

Abstract
Transitional cell carcinoma (TCC), a neoplasm of urinary bladder urothelial cells, generally appears in either of two forms, papillary non-invasive or invasive TCC, although intermediate forms can occur. Each has a distinctive morphology and clinical course. Altered expression of the p53 and pRb genes has been associated with the more serious invasive TCC, suggesting that the loss of activity of these tumor suppressor proteins may have a causal role in this disease. To test this hypothesis directly, transgenic mice were developed that expressed the simian virus 40 large T antigen (TAg) in urothelial cells under the control of the cytokeratin 19 gene (CK19) regulatory elements. In one CK19-TAg lineage, all transgenic mice developed highly invasive bladder neoplasms that resembled invasive human bladder TCCs. Stages of disease progression included development of carcinoma in situ, stromal invasion, muscle invasion, rapid growth, and, in 20% of affected mice, intravascular lung metastasis. Papillary lesions never were observed. Western blot analysis indicated that TAg was bound to both p53 and pRb, which has been shown to cause inactivation of these proteins. Our findings support suggestions that (i) inactivation of p53 and/or pRb constitutes a causal step in the etiology of invasive TCC, (ii) papillary and invasive TCC may have different molecular causes, and (iii) carcinoma in situ can represent an early stage in the progression to invasive TCC.
Transitional cell carcinoma (TCC) is the most frequently diagnosed malignancy of the urinary bladder in humans, comprising more than 90% of all neoplasms identified at this site. 1-6 Approximately 80% of TCC are papillary, and these generally are low grade and non-invasive. Most patients with papillary TCC respond well to resection and bacillus Calmette Guérin (BCG) treatment, although in 50 to 80% the low-grade papillary cancer will reoccur. 4,6,7 Despite this fact, the overall prognosis is good. The more serious form of TCC is nonpapillary, and up to 90% display local to distant invasion at the time of diagnosis. For this reason the prognosis is poor and the 5-year survival rate is less than 50%. 6 Due primarily to the severity of invasive TCCs, bladder cancer represents the twelfth leading cause of cancer mortality in humans. 8 Because of their markedly different presentation and course, papillary and nonpapillary TCC have been suggested to be the products of different molecular pathways of neoplastic progression, in effect representing different diseases. 5,9-11
Several molecular alterations have been associated with TCC. 2,3,11-15 These include mutation or reduced/lost expression of p53 or the retinoblastoma gene product pRb, mutation of H-ras, increased expression of c-myc and/or epidermal growth factor receptor family members, loss of certain cell-surface adhesion molecules, and deletion or duplication of specific chromosomal regions. 16,17 Of these, the status of p53 and pRb are considered to provide the best prognostic information. 18-21 In most reports, altered expression of either gene has been identified more frequently in high-grade compared to low-grade neoplasms, and has been associated with reduced patient survival. Altered expression of both has the worst prognosis. Based on these observations, numerous investigators have proposed that loss of activity of the tumor suppressor genes p53 and/or pRb represent important steps in the etiology of bladder cancer, and specifically invasive TCC, with other molecular alterations variably contributing to the development of the disease.
Several animal models have been developed to study bladder carcinogenesis, most involving administration of carcinogens to rats or mice. 1,5 Resulting lesions identified in rats tend to progress from simple hyperplasia to papilloma to low-grade papillary carcinoma, and these can progress further to high-grade, invasive papillary carcinoma. Similar lesions appear in certain mouse strains. This sequence of changes resembles the pathogenesis of the most common form of TCC in humans, although as noted above human papillary neoplasms typically remain non-invasive. In contrast, administration of N-butyl-N-(4-hydroxybutyl)-nitrosamine (BHBN) to B6D2F1 hybrid mice induces focal dysplasia, carcinoma in situ (CIS), and high-grade, invasive TCC with occasional metastasis. 22,23 These lesions mimic those observed in the more serious invasive TCC in humans, although mouse neoplasms typically display squamous changes or a transition to squamous cell carcinoma, only rarely observed in humans. Interestingly, recent studies have reported identification of (i) a high frequency of p53 mutations in BHBN-induced TCC 24 and (ii) increased susceptibility of heterozygous (+/−) p53 knockout mice to BHBN-induced bladder carcinogenesis, although without frequent mutation of the normal allele. 25 These findings reproduce the association between p53 alterations and TCC observed in humans, and reinforce the suggestion that altered tumor suppressor protein function may be linked functionally to the development of urinary bladder neoplasia.
To test the hypothesis that altering function of p53 and pRb in urothelial cells can have a causative role in urinary bladder carcinogenesis, we generated transgenic mice expressing the simian virus 40 (SV40) T-antigen (TAg) in urothelium under control of the cytokeratin 19 (CK19) gene regulatory elements. The TAg protein binds to and inactivates both p53 and pRb, 26 and is a potent transforming agent when targeted to cultured human urothelial cells 27 and to multiple cell types in vivo in mice and rats. 28 Our findings support a primary role for decreased function of one or both of these tumor suppressor genes in the etiology of invasive TCC.
Materials and Methods
Transgene Construction and Generation of Transgenic Mice
Gene regulatory elements from the human cytokeratin 19 (CK19) gene were combined with coding sequences from either human placental alkaline phosphatase (hPAP) or SV40 large TAg (Figure 1) ▶ . The CK19 gene is expressed primarily in simple epithelia, but also in urothelial cells. 29 The 5.5-kb CK19 promoter element, flanked by ClaI and XhoI restriction endonuclease sites, was received from Dr. Bernhard Bader 30 (Max Planck Institute, Martinsried, Germany) in pBLCAT3. It included 2.2 kb of DNA from −9 to −6.8 kb and 3.6 kb of DNA immediately upstream relative to the first exon. The XhoI site was converted to ClaI, and the resulting ClaI-flanked CK19 promoter fragment was cloned into the ClaI site of pBluescript (creating pBSK19p). To generate the CK19-hPAP transgene, the hPAP coding sequence together with an attached SV40 polyadenylation signal site (provided by Dr. Richard Palmiter, University of Washington, Seattle, WA) was subcloned into EcoRI and XbaI sites of the pSp72 plasmid vector. A HindIII site in the polylinker was converted to BglII, and BglII-flanked hPAP was cloned into the BamHI site of pBSK19p (generating pBSK19p-hPAP). A 2.2-kb CK19 3′ enhancer element in pBLCAT3, shown to be necessary for cell-appropriate CK19 transgene expression, 30,31 was received from Dr. Loraine Gudas (Cornell University, Ithaca, NY). 31 The KpnI site in this plasmid was converted to SpeI, and the SpeI-flanked enhancer element was cloned into the SpeI site downstream of hPAP in pBSK19p-hPAP (generating pBSK19p-hPAP-K19e). To generate the CK19-TAg transgene, the TAg coding sequence together with its polyadenylation signal sequence was removed from the vector pBXΔ (provided by Dr. Richard Palmiter) via a BamHI digest. This sequence was cloned into the BamHI site of pBSK19p (generating pBSK19p-TAg). The SpeI-flanked 3′ enhancer element was cloned into the SpeI site of pBSK19p-TAg downstream of the TAg coding sequence (generating pBSK19p-TAg-K19e). Both transgenes were isolated from plasmid DNA by digesting with SalI, followed by electrophoretic separation on 1% agarose gels, electro-elution from the gel fragment, phenol/chloroform extraction, alcohol precipitation, and resuspension in distilled water.
Figure 1.

CK19-hPAP and CK19-TAg transgene constructs. E, enhancer.
Transgenic mice were generated by microinjection of transgene DNA into pronuclei of fertilized FVB-strain mouse eggs. 32 Founder mice and their offspring were identified via polymerase chain reaction (PCR) analysis of tail DNA. A 2-mm piece of tail was digested with 500 μl of a 200 μg/ml proteinase K (Fisher Scientific, Pittsburgh, PA) solution in GNT-K buffer (50 mmol/L KCl, 1.5 mmol/L MgCl2, 10 mmol/L Tris-HCl, pH 8.5, 0.01% gelatin, 0.45% Nonidet P-40, 0.45% Tween) at 56°C, while shaken at 250 rpm for 4 to 18 hours. The proteinase K was heat-inactivated at 95°C for 15 minutes, and the digest was centrifuged at 14,000 rpm for 5 minutes. Two microliters of the supernatant were used in a PCR reaction mix with the primers 5′ CCATTGTTTGCAGTACATTGCATC 3′ and 5′ GGACCTTCTAGGTCTTGAAAGGAG 3′ (specific for the TAg coding region). hPAP primers were as described. 33 Thermocycler conditions were 1 cycle of 5 minutes at 92°C; 25–35 cycles of 45 seconds at 92°C, 1 minute at 60°C, and 1 minute at 72°C; then 1 cycle of 5 minutes at 72°C. A 10-μl aliquot was electrophoresed through a 2% agarose gel. Samples yielding a 258-bp product were considered positive for the transgene. For some studies, blood was collected from deeply anesthetized mice by cardiac puncture. Clinical chemistry analysis was performed on serum samples using a Vitros 250 Blood Chemistry Analyzer (Ortho Clinical Diagnostics, Raritan, NJ). Mice were housed in an AAALAC-approved facility under conditions that conformed to the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the University of Wisconsin-Madison School of Veterinary Medicine Animal Care and Use Committee. Certain mice used in these studies have been assigned the following genetic designations: CK19-hPAP line 1281–4, TgN(Ck19ALPP)6Eps; and CK19-TAg line 1244–3, TgN(Ck19SV)7Eps.
Microscopic Analysis and Immunohistochemistry
To label cells undergoing DNA synthesis, mice were injected with 200 mg/kg BrdU (Sigma, St. Louis, MO), a nucleotide analogue that is incorporated into DNA, and sacrificed 1 to 2 hours later. Following euthanasia, mice were examined grossly for the presence of lesions. Selected tissues were fixed in 10% neutral buffered formalin at room temperature overnight, in 4% paraformaldehyde in 0.1 mol/L NaP buffer at 4°C for 1 to 4 hours, or in Carnoy’s fixative at room temperature for 15 to 60 minutes. Tissues were transferred to 70% EtOH, paraffin-embedded, sectioned at 5 μ, mounted on a slide, and stained with hematoxylin and eosin. For immunohistochemistry, unstained sections were hydrated, blocked with 0.5% H2O2 in methanol, then exposed to 4N HCl for 20 minutes (BrdU only) or boiled in 0.1 Mol/L Tris pH 9.0 for 7–10 minutes in a microwave. Samples were incubated for 2 to 18 hours in a hydrated chamber with primary antibody diluted in PBS containing 0.5% nonfat dry milk. The anti-BrdU rat monoclonal (Accurate Scientific, Westbury, NY) was applied at a dilution of 1:40. The anti-TAg mouse monoclonal (Pab101, Santa Cruz Biotechnology, Santa Cruz, CA) was applied at a dilution of 1:200. The anti-cytokeratin 19 rat monoclonal TROMA 3 (kindly provided by Dr. Rolf Kemler, Max Planck Institute, Freiburg, Germany) was applied at a dilution of 1:100. The anti-uroplakin rabbit polyclonal antiserum (kindly provided by Dr. Tung-Tien Sun, New York Univ. Medical Center, New York, NY) was applied at a dilution of 1:500. Sections were rinsed and exposed for 30 minutes to the appropriate species-specific link antibody (BioGenex, San Ramon, CA), rinsed again, then exposed for 30 minutes to peroxidase- or alkaline phosphatase-conjugated streptavidin (BioGenex). After a final rinse, tissues were incubated for 5–10 minutes with diaminobenzidine (DAB, Sigma) or 10 to 30 minutes with New Fuchsin (BioGenex). Sections then were counterstained with hematoxylin (Polysciences, Inc, Warrington, PA) or nuclear fast red (PolyScientific, Bay Shore, NY), dehydrated through graded alcohols and xylene, and coverslipped.
hPAP Staining
Paraformaldehyde-fixed tissues collected from CK19-hPAP and nontransgenic control mice were incubated in preheated AP buffer (0.1 mol/L NaCl, 5 mmol/L MgCl2, 0.1 mol/L Tris-HCl, pH 9.5) at 65°C for 45 minutes. Tissues then were incubated 18 to 40 hours in 0.17 mg/ml 5-Bromo-4-chloro-3-indolyl phosphate (BCIP; Sigma) in AP buffer at 37°C with gentle agitation. This produced a blue precipitate over cells expressing hPAP. Similarly, paraffin-embedded tissue sections were heat-inactivated at 65°C for 30 to 40 minutes, then exposed to BCIP solution for 18 to 40 hours. Tissue sections were counterstained with nuclear fast red for 1 to 2 minutes, dehydrated, and coverslipped.
Tumor Cell Transplantation
CK19-TAg mouse tumors were resected and minced with scissors in sterile phosphate buffered saline at approximately 0.25 g/ml. Between 0.1 and 0.3 ml of the resulting suspension was injected under the interscapular skin of syngeneic FVB nontransgenic mice. Animals were monitored daily to identify transplant growth latency. At the time of recipient sacrifice, portions of each tumor were fixed, sectioned, and stained as described above.
Immunoprecipitation and Western Analysis
Lysate Preparation
Kidney from a nontransgenic mouse, primary bladder tumors, and transplanted tumors from CK19-TAg transgenic mice were frozen in liquid nitrogen and stored at −80°C. Tissues were thawed at 4°C in 0.33 g of tissue per ml of lysis buffer (50 μl of phenylmethylsulfonyl fluoride (PMSF), 24 μl aprotinin, and 10 μl leupeptin added to 5 ml ECB buffer, which contains 100 mmol/L NaF, 0.5% NP-40, 120 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 8.0, and 200 μmol/L Na3VO4). The tissues were gently dounce homogenized for 12 to 15 strokes and/or homogenized with a spinning dounce head for 1 to 2 minutes at ∼6000 rpm at 4°C. Then 30 μl of PMSF per gram of tissue were added to each homogenate, and samples were incubated at 4°C for at least 30 minutes. The homogenates were centrifuged in 1.5-ml aliquots at 10,000 rpm for 20 minutes, and the resulting protein concentration of each supernatant was determined using Bradford analysis and UV spectrophotometry.
Immunoprecipitation
Approximately 1 mg total protein was incubated at 4°C for 1 hour with 2 μg of a relevant mouse monoclonal antibody (anti-pRb, PharMingen, San Diego, CA, #14001A; anti-TAg Pab101, Santa Cruz, Santa Cruz, CA, #SC-147; and anti-p53, CalBiochem, La Jolla, CA, #OP03). Fresh lysis buffer was added to bring the total volume to 150 to 200 μl, then 40 μl of Protein G-Agarose (Gibco BRL, Rockville, MD) were added to each sample. Samples were inverted continuously overnight at 4°C, then centrifuged at 5200 rpm for 5 minutes at 4°C to pellet the agarose beads. Beads were washed four times by centrifuging at 5200 rpm 5 minutes at 4°C, each time decanting and adding 100 to 150 μl fresh RIPA buffer (50 μl PMSF, 24 μl aprotinin, and 10 μl leupeptin added to 5 ml of 1× PBS, 1% NP-40, and 0.5% sodium deoxycholate). Following the last wash, the Protein G-Agarose from each sample was frozen at -−80°C until analyzed via Western Blot Analysis.
Western Blot Analysis
To each protein G-agarose pellet, 80 μl 1× TGE (125 mmol/L Tris, 1.25 mol/L glycine, 0.5% sodium dodecyl sulfate) and 20 μl loading dye were added, then each sample was boiled for 5 to 10 minutes and placed on ice. Thirty microliters of each sample were loaded onto a 10% stacking/8–12% resolving gel and electrophoresed at 120V to 160V for several hours. Colored molecular weight standards (Novex, Frankfurt, Germany, #LC5725) were loaded in separate wells. In general, electrophoresis was continued until a 26- to 30-kd marker was near the bottom of the gel. Proteins were transferred to PDVF membranes (Millipore, Bedford, MA, #IPVH15150) via electro-transfer in transfer buffer (25 μmol/L Tris-base, pH 8.3, 192 μmol/L glycine, and 20% methanol) at 30V overnight or 100V for 1 hour. Membranes were rinsed in water, dipped in methanol, then dried for 30 minutes (or stored at room temperature) before exposure to 20% methanol to allow visualization of protein bands. Membranes then were dipped in methanol, washed in water, and blocked in 5% low-fat milk dissolved in PBS-T (1× PBS and 0.1% Tween-20) for 30–90 minutes. Membranes were incubated with the anti-TAg antibody (2 μg antibody in 10 ml of the blocking buffer) for 1 hour at room temperature, washed in PBS-T once for 15 minutes and twice for 5 minutes, then incubated with secondary antibody (2 μl anti-mouse IgG saturated with human serum proteins (Pierce, Rockford, IL) in 10 ml blocking buffer) for 1 hour at room temperature. Membranes were washed once in PBS-T for 15 minutes and four times for 5 minutes, then incubated with Supersignal Substrate (Pierce) for 5 minutes. Finally, membranes were exposed to Kodak X-OMAT AR film for 30 seconds to 4 minutes.
Results
CK19-hPAP Transgenic Mice
To determine the organ- and cell-specific pattern of expression of transgenes containing CK19 gene regulatory elements, six founder mice were generated that carried the CK19-hPAP transgene. hPAP provides a good marker because it remains active after fixation in 4% paraformaldehyde and paraffin embedding. 33 Multiple tissues were collected from CK19-hPAP founder mice or their offspring, fixed, and stained as either whole tissue or paraffin-embedded tissue mounted on a slide. hPAP protein activity was observed in 3 lineages, and staining generally conformed to the pattern expected for CK19 (Table 1) ▶ . In particular, strong staining was observed in both basal and suprabasal urothelial cells (Figure 2A) ▶ . Staining was observed consistently in some unexpected sites, including arterial endothelium and stroma underlying urothelium (Figure 2A) ▶ . In the highest-expressing line, 1281–4, staining also was observed in urinary bladder smooth muscle. CK19-hPAP mice reproduced normally and displayed no gross or microscopic lesions in any tissue examined.
Table 1.
hPAP Staining in CK19-hPAP Transgenic Mice
| Organ | Cell type | Lineage | ||
|---|---|---|---|---|
| 1281-4 | 1282-5 | 1282-2 | ||
| Bladder | urothelial; stromal | ++++* | ++ | ND |
| Salivary gland | ductal and acinar epithelium | ++++ | ++ | ++ |
| Skin | follicular epithelium | ++++ | ++++ | ++++ |
| Kidney | distal tubules; collecting ducts | ++++ | ++ | +++ |
| Mesothelium | mesothelial cells | ++++ | + | + |
| Lung | airway epithelium | +++ | +/0 | ND |
| Pancreas | ductal epithelium | +++ | ++ | +++ |
| Prostate | secretory epithelium; stromal | +++ | ++ | ND |
| Mammary | ductal epithelium | ++ | +++ | +++ |
| Adrenal gland | medullary epithelium | ++ | ND | ND |
| Liver | biliary epithelium | ++ | 0 | ++ |
| Intestine | enterocytes; goblet cells | ++ | +/0 | +/0 |
| Spleen | arterial endothelium | + | ++ | + |
Organs were embedded in paraffin, sectioned at 5 μm, incubated with BCIP, and examined microscopically. The predominant cell type displaying blue reaction product is noted. Most tissues also displayed staining of arterial endothelium.
*Relative staining intensity in the listed cell types, ranging from intense (++++) to faint (+). (0) represents no staining, and (+/0) represents variable staining within a cell type. ND, not determined.
Figure 2.

CK19-mediated transgene expression. A: Section of bladder from a 18-week-old CK19-hPAP transgenic mouse incubated with BCIP showing diffuse and uniform staining in urothelium and adjacent stroma. Muscle is not stained. B: Somniferous tubules and bladder from a nontransgenic mouse (top; arrow indicates bladder) and a CK19-TAg transgenic male at 13 weeks of age (bottom). The transgenic mouse bladder is distended and the basal portion is diffusely infiltrated by neoplastic cells. Original magnifications, ×100 (A) and ×1 (B).
CK19-TAg Mice
Five CK19-TAg transgenic founder mice were generated, but 3 were found dead shortly after birth. Of 2 surviving founder mice, one remained healthy for 16 months of age, did not develop lesions, and did not pass the transgene to any of 8 offspring. The other founder mouse (designated 1244–3) developed a large urinary bladder neoplasm and was sacrificed at 80 days of age. Transgenic offspring of this mouse displayed significantly reduced weight gains (transgenic mice weighed approximately 70% as much as nontransgenic mice at 9 weeks of age; P < 0.003). Beginning at 10 weeks of age, both male and female CK19-TAg mice developed ruffled coats and lethargy, and they required sacrifice at a median age of 12 weeks of age (range, 11–14 weeks; n = 35).
Gross examination of diseased mice revealed the presence in all transgenic mice of a moderately firm, tan to red mass, 1 to 3 cm in diameter, in the pelvic region within and/or surrounding the urinary bladder (Figure 2B ▶ and Table 2 ▶ ). In bitransgenic mice carrying both CK19-TAg and CK19-hPAP transgenes, neoplastic cells stained intensely blue, indicating maintenance of CK19 promoter activity by neoplastic urothelium. This was confirmed by immunohistochemical detection of endogenous CK19 using the TROMA 3 monoclonal antibody (data not shown). Mice also displayed twofold adrenal and kidney enlargement and occasional hydronephrosis. In some mice, abdominal masses were associated with the adrenal gland or peritoneal surfaces (Table 2) ▶ . Other organs appeared normal. Microscopic examination of pelvic masses indicated an origin in the urinary bladder epithelium (Figure 3A) ▶ . Neoplastic cells uniformly displayed nuclear TAg protein (Figure 3B) ▶ . Masses were solid and composed of sheets of anaplastic cells with large irregular nuclei, prominent nucleoli, high nuclear to cytoplasmic ratio, and frequent mitotic figures (Figure 3, C and D) ▶ . They did not, however, maintain detectable expression of uroplakin, an integral membrane protein present in normal urothelial cells (Figure 3E) ▶ . In all masses, the cells were highly invasive, as evidenced by their presence under the urothelial stroma and within bladder smooth muscle (Figure 3F) ▶ , and their frequent extension in males into periprostatic connective tissue. Masses often were highly vascularized. In 5 of 20 mouse lungs examined microscopically, neoplastic cells were identified in blood vessels (Figure 3G) ▶ . These cells expressed TAg (Figure 3H) ▶ , and in 4 mice morphologically resembled the bladder cancer cells although the origin of these cells could not be confirmed by immunohistochemistry, because urinary bladder neoplasms and the metastases did not maintain uroplakin expression. Metastatic cells in the fifth mouse morphologically resembled cells in an adrenal neoplasm also present in that mouse. There was no evidence of TAg expression in airway epithelium nor of primary lung abnormalities in these mice. Collectively, these features indicated a diagnosis of invasive TCC. Interestingly, blood chemistry was only mildly abnormal in mice with large neoplasms: blood urea nitrogen (BUN) was elevated 2.2-fold in transgenic mice relative to controls (P = 0.02, n = 5) but creatinine was unchanged (P = 0.24, n = 5; Mann-Whitney test).
Table 2.
Incidence of Neoplasia in CK19-TAg Transgenic Mice
| Mice | Fraction of mice with the following lesions* | ||||
|---|---|---|---|---|---|
| Bladder neoplasia | Adrenal neoplasia | Prostate/CG neoplasia | Mesothelial neoplasia | Lung metastasis† | |
| Male | 17 /17 | 3 /17 | 4 /17 | 4 /17 | 2 /11 |
| Female | 11 /11 | 4 /11 | NA | 2 /11 | 3 /9 |
| Total | 28 /28 | 7 /28 | 4 /17 | 6 /28 | 5 /20 |
*Analysis based on microscopic examination of fixed, H&E-stained tissue sections.
†Lesions were restricted to vascular and perivascular areas in lung. No lesions were observed that resembled primary lung neoplasms, which generally appear as papillary or tubulo-papillary and are associated with airway epithelium.
CG, coagulation gland; NA, not applicable.
Figure 3.
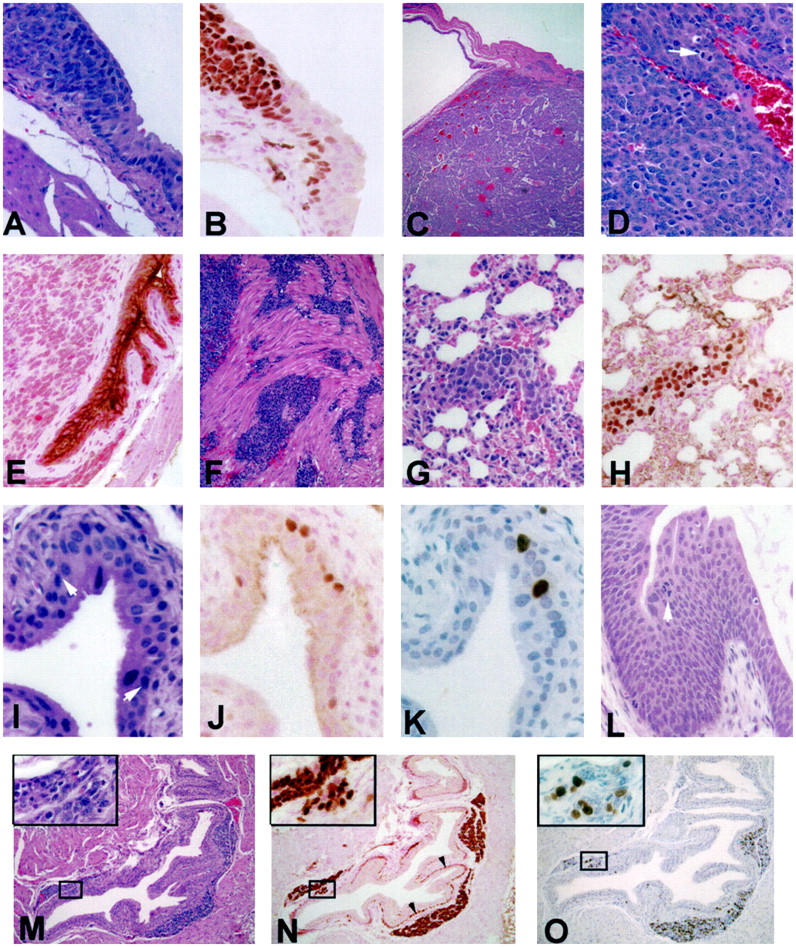
Bladder lesions and their progression in CK19-TAg transgenic mice. A: Transition from normal-appearing urothelium to neoplastic urothelium in a 12-week-old CK19-TAg transgenic mouse. B: Tissue section adjacent to A displaying immunohistochemical localization of TAg protein. C: Bladder tumor from a 12-week-old CK19-TAg transgenic mouse. D: Higher power photomicrograph of a bladder neoplasm illustrating typical cellular characteristics, including the presence of mitotic figures (arrow). E: Transgenic mouse urothelium displaying localization of uroplakin protein, which is excluded from neoplastic cells. F: Invasion of neoplastic urothelial cells into muscle. G: H&E stain of lung metastasis in the vessel of a 12-week-old CK19-TAg transgenic mouse, which expresses TAg protein (H). I−K: Earliest urothelial lesion visualized in a 3-week-old mouse. In I, arrowheads indicate the approximate lesion margins. There is an increase of nuclear TAg protein in some cells in the lesion (J), and an associated increase in BrdU labeling (darkly labeled cells in K). L: Large CIS present in a 6-week-old transgenic mouse. Arrowhead indicates a tripolar mitosis. M−O: H&E stain (M), TAg protein localization (N), and BrdU localization (O) in bladder of a 9-week-old transgenic mouse displaying characteristic nests of neoplastic cells between the suburothelial stroma and muscle. Insets display magnified views of the boxed areas in which a focal collection of cells extends through the stroma, appearing to connect urothelium and the substromal neoplastic focus of cells. Arrowheads in N indicate TAg-expressing hypertrophied basal urothelial cells in this mouse. For TAg and uroplakin immunohistochemistry, counterstain is nuclear-fast red; for BrdU immunohistochemistry, counterstain is hematoxylin. All others are H&E. Original magnifications, ×400 (A, B, D, E, G−L, and insets for M−O); ×40 (C); and ×100 (F, M−O).
Four of 5 neoplasms transplanted under the skin of syngeneic recipient mice were able to grow, becoming palpable with a latency of 4 to 5 weeks. Transplantability further demonstrated the malignant character of these cells. Transplanted neoplasms microscopically resembled the original TCCs, maintained uniform expression of TAg, and could be serially passaged in vivo.
Immunoprecipitation and Western blot analysis indicated that TAg protein was in a complex with pRb and p53 in bladder neoplasms. Protein was immunoprecipitated (IP) from a nontransgenic mouse kidney, a primary bladder neoplasm, and a transplanted bladder neoplasm with pRb, p53, or TAg antibodies. All IP samples except those from the nontransgenic mouse kidney displayed a labeled protein band at 96 kd on a Western blot when probed with TAg antibody (Figure 4) ▶ . Samples from one additional primary neoplasm displayed a similar pattern of protein binding.
Figure 4.

Immunoprecipitation and Western blot analysis of protein lysates isolated from nontransgenic mouse kidney (kid), a primary TAg-induced bladder neoplasm (9–5), and a transplanted bladder neoplasm (3–6). Lysates were immunoprecipitated as indicated at the bottom of the figure. Western blots were probed with anti-TAg antibody. p55 represents mouse immunoglobulin heavy chain, which is precipitated and then visualized on the blot by virtue of its reaction with the anti-mouse Ig secondary antibody reagents.
To identify stages of bladder lesion progression, mice were sacrificed at 3, 6, 9, and 12 weeks of age. One hour before sacrifice, each was administered 200 mg/kg BrdU to label cells undergoing DNA synthesis. Bladder lesions were identified, and for each lesion type the BrdU labeling index was determined (Table 3) ▶ . At all ages, nontransgenic mouse urothelium and normal-appearing transgenic mouse urothelium displayed similar and relatively low BrdU labeling indices. In 3-week-old mice, there was immunohistochemically detectable TAg expression in single or small collections of mildly hypertrophied basal urothelial cells, and these areas displayed a moderate increase in BrdU label (Figure 3, I ▶ -K). By 6 weeks of age, 4 of 7 transgenic mice displayed CIS, small foci of dysplastic urothelial cells with increased BrdU labeling relative to lesion-free urothelium. Occasional mice exhibited much larger focal CIS (Figure 3L) ▶ . At this and all subsequent ages, neoplastic cells always were visible between stroma and muscle (Figure 3, M ▶ -O). Cell nuclei in substromal foci stained uniformly for TAg, and multiple hypertrophied basal urothelial cells often displayed TAg immunoreactivity (Figure 3N) ▶ . Five of 7 mice examined at 6 weeks and all 7 mice examined at 9 weeks exhibited neoplastic cell invasion into muscle. By 12 weeks of age, each of 28 transgenic mice examined displayed invasive TCC. Most bladder carcinoma cells expressed TAg, and the BrdU labeling index in invasive neoplasms was approximately 30% (Table 3) ▶ . Interestingly, substromal-invasive foci and muscle-invasive neoplasms displayed nearly identical labeling indices. The finding of substromal lesions raises the possibility that some neoplasms may develop from simple epithelia, such as Brunn’s nests, present within the lamina propria. However, the clear presence of urothelial CIS and identifiable transitions in the urothelium from normal urothelium to neoplasm (Figure 3, A and B) ▶ indicates a urothelial origin for many neoplasms.
Table 3.
BrdU Labeling Index in CK19-TAg Transgenic Mouse Lesions
| Urothelium | CIS | Invasive TCC | ||
|---|---|---|---|---|
| Non-transgenic | Transgenic | Substromal | Muscle | |
| 0.5 ± 0.3% | 0.5 ± 0.5% | 10.3 ± 4.7% | 33 ± 6% | 29 ± 6% |
CIS, carcinoma in situ; TCC, transitional cell carcinoma. See text for description of lesions. Calculated as percentage of examined nuclei that were immunohistochemically positive for BrdU staining. For nontransgenic urothelium at each age, n = 2 mice. For all others, n ≥ 5 mice. For each lesion type except CIS, multiple microscopic fields were counted in each mouse, totaling at least 1000 nuclei examined. For CIS, all visible nuclei were examined.
The occasional presence of adrenal and mesothelial neoplasms (Table 2) ▶ was confirmed by microscopic examination (Figure 5) ▶ . Most male mice displayed focal dysplasia and hyperplasia in prostatic/coagulation gland and seminal vesicle epithelium associated with TAg immunoreactivity and increased BrdU labeling. Four males displayed small prostate/coagulation gland neoplasms (Table 2) ▶ . Finally, all transgenic mice displayed hyperplasia and dysplasia of distal convoluted renal tubules associated with increased TAg expression. This pattern of lesion localization is consistent with the pattern of CK19-hPAP expression observed in kidney. Despite the resemblance of some of these lesions to renal tubular CIS, renal neoplasms have not been observed in these mice.
Figure 5.

Adrenal gland and mesothelial neoplasms in CK19-TAg transgenic mice. A: Adrenal neoplasm (inset, normal adrenal gland). B: Mesothelial neoplasm (inset, normal mesothelium). Original magnifications, ×40 (A and A inset), ×100 (B), and ×400 (B, inset).
Discussion
The findings reported above demonstrate that urothelial-targeted expression of TAg, a potent viral transforming protein that inactivates cellular p53 and pRb, induces the rapid development of highly invasive TCCs in transgenic mice. TAg is a 708-amino acid protein product of SV40 and contains several biologically active domains. 26,34,35 In addition to binding and inactivating p53 and pRb, the TAg protein has other activities in eukaryotic cells, including ATPase activity, RNA and DNA helicase ability, binding to specific DNA sequences, and altering the properties of other cellular proteins. 26,34-37 However, TAg mutants lacking p53 and pRb binding sites are not transforming in vivo, suggesting that inactivation of these tumor suppressor genes represents the principal mechanism by which TAg induces tumors in transgenic mice. 38,39 Our findings complement those recently reported by Zhang et al, 40 who targeted TAg to urothelial suprabasal and some basal cells using the uroplakin II gene promoter. In that study, 2 founder mice developed TCC at 3 and 5 months of age, and offspring of mice in 3 lineages displayed evidence of CIS between 1 and 8 months of age. TAg-induced TCC also morphologically resembles BHBN-induced TCC in the mouse, with the exception that the latter treatment regimen frequently induces squamous cell carcinoma. 1,5 Furthermore, BHBN-induced neoplasms display a high frequency of p53 mutation or loss of heterozygosity, 24 and TCC latency is reduced in BHBN-treated mice lacking one p53 allele. 25 Collectively, these data suggest that loss of p53 and/or pRb activity can have a primary causal role in the etiology of nonpapillary TCC.
The resemblance of the disease in CK19-TAg transgenic mice to the most severe form of TCC in humans is striking. In both, disease pathogenesis is associated with (i) the presence of flat (nonpapillary) CIS, (ii) high frequency invasion of neoplastic cells (in CK19-TAg mice, nests of neoplastic cells are found between the suburothelial stroma and muscle, which is less common in the human disease; see Figure 3, M ▶ -O), and (iii) occasional metastasis. Furthermore, human nonpapillary TCC frequently is associated with loss of p53 and/or pRb activity, and these molecular alterations form the basis of the disease etiology in CK19-TAg transgenic mice. The similarities between human invasive TCC and CK19-TAg-induced TCC suggest that the model can be used to provide additional insight into disease initiation and progression, in particular the rapid development of the highly invasive phenotype, and to test novel, molecularly based therapies.
The pathogenesis described above contrasts with that of papillary TCC in humans, which displays a different morphology, is less often invasive, and does not commonly display pRb alterations, although frequent loss of chromosome 9 has been reported. 41 Papillary lesions never were observed in CK19-TAg transgenic mice, supporting suggestions that the molecular etiologies of invasive versus papillary, non-invasive TCCs are distinct. 5,9-11 However, it remains possible that the differences in appearance of these neoplasms reflect the order in which genetic alterations accumulate, and that either type of tumor cell can become invasive if it develops a sufficient complement of genetic changes (in particular, loss of p53 and pRb). In this sense, these pathways may converge.
Based on the CK19-TAg model, we propose the following stepwise pathogenesis for the development of invasive TCC in mice. In step 1, expression of TAg in basal urothelial cells and subsequent binding and inactivation of p53 and pRb produces focal CIS. This step presumably requires other cellular alterations, since all TAg-expressing urothelial cells do not display the altered morphology and growth characteristics associated with CIS. It also may involve up-regulation of TAg expression. In view of the universal and near-simultaneous progression to subsequent stages of pathogenesis, this step may be rate-limiting for the development of disease in this model. In step 2, CIS cells rapidly develop the ability to invade into and under the suburothelial stroma, and the resulting lesions spread quickly within the bladder wall. Substromal lesions already display a BrdU labeling index (a measure of the fraction of cells undergoing DNA synthesis) equivalent to that of neoplasms. In step 3, invasive urothelial cells enter and spread within muscle. In step 4, final stages of TCC development include rapid growth of the neoplastic mass within and outside of the urinary bladder with occasional metastasis. Incidence is 100%, and the rate at which this sequence of events proceeds is remarkable: all mice require sacrifice due to the presence of high-grade disease by 3 months of age.
Note that the transgene targeting strategy used in this study has several consequences. CK19 is expressed primarily in simple epithelial cells in salivary glands, esophagus, gastrointestinal tract, pancreas, liver, kidney, mammary gland, bladder, and lung, although other cell types also express this gene, including adrenal and mesothelial cells. 29 The overall similarity between transgene expression and endogenous CK19 expression was confirmed in this report by our analysis of CK19-hPAP transgenic mice, although some differences were identified. Because CK19 is expressed in multiple cell types, use of this promoter to target oncogene expression is likely to induce lesions in tissues that are most susceptible to transformation by the transgene coding region being expressed. In the CK19-TAg 1244–3 lineage, this included predominantly urothelium, but also adrenal gland, renal tubule epithelium, prostate/coagulation gland and seminal vesicle epithelia, and mesothelium. These latter tissues may be less sensitive than bladder epithelium to transformation by TAg or may express the transgene at lower levels. Nevertheless, lesions did not appear in several tissues known to be responsive to TAg expression. For example, Furth and colleagues 28,42 have demonstrated that salivary gland, a site of endogenous CK19 expression, is susceptible to TAg-induced transformation. Lack of lesions and immunohistochemically detectable TAg in salivary gland of our mice suggests that TAg is not expressed in this tissue. Similarly, mammary gland and lung epithelium can be transformed by TAg, and both express endogenous CK19, yet primary neoplasms did not develop at these sites in our CK19-TAg transgenic mice. These observations suggest that the pattern of TAg expression in the 1244–3 lineage does not reflect that of normal CK19. TAg expression may be influenced by the site of transgene integration and, in this lineage, limited to a restricted set of tissues. This suggestion could explain why these mice live, whereas most CK19-TAg founder mice died shortly after birth. As an alternative explanation for the line 1244–3 phenotype, the transgene may have inserted into and inactivated an endogenous gene, thereby influencing tumor phenotype. This possibility would be excluded by observations of the same phenotype in multiple lines, representing multiple independent transgene insertions. Although our findings are restricted to one line of CK19-TAg transgenic mice, the description by Zhang and colleagues of lesions in urothelium in three lines of transgenic mice expressing a UPII-TAg transgene provides supporting evidence that the bladder lesions we describe are caused by TAg expression and not by the site of integration. 40 Thus, in multiple lines of transgenic mice employing two different targeting strategies, expression of TAg in urothelium induces invasive TCC.
Finally, the maintenance of CK19 transgene promoter activity in bladder neoplasms indicates the practicality of targeting multiple transgenes simultaneously to urothelial cells, thereby permitting a detailed genetic analysis of factors involved in progression of cancer in this organ.
Acknowledgments
We thank Dr. Catherine Reznikoff, Dr. Somdatta Sarkar, and Melissa Burger for advice and helpful discussions during all stages of this study; Dr. Bernhard Bader, Dr. Lorraine Gudas, and Dr. Richard Palmiter for providing DNA clones; Dr. Tung-Tein Sun for providing anti-uroplakin antiserum; Dr. Matthew Schroeder for assistance with Western blotting; and Renee Szakaly for assistance with figure preparation.
Footnotes
Address reprint requests to Dr. Eric P. Sandgren, School of Veterinary Medicine, University of Wisconsin-Madison, 2015 Linden Drive West, Madison, WI 53706. E-mail: sandgren@svm.vetmed.wisc.edu.
Supported by National Institutes of Health grant RO1-CA76361 (to E. P. S.).
References
- 1.Oyasu R: Epithelial tumours of the lower urinary tract in humans and rodents. Food Chem Toxicol 1995, 33:747-755 [DOI] [PubMed] [Google Scholar]
- 2.Liebert M, Seigne J: Characteristics of invasive bladder cancers: histological and molecular markers. Semin Urol Oncol 1996, 14:62-72 [PubMed] [Google Scholar]
- 3.Foresman WH, Messing EM: Bladder cancer: natural history, tumor markers, and early detection strategies. Semin Surg Oncol 1997, 13:299-306 [DOI] [PubMed] [Google Scholar]
- 4.Lapham RL, Ro JY, Staerkel GA, Ayala AG: Pathology of transitional cell carcinoma of the bladder and its clinical implications. Semin Surg Oncol 1997, 13:307-318 [DOI] [PubMed] [Google Scholar]
- 5.Cohen SM: Urinary bladder carcinogenesis. Toxicol Pathol 1998, 26:121-127 [DOI] [PubMed] [Google Scholar]
- 6.van der Meijden AP: Bladder cancer. Br Med J 1998, 317:1366-1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein JP, Grossfeld GD, Ginsberg DA, Esrig D, Freeman JA, Figueroa AJ, Skinner DG, Cote RJ: Prognostic markers in bladder cancer: a contemporary review of the literature. J Urol 1998, 160:645-659 [DOI] [PubMed] [Google Scholar]
- 8.Arak R: American Cancer Society: Cancer Facts and Figures. Atlanta, GA, American Cancer Society, 1998, 6:12–14
- 9.Spruck CH 3rd, Ohneseit PF, Gonzalez-Zulueta M, Esrig D, Miyao N, Tsai YC, Lerner SP, Schmutte C, Yang AS, Cote R, Dubeau L, Nichols PW, Hermann GG, Steven K, Horn T, Skinner DG, Jones PA: Two molecular pathways to transitional cell carcinoma of the bladder. Cancer Res 1994, 54:784–788 [PubMed]
- 10.Reznikoff CA, Belair CD, Yeager TR, Savelieva E, Blelloch RH, Puthenveettil JA, Cuthill S: A molecular genetic model of human bladder cancer pathogenesis. Semin Oncol 1996, 23:571-584 [PubMed] [Google Scholar]
- 11.Knowles MA: Molecular genetics of bladder cancer: pathways of development and progression. Cancer Surv 1998, 31:49-76 [PubMed] [Google Scholar]
- 12.Cordon-Cardo C, Sheinfeld J, Dalbagni G: Genetic studies and molecular markers of bladder cancer. Semin Surg Oncol 1997, 13:319-327 [DOI] [PubMed] [Google Scholar]
- 13.Orntoft TF, Wolf H: Molecular alterations in bladder cancer. Urol Res 1998, 26:223-233 [DOI] [PubMed] [Google Scholar]
- 14.Qureshi KN, Lunec J, Neal DE: Molecular biological changes in bladder cancer. Cancer Surv 1998, 31:77-97 [PubMed] [Google Scholar]
- 15.Ozen H: Bladder cancer. Curr Opin Oncol 1998, 10:273-278 [PubMed] [Google Scholar]
- 16.Hovey RM, Chu L, Balazs M, DeVries S, Moore D, Sauter G, Carroll PR, Waldman FM: Genetic alterations in primary bladder cancers and their metastases. Cancer Res 1998, 58:3555-3560 [PubMed] [Google Scholar]
- 17.Richter J, Beffa L, Wagner U, Schraml P, Gasser TC, Moch H, Mihatsch MJ, Sauter G: Patterns of chromosomal imbalances in advanced urinary bladder cancer detected by comparative genomic hybridization. Am J Pathol 1998, 153:1615-1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grossman HB, Liebert M, Antelo M, Dinney CP, Hu SX, Palmer JL, Benedict WF: p53 and RB expression predict progression in T1 bladder cancer. Clin Cancer Res 1998, 4:829-834 [PubMed] [Google Scholar]
- 19.Cordon-Cardo C, Zhang ZF, Dalbagni G, Drobnjak M, Charytonowicz E, Hu SX, Xu HJ, Reuter VE, Benedict WF: Cooperative effects of p53 and pRB alterations in primary superficial bladder tumors. Cancer Res 1997, 57:1217-1221 [PubMed] [Google Scholar]
- 20.Keegan PE, Lunec J, Neal DE: p53 and p53-regulated genes in bladder cancer. Br J Urol 1998, 82:710-720 [DOI] [PubMed] [Google Scholar]
- 21.Cote RJ, Dunn MD, Chatterjee SJ, Stein JP, Shi SR, Tran QC, Hu SX, Xu HJ, Groshen S, Taylor CR, Skinner DG, Benedict WF: Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Res 1998, 58:1090-1094 [PubMed] [Google Scholar]
- 22.Becci PJ, Thompson HJ, Strum JM, Brown CC, Sporn MB, Moon RC: N-butyl-N-(4-hydroxybutyl)nitrosamine-induced urinary bladder cancer in C57BL/6 X DBA/2 F1 mice as a useful model for study of chemoprevention of cancer with retinoids. Cancer Res 1981, 41:927-932 [PubMed] [Google Scholar]
- 23.Ohtani M, Kakizoe T, Nishio Y, Sato S, Sugimura T, Fukushima S, Niijima T: Sequential changes of mouse bladder epithelium during induction of invasive carcinomas by N-butyl-N-(4-hydroxybutyl)nitrosamine. Cancer Res 1986, 46:2001-2004 [PubMed] [Google Scholar]
- 24.Ogawa K, Uzvolgyi E, St John MK, de Oliveira ML, Arnold L, Cohen SM: Frequent p53 mutations and occasional loss of chromosome 4 in invasive bladder carcinoma induced by N-butyl-N-(4-hydroxybutyl)nitrosamine in B6D2F1 mice. Mol Carcinog 1998, 21:70-79 [DOI] [PubMed] [Google Scholar]
- 25.Ozaki K, Sukata T, Yamamoto S, Uwagawa S, Seki T, Kawasaki H, Yoshitake A, Wanibuchi H, Koide A, Mori Y, Fukushima S: High susceptibility of p53(+/−) knockout mice in N-butyl-N-(4-hydroxybutyl)nitrosamine urinary bladder carcinogenesis and lack of frequent mutation in residual allele. Cancer Res 1998, 58:3806-3811 [PubMed] [Google Scholar]
- 26.Ludlow JW: Interactions between SV40 large-tumor antigen and the growth suppressor proteins pRB and p53. FASEB J 1993, 7:866-871 [DOI] [PubMed] [Google Scholar]
- 27.Kao C, Huang J, Wu SQ, Hauser P, Reznikoff CA: Role of SV40 T antigen binding to pRB and p53 in multistep transformation in vitro of human uroepithelial cells. Carcinogenesis 1993, 14:2297-2302 [DOI] [PubMed] [Google Scholar]
- 28.Furth PA: SV40 rodent tumour models as paradigms of human disease: transgenic mouse models. Devel Biol Standardiz 1998, 94:281-287 [PubMed] [Google Scholar]
- 29.Moll R, Franke WW, Schiller DL, Geiger B, Krepler R: The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982, 31:11-24 [DOI] [PubMed] [Google Scholar]
- 30.Bader BL, Franke WW: Cell type-specific and efficient synthesis of human cytokeratin 19 in transgenic mice. Differentiation 1990, 45:109-118 [DOI] [PubMed] [Google Scholar]
- 31.Hu L, Gudas LJ: Activation of keratin 19 gene expression by a 3′ enhancer containing an AP1 site. J Biol Chem 1994, 269:183-191 [PubMed] [Google Scholar]
- 32.Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD: Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci USA 1985, 82:4438-4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kisseberth W, Brettingen N, Lohse J, Sandgren E: Ubiquitous expression of marker transgenes in mice and rats. Devel Biol 1999, 214:128-138 [DOI] [PubMed] [Google Scholar]
- 34.Moens U, Seternes OM, Johansen B, Rekvig OP: Mechanisms of transcriptional regulation of cellular genes by SV40 large T- and small T-antigens. Virus Genes 1997, 15:135-154 [DOI] [PubMed] [Google Scholar]
- 35.Pipas JM: Molecular chaperone function of the SV40 large T antigen. Devel Biol Standardiz 1998, 94:313-319 [PubMed] [Google Scholar]
- 36.Zalvide J, Stubdal H, DeCaprio JA: The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol Cell Biol 1998, 18:1408-1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Damania B, Mital R, Alwine JC: Simian virus 40 large T antigen interacts with human TFIIB-related factor and small nuclear RNA-activating protein complex for transcriptional activation of TATA-containing polymerase III promoters. Mol Cell Biol 1998, 18:1331-1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Tobin GJ, Pipas JM, Van Dyke T: T-antigen mutant activities in vivo: roles of p53 and pRB binding in tumorigenesis of the choroid plexus. Oncogene 1992, 7:1167-1175 [PubMed] [Google Scholar]
- 39.Saenz Robles MT, Symonds H, Chen J, Van Dyke T: Induction versus progression of brain tumor development: differential functions for the pRB- and p53-targeting domains of simian virus 40 T antigen. Mol Cell Biol 1994, 14:2686–2698 [DOI] [PMC free article] [PubMed]
- 40.Zhang Z-T, Pak J, Shapiro E, Sun T-T, Wu X-R: Urothelium-specific expression of an oncogene in transgenic mice induced the formation of carcinoma in situ and invasive transitional cell carcinoma. Cancer Res 1999, 59:3512-3517 [PubMed] [Google Scholar]
- 41.Orlow I, LaRue H, Osman I, Lacombe L, Moore L, Rabbani F, Meyer F, Fradet Y, Cordon-Cardo C: Deletions of the INK4A gene in superficial bladder tumors. Association with recurrence. Am J Pathol 1999, 155:105–113 [DOI] [PMC free article] [PubMed]
- 42.Furth PA, Li M, Hennighausen L: Studying development of disease through temporally controlled gene expression in the salivary gland. Ann NY Acad Sci 1998, 842:181-187 [DOI] [PubMed] [Google Scholar]