PCR analysis was required for specific determination of RNA expression.
The TaqMan RT-PCR RNA and immunohistochemical protein expression data on the eight examined UC sites and non-UC controls are shown in Table 2 ▶ and in the spatial colonic maps in Figure 1 ▶ . The regression analysis of this RNA versus protein data are graphed in Figure 3 ▶ . There was a strong positive correlation (Pearson correlation, 0.97; Spearman correlation, 0.74).
Table 2.
COX-2 Immunohistochemical and TaqMan RT-PCR Results
| Patient | Histology | Inflammation | COX-2 IHC | COX-2 RNA PCR CT* |
|---|---|---|---|---|
| UC-1B | IND | 2+ | 1+ focal | 40† |
| UC-1B | NEG | 0 | 0 | 40 |
| UC-1B | IND | 2+ | 1+ focal | 35.3 |
| UC-1B | NEG | 0 | 1+ | 34.7 |
| UC-2S | IND | 2+ | 1+ focal | 38.3 |
| UC-2S | IND | 3+ | 1+ focal | 40 |
| UC-4R | NEG | 0 | 0 | 40 |
| UC-4R | IND | 0 | 0 | 40 |
| Control 1a | CA sporadic-1 | 0 | 3+ | 26.4 |
| Control 1b | N1 colon-1 | 0 | 0 | 40 |
| Control 2a | CA sporadic-2 | 0 | 3+ | 26.4 |
| Control 2b | N1 colon-2 | 0 | 0 | 39.8 |
| Control 3 | N1 colon | 0 | 0 | 40 |
*CT refers to the TaqMan ABI RT-PCR cycle that produced fluorescent signal above the threshold level.
†Cycle number 40 is the maximum cycle number generated, and indicates absence of RNA in the starting sample. Cycle numbers progressively less than 40 indicate the cycle number where generated fluorescent signal crossed the threshold. Lower cycle numbers reflect more RNA in the starting sample. One cycle decrease indicates doubling of RNA template.
Abbreviations: NEG, negative for dysplasia; IND, indefinite for dysplasia; CA, carcinoma; N1, matched normal colonic mucosa from the same patient as the carcinoma with the same number.
Inflammation score: 0, inactive, 1+, cryptitis; 2+, crypt abscess; 3+, more than 3 crypt abscesses/section; 4+, granulation tissue. COX-2 score: 0, none; 1+, mild; 2+, moderate; 3+, marked overexpression.
Figure 3.
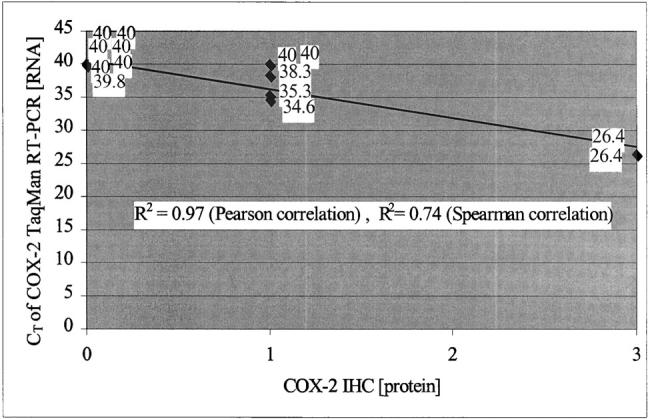
Correlation of COX-2 protein expression by immunohistochemistry and RNA expression by TaqMan RT-PCR on epithelial isolated mucosal samples. CT refers to the TaqMan ABI RT-PCR cycle that produced fluorescent signal above the threshold level. Cycle number 40 is the maximum cycle number generated, and indicates absence of RNA in the starting sample. Cycle numbers progressively less than 40 indicate the cycle number where generated fluorescent signal crossed the baseline. Lower cycle numbers reflect more RNA in the starting sample. One cycle decrease indicates doubling of RNA template.
cDNA-representational difference analysis revealed COX-2 to be an up-regulated difference product \ when a site negative for dysplasia was used as a driver against tester sites that were indefinite for dysplasia or had low-grade dysplasia. Again, these sites had equivalent inflammation scores.
All aneuploid samples (n = 8) showed COX-2 overexpression, compared to 38% (6 of 16) of diploid samples (P = 0.0074). Aneuploidy usually corresponded to neoplastic foci (four cancer sites, two dysplastic sites, versus one indefinite and one negative site) (Figure 1) ▶ ; however, six diploid neoplastic sites (four indefinite, one high-grade dysplasia, and one cancer) also showed overexpression (Figure 1) ▶ .
Of the 36 samples with COX-2 overexpression, 23 had active inflammation, whereas 13 had inactive disease. Further, four of the 25 sites negative for COX-2 overexpression had active inflammation. Overall, there was a moderate correlation between inflammatory activity and COX-2 overexpression with a correlation coefficient of 0.31 (P = 0.013, R 2 = 0.10). Inflammation alone explained only 11% of the variation in COX-2 expression.
Discussion
COX-2 in UC-Associated Neoplasia
This study demonstrates that COX-2 overexpression in UC-associated neoplasia occurs early, beginning in mucosa that is diploid and negative for dysplasia, and in mucosa that is noninflamed. This is true of both COX-2 protein and its mRNA. We have demonstrated that COX-2 protein overexpression by immunohistochemistry in >60 mapped mucosal samples occurs early in UC-associated neoplastic progression. This observation was confirmed in selected samples by demonstrating increased COX-2 mRNA, using isolated epithelium in a novel TaqMan RT-PCR assay. To specifically evaluate epithelial COX-2 RNA expression, we purified epithelial cells from the stromal elements of the lamina propria by an ethylenediaminetetraacetic acid shake-off technique. We have previously shown, by flow cytometric cytokeratin sorting of aneuploid colonic biopsies, that this shake-off technique yields a >90% pure epithelial cell population. 26 To our knowledge, no other investigations of COX-2 RNA from whole tissues have been done on purified epithelium separated from stromal components. This may be important, as mononuclear cells in mucosal lamina propria normally express COX-2, and in fact, serve as internal positive controls in all reported immunocytochemical studies of COX-2 expression, including our own. Thus, separating the epithelial and stromal components in the analysis of mRNA expression may be critical for the accurate determination of COX-2 mRNA from tissue.
The correlation between COX-2 protein and mRNA using the methods described was excellent, with a Spearman correlation of 0.74 and a Pearson correlation of 0.97. This is particularly impressive because the samples for correlation were all specifically chosen from the low end of expression and from samples with early neoplastic change (negative or indefinite histology). This is the end of the spectrum where false-positive and -negative readings are more likely, and where intervention to prevent neoplastic progression is more likely to succeed.
Spatial mapping throughout the neoplastic spectrum provides important insight into whether abnormalities occur early or late during neoplastic progression. UC neoplasia is a particularly well-suited model to assess this issue, based on the broad fields of abnormalities that develop within single patients that can be spatially mapped. In fact, we have recently shown that genetic abnormalities are pancolonic in UC, as determined by fluorescent in situ hybridization. 26 Thus, as opposed the very focal changes in sporadic colorectal tumorigenesis, UC provides a useful model for determining the timing of individual events, such as COX-2, at specified steps in the morphological and biological neoplastic continua.
Using this approach, we have found COX-2 overexpression at both the protein and mRNA levels occurs early. We showed mild to focally moderate overexpression of COX-2 in 25% of UC samples that were negative for dysplasia, in 60% of samples that were indefinite for dysplasia, and in all dysplastic samples. COX-2 overexpression often correlated with DNA aneuploidy, but was also seen in six diploid sites. These observations, along with the corresponding up-regulation of COX-2 expression at the mRNA level in multiple corresponding mucosal samples that were negative or indefinite for dysplasia, indicate that COX-2 overexpression occurs early in UC neoplastic progression. In view of the therapeutic potential of COX inhibitors, this early development of COX-2 overexpression is of great interest.
COX-2 overexpression has been described previously in actively inflamed, nondysplastic mucosa in both UC and Crohn’s disease. 31,32 We have demonstrated it in both inflamed and noninflamed mucosa in all stages of neoplastic progression in UC (Figure 1) ▶ . In addition, some of the actively inflamed nondysplastic areas showed no COX-2 expression (Figure 1) ▶ . These data indicate that COX-2 overexpression does not simply reflect inflammation, because inflammation alone explains only 11% of the variation in COX-2 expression. Rather, our data indicate that a much higher percentage (53%) of the variation in COX-2 expression is associated with neoplastic progression.
Two potential mechanisms may explain the relationship between COX-2 overexpression and neoplastic progression in UC: 1) it may increase malondialdehyde levels, and 2) it may up-regulate bcl-2. The first hypothesis contends that increased COX-2 activity, in part brought about by the normal physiological response to injury and inflammation, may accelerate genetic damage through increased production of malondialdehyde, a mutagenic by-product of COX-mediated prostaglandin synthesis and lipid peroxidation. 33,34 This malondialdehyde production would be in addition to that produced by the constitutive activity of COX-1, which is thought to be important in sporadic colorectal neoplasia. 1 In support of this hypothesis, elevated levels of malondialdehyde have been detected both in sporadic colon cancer and in inflammatory bowel disease. 35-38
After the initiation of neoplasia, COX-2 may promote tumor progression by increasing expression of bcl-2. 39,40 bcl-2 generates resistance to apoptosis, and bcl-2 up-regulation has also been demonstrated in UC-associated neoplasia. 17 Of further interest, overexpression of bcl-2 is reversible by both nonspecific COX inhibitors 40 and by highly selective COX-2 inhibitors. 41
COX-2 Selective Inhibitor Therapy in Sporadic and UC Neoplasia
The overexpression of COX-2 in colorectal adenomas and adenocarcinomas suggests that treatment of individuals without colorectal neoplasms with selective COX-2 inhibitors might lower their risk of developing them. Promising preliminary studies in rodents treated with selective COX-2 inhibitors show suppression of neoplastic development with minimal toxic side effects. 9,10,41 Specifically, the prevalence of gastrointestinal side effects, such as ulceration and bleeding, are lower in animals treated with selective COX-2 inhibitors than those given nonspecific cyclooxygenase inhibitors, such as aspirin and other NSAIDs. 42,43
A treatment that could prevent colorectal neoplasia in UC would be of great benefit by obviating the need for surveillance and, in some patients, the need for total colectomy for dysplasia or carcinoma. Caution is warranted, however, as COX-2 selective inhibition has been shown to impair healing of gastric ulcers in some studies, 43 and NSAID use may exacerbate the activity of idiopathic inflammatory bowel disease. 44 There are, however, few published data on NSAID exacerbation of idiopathic inflammatory bowel disease, either Crohn’s disease or UC. A recent retrospective cohort study of 1,940 inflammatory bowel disease patients (881 had Crohn’s disease and 1,059 had UC), followed for an average of 3.1 years, demonstrated that the majority of inflammatory bowel disease patients use NSAID analgesics, and that their use is not associated with disease flares, as analyzed by multivariate Cox proportional hazards models (hazard ratio, 0.93; 95% confidence interval, 0.68 to 1.27). 45 Another study using COX-2-specific inhibitors in rodents with a chemically induced colitis showed exacerbation with colonic perforation after 1 week of treatment. 46 This rodent model of colitis may not be representative of human UC, however, because it is induced by trinitrobenzene sulfonic acid and yields a different histological picture than typical UC, making direct comparisons tenuous.
Studies addressing the effects of highly selective COX-2 inhibitors in human UC are needed to determine whether these new agents will impair colonic ulcer healing or exacerbate UC inflammatory activity. The fact that COX-2 overexpression occurs early in UC neoplasia and that inhibition of this overexpression may inhibit neoplastic progression emphasizes the importance of answering these questions.
Acknowledgments
We thank Dr. John Potter for his review of this manuscript and helpful advice and Dr. Jason Dominitz for his recent data on NSAID use in inflammatory bowel disease patients. We also thank Dr. Raymond DuBois for his technical advice, and the members of the Immunocytochemistry Laboratory at the University of Washington Medical Center, directed by Dr. Rodney Schmidt, for their expert technical assistance with this project.
Footnotes
Address reprint requests to Mary P. Bronner, M.D., University of Washington Medical Center, Department of Pathology, BB220, 1959 NE Pacific St., Box 356100, Seattle, WA 98195-6100. E-mail: bronner@u.washington.edu.
Supported by National Institutes of Health grant RO1 CA68124-01.
References
- 1.Watson AJM: Chemopreventive effects of NSAIDs against colorectal cancer: regulation of apoptosis and mitosis by COX-1 and COX-2. Histol Histopathol 1998, 13:591-597 [DOI] [PubMed] [Google Scholar]
- 2.Sakamoto C: Roles of COX-1 and COX-2 in gastrointestinal pathophysiology. J Gastroenterol 1998, 33:618-624 [DOI] [PubMed] [Google Scholar]
- 3.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN: Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107:1183-1188 [DOI] [PubMed] [Google Scholar]
- 4.Williams CS, Luongo C, Radhika A, Zhang T, Lamps LW, Nanney LB, Beauchamp RD, DuBois RN: Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology 1996, 111:1134-1140 [DOI] [PubMed] [Google Scholar]
- 5.Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M, Hla T: Expression of cyclooxygenase-1 and 2 in human colorectal cancer. Cancer Res 1995, 55:3785-3789 [PubMed] [Google Scholar]
- 6.Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, Booker SV, Robinson CR, Offerhaus GJA: Treatment of colonic and rectal adenomas with Sulindac in FAP. N Engl J Med 1993, 328:1313-1316 [DOI] [PubMed] [Google Scholar]
- 7.Kune GA, Kune S, Watson LF: Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res 1988, 48:4399-4404 [PubMed] [Google Scholar]
- 8.Thun MJ, Namboodiri MM, Heath CW: Aspirin use and reduced risk of fatal colon cancer. N Engl J Med 1991, 325:1593-1596 [DOI] [PubMed] [Google Scholar]
- 9.Jacoby RF, Marchall DJ, Newton MA, Novakovic K, Tutsch K, Cole CE, Lubet RA, Kelloff GJ, Verma A, Moser AR, Dove WF: Chemoprevention of spontaneous intestinal adenomas in the APC Min mouse model by the nonsteroidal anti-inflammatory drug Piroxicam. Cancer Res 1996, 56:710-714 [PubMed] [Google Scholar]
- 10.Oshima M, Dinchuck JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM: Suppression of intestinal polyposis in APC knockout mice by inhibition of COX-2. Cell 1996, 87:803-809 [DOI] [PubMed] [Google Scholar]
- 11.Morson BC: Precancer and cancer in inflammatory bowel disease. Pathology 1985, 17:173-180 [DOI] [PubMed] [Google Scholar]
- 12.Brentnall TA: Risk factors for development of colorectal cancer in inflammatory bowel disease. Norwell MA eds. Advances in Inflammatory Bowel Disease. 1998, :pp 159-167 Kluwer Academic Publishers, Dordrecht/Boston/London [Google Scholar]
- 13.Goldgraser MB, Humphreys EM, Kirschner JB, Palmer WL: Carcinoma and ulcerative colitis. Gastroenterology 1958, 34:809-839 [PubMed] [Google Scholar]
- 14.Kern SE, Redston M, Seymour AB, Caldas C, Powell SM, Kornacki S, Kinzler KW: Molecular genetic profiles of colitis-associated neoplasms. Gastroenterology 1994, 107:420-428 [DOI] [PubMed] [Google Scholar]
- 15.Park WS, Pham T, Wang C, Pack S, Mueller E, Mueller J, Vortmeyer A, Zhuang Z, Fogt F: Loss of heterozygosity and microsatellite instability in non-neoplastic mucosa from patients with chronic ulcerative colitis. Int J Mol Med 1998, 2:221-224 [PubMed] [Google Scholar]
- 16.Brentnall TA, Crispin DA, Rabinovitch PS, Haggitt RC, Rubin CE, Stevens AC, Burmer GC: Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994, 107:369-378 [DOI] [PubMed] [Google Scholar]
- 17.Bronner MP, Culin C, Reed JC, Furth EE: The bcl-2 proto-oncogene and the gastrointestinal epithelial tumor progression model. Am J Pathol 1995, 146:20-26 [PMC free article] [PubMed] [Google Scholar]
- 18.Burmer GC, Levine DS, Kulander BG, Haggitt RC, Rubin CE, Rabinovitch PS: C-Ki-ras mutations in chronic ulcerative colitis and sporadic colon carcinoma. Gastroenterology 1990, 99:416-420 [DOI] [PubMed] [Google Scholar]
- 19.Tarmin L, Yin J, Harpaz N, Kozam M, Noordzij J, Antonio LB, Jiang HY, Chan O, Cymes K, Meltzer SJ: Adenomatous polyposis coli gene mutations in ulcerative colitis-associated dysplasias and cancers versus sporadic colon neoplasms. Cancer Res 1995, 55:2035-2038 [PubMed] [Google Scholar]
- 20.Rubin CE, Haggitt RC, Burmer GC, Brentnall TA, Stevens AC, Levine DS, Dean PJ, Kimmey M, Perera DR, Rabinovitch PS: DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992, 103:1611-1620 [DOI] [PubMed] [Google Scholar]
- 21.Brentnall TA, Crispin DA, Bronner MP, Cherian SP, Hueffed M, Rabinovitch PS, Rubin CE, Haggitt RC, Boland CR: Microsatellite instability in non-neoplastic mucosa from patients with chronic ulcerative colitis. Cancer Res 1996, 56:1237-1240 [PubMed] [Google Scholar]
- 22.Burmer GC, Ravinovitch PS, Haggitt RC, Crispin DA, Brentnall TA, Kolli VR, Stevens AC, Rubin CE: Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology 1992, 103:1602-1610 [DOI] [PubMed] [Google Scholar]
- 23.Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC, Sommers SC, Yardley JH: Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical application. Hum Pathol 1983, 14:931-968 [DOI] [PubMed] [Google Scholar]
- 24.Gown AM, de Wever N, Battifora H: Microwave based antigenic unmasking: a revolutionary new technique for routine immunohistochemistry. Appl Immunohistol 1993, 22:934-944 [Google Scholar]
- 25.Hao X, Bishop AE, Wallace M, Wang H, Willcocks TC, MacLouf J, Polak JM, Knight S, Talbot IC: Early expression of cyclooxygenase-2 during sporadic colorectal carcinogenesis. J Pathol 1999, 187:295-301 [DOI] [PubMed] [Google Scholar]
- 26.Rabinovitch PS, Dziadon S, Brentnall TA, Emond M, Crispin DA, Haggitt RC, Bronner MP: Pancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res 1999, 59:5148-5153 [PubMed] [Google Scholar]
- 27.Holland PM, Abramson RD, Watson R, Gelfand DH: Detection of specific PCR reaction product by utilizing the 5′ to 3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Nat Acad Sci USA 1991, 88:7276-7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, Singer MJ, Walburger DK, Lokhov SG, Gall AA, Dempcy R, Reed MW, Meyer RB, Hedgpeth J: 3′-Minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res 2000, 28:655-661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lisitsyn N, Lisitsyn N, Wigler M: Cloning the difference between two complex genomes. Science 1993, 25:9946-9951 [DOI] [PubMed] [Google Scholar]
- 30.Chu CC, Paul WE: Expressed genes in interleukin-4 treated B cells identified by cDNA representational difference analysis. Mol Immunol 1998, 35:487-502 [DOI] [PubMed] [Google Scholar]
- 31.Singer II, Kawka DW, Schloemann S, Tessner T, Riehl T, Stenson WF: Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 1998, 115:297-306 [DOI] [PubMed] [Google Scholar]
- 32.Hendel J, Nielsen OH: Expression of cyclooxygenase-2 mRNA in active inflammatory bowel disease. Am J Gastroenterol 1997, 92:1170-1173 [PubMed] [Google Scholar]
- 33.Basu AK, Marnett LJ: Unequivocal demonstration that malondialdehyde is a mutagen. Carcinogenesis 1983, 4:331-333 [DOI] [PubMed] [Google Scholar]
- 34.Mukia FH, Goldstein BD: Mutagenicity of malondialdehyde, a decomposition product of peroxidized polyunsaturated fatty acids. Science 1976, 191:868-869 [DOI] [PubMed] [Google Scholar]
- 35.Baur G, Wendel A: The activity of the peroxide-metabolizing system in human colon carcinoma. J Cancer Res Clin Oncol 1980, 97:267-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendrickse CW, Kelly RW, Radley S, Donovan IA, Keighley MR, Neoptolemos JP: Lipid peroxidation and prostaglandins in colorectal cancer. Br J Surg 1994, 81:1219-1223 [DOI] [PubMed] [Google Scholar]
- 37.Oliva MR, Ripoll F, Muniz P, Iradi A, Trullenque R, Valls V, Drehmer E, Saez GT: Genetic alteration and oxidative metabolism in sporadic colorectal tumors from a Spanish community. Mol Carcinog 1997, 18:232-243 [PubMed] [Google Scholar]
- 38.Chiarpotto E, Scavazza A, Leonaruzzi G, Camandola S, Biasi F, Teggia PM, Garavoglia M, Robecchi A, Roncari A, Poli G: Oxidative damage and transforming growth factor beta-1 expression in pretumoral and tumoral lesions of human intestine. Free Radic Biol Med 1997, 22:889-894 [DOI] [PubMed] [Google Scholar]
- 39.Tsujii M, DuBois RN: Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase-2. Cell 1995, 83:493-501 [DOI] [PubMed] [Google Scholar]
- 40.Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN: Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res 1998, 58:362-366 [PubMed] [Google Scholar]
- 41.Kawamori T, Rao CV, Seibert K, Reddy BS: Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res 1998, 58:409-412 [PubMed] [Google Scholar]
- 42.Mansferrer JL, Zweifel BS, Manning SD: Selective inhibition of inducible cyclooxygenase-2 in vivo is anti-inflammatory and non-ulcerogenic. Proc Natl Acad Sci USA 1994, 91:3228-3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ukawa H, Yamakuni H, Kato S, Takeuchi K: Effects of cyclooxygenase-2 selective and nitric oxide-releasing nonsteroidal antiinflammatory drugs on mucosal ulcerogenic and healing responses of the stomach. Dig Dis Sci 1998, 43:2003-2011 [DOI] [PubMed] [Google Scholar]
- 44.Kaufmann HJ, Taubin HL: Nonsteroidal anti-inflammatory drugs activate quiescent inflammatory bowel disease. Ann Intern Med 1987, 107:513-516 [DOI] [PubMed] [Google Scholar]
- 45.Dominitz JA, Koepsell TD, Boyko EJ: Association between analgesic use and inflammatory bowel disease flares: a retrospective cohort study. Presented at the 101th Annual Meeting of the American Gastroenterology Association, San Diego, CA, May 23, 2000; Gastroenterology (abstract in press)
- 46.Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL: Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J Clin Invest 1996, 98:2076-2085 [DOI] [PMC free article] [PubMed] [Google Scholar]