

Abstract
Alport syndrome is a genetic disorder resulting from mutations in type IV collagen genes. The defect results in pathological changes in kidney glomerular and inner-ear basement membranes. In the kidney, progressive glomerulonephritis culminates in tubulointerstitial fibrosis and death. Using gene knockout-mouse models, we demonstrate that two different pathways, one mediated by transforming growth factor (TGF)-β1 and the other by integrin α1β1, affect Alport glomerular pathogenesis in distinct ways. In Alport mice that are also null for integrin α1 expression, expansion of the mesangial matrix and podocyte foot process effacement are attenuated. The novel observation of nonnative laminin isoforms (laminin-2 and/or laminin-4) accumulating in the glomerular basement membrane of Alport mice is markedly reduced in the double knockouts. The second pathway, mediated by TGF-β1, was blocked using a soluble fusion protein comprising the extracellular domain of the TGF-β1 type II receptor. This inhibitor prevents focal thickening of the glomerular basement membrane, but does not prevent effacement of the podocyte foot processes. If both integrin α1β1 and TGF-β1 pathways are functionally inhibited, glomerular foot process and glomerular basement membrane morphology are primarily restored and renal function is markedly improved. These data suggest that integrin α1β1 and TGF-β1 may provide useful targets for a dual therapy aimed at slowing disease progression in Alport glomerulonephritis.
Alport syndrome is a hereditary basement membrane disease affecting approximately one in 5,000 people. 1 The disease is manifest by juvenile to adult onset progressive glomerulonephritis usually associated with a high-frequency-specific sensorineural hearing loss, dot and fleck retinopathy, and lens abnormalities. No effective drug therapy exists for this disease, which is currently treated by dialysis and renal transplant. 1,2 The most common form of the disease is X-linked, and caused primarily by mutations in the collagen α5(IV) gene, 3 accounting for ∼80% of the cases. Mutations in the collagen α3(IV) or α4(IV) genes lead to the recessive forms of the disease. 4,5 The absence of any one of these type IV collagen chains can result in the absence of all three chains in the glomerular basement membrane (GBM), presumably due to an obligatory association of the three chains in forming the type IV collagen superstructure. 6,7 Normal distribution of the three α chains is observed in approximately one third of patients. 8
The adult GBM contains a thin subendothelial network of collagen α1(IV) and α2(IV) chains, and a thick subepithelial network of collagen α3(IV), α4(IV), and α5(IV) chains. 9 These networks are thought to be physically separate from one another. 10,11 In Alport syndrome the entire width of the GBM is comprised of collagen α1(IV) and α2(IV) chains, which is the normal collagen composition of the embryonic GBM. 12,13 These changes result in progressive loss of glomerular function because of alterations in the GBM, podocyte effacement, and mesangial matrix expansion.
Type IV collagen networks comprised of only α1(IV) and α2(IV) chains are more susceptible to endoproteolysis than GBM containing all five type IV collagen chains, 13 which is likely because of the greater number of crosslinks formed in a network of collagen α3(IV), α4(IV), and α5(IV) chains. 11 Based on these observations, it has been proposed that the irregular ultrastructure of Alport GBM might be attributed to focal endoproteolysis of the GBM.
Two independently produced gene knockout murine models for Alport syndrome have been described, 14,15 as well as one resulting from a random transgene insertion event. 16 These models have proven to have progressive renal disease that is remarkably similar to that in humans.
Expansion of the mesangial matrix occurs early in Alport renal pathogenesis. The most abundant integrin on mesangial cells is the α1β1 heterodimer. 17,18 An α1 integrin knockout has been produced that shows no renal abnormalities and no phenotype detrimental to the survival of the animal. 19 Considering the recently described roles for α1β1 integrin in collagen-dependent cell proliferation, cell adhesion, mesangial matrix remodeling, and mesangial cell migration, 19-21 we suspected that integrin α1β1 might play a specific role in Alport renal disease progression. To test this notion, we produced a mouse null at both the collagen α3(IV) gene (Alport mouse) and the α1 integrin gene. These double-knockout mice have delayed onset and slowed progression of glomerular disease, attenuated expansion of the mesangial matrix, and markedly improved foot process architecture, illustrating a major role for α1β1 integrin in Alport glomerular disease progression.
Transforming growth factor (TGF)-β has been shown to promote accumulation of extracellular matrix in both wound repair and fibrotic diseases, including glomerulonephritis. 22 In recent studies, we demonstrated a likely role for TGF-β1 in Alport glomerular and tubulointerstitial disease. 23 Herein, we extend these earlier studies by illustrating that inhibition of TGF-β1, by injecting a type II TGF-β soluble receptor as a competitive inhibitor, prevents the irregular thickening of the GBM.
Treating the double knockouts with the TGF-β1 soluble receptor provides synergistic benefits, restoring podocyte foot process architecture, inhibiting matrix deposition in the GBM, and slowing mesangial matrix expansion. Based on this new evidence, we conclude that renal pathogenesis in Alport syndrome involves biochemical pathways modulated by TGF-β1 and integrin α1β1, and that the two pathways affect distinct aspects of glomerular pathology.
Materials and Methods
Mice
The collagen α3(IV) knockout mice were described previously. 14 These mice, which were originally in the 129 Sv/J background, were successively back-crossed 10 times with 129 Sv mice. Heterozygotes were then crossed with homozygote integrin α1 knockouts, also described previously, 19 which were on a pure 129 Sv background. A breeding stock of mice heterozygous for the collagen α3(IV) mutation and homozygous for the integrin α1 mutation was established. No difference in the kinetics of renal pathogenesis was observed for Alport mice on the 129 Sv/J versus 129 Sv backgrounds.
Urinary Protein Analysis
Semiquantitative measurements of urinary protein were performed using Albustix reagent strips (Bayer Corporation, Elkhart, IN), and reading the relative amounts from the color chart provided with the kit. Successive readings were performed weekly on fresh urine from the same experimental animals. Microhematuria was assessed using Hemastix reagent strips (Bayer Corporation).
Fractions of the above urine samples were subjected to gel analysis. Samples (the equivalent of 0.5 μl of undiluted urine) were fractionated by electrophoresis through 10% denaturing acrylamide gels. Urine creatinine levels were measured when sufficient urine was collected for meaningful readings (>100 μl). We observed no significant effect of the TGF-β1 inhibitor on urine creatinine levels, and these levels fluctuated by no more than 20% throughout the time course. The protein in the gels was stained with Coomassie blue and photographed. Bovine serum albumin was used as a molecular weight standard.
Determination of End-Stage Renal Disease
End-stage renal disease was defined as the point in the disease progression where a weight loss of >15% of the body mass was observed. Animals were euthanized at this point and end stage was confirmed by visual examination. Blood urea nitrogen levels at this stage were consistently >200 mg/dl. The kidneys are visibly smaller with a granular surface and are pale in color. When such visual characteristics were present, histological examination invariably confirmed advanced fibrosis.
Transmission Electron Microscopy
Fresh external renal cortex was minced in 4% paraformaldehyde, allowed to fix for 2 hours, and stored at 5°C in phosphate-buffered saline (PBS). The tissue was washed extensively (5 times for 10 minutes each at 4°C) with 0.1 mol/L Sorenson’s buffer (Sorenson’s buffer was made by combining 100 ml of a 200 mmol/L monobasic sodium phosphate with 400 ml of 200 mmol/L dibasic sodium phosphate, with 500 ml of water, and adjusted to pH 7.4), and postfixed in 1% osmium tetroxide in Sorenson’s buffer for 1 hour. The tissue was then dehydrated in graded ethanol (70%, then 80%, then 90%, then 100% for 10 minutes each), and finally in propylene oxide and embedded in Poly/Bed 812 epoxy resin (Polysciences, Inc., Warrington, PA) following the procedures described by the manufacturer. Glomeruli were identified in 1-μm sections stained with toluidine blue, and thin sections were cut at 70-nm thickness using a Reichert Jung Ultracut E ultramicrotome (Cambridge Instrument Co., Vienna, Austria). Sections were mounted onto grids and stained with uranyl acetate and lead citrate. Grid-mounted sections were examined and photographed using a Phillips CM10 electron microscope.
Scanning Electron Microscopy
Small pieces (approximately 2-mm cubes) of kidney cortex were fixed in 3% phosphate-buffered glutaraldehyde, then postfixed in 1% phosphate-buffered osmium tetroxide. Samples were then dehydrated in graded ethanols, and critical point-dried in carbon dioxide. The cubes were then cracked in pieces by stressing them with the edge of a razor blade, and mounted with glue onto stubs with the cracked surface facing upward. The surface was sputter-coated using gold/palladium and visualized with a scanning electron microscope.
Immunofluorescence Analysis
Fresh kidneys were removed and embedded in Tissue Tek OCT aqueous compound. They were frozen at −150°C and sectioned at 4 μm on a Microm cryostat. Slides were fixed in cold 100% acetone for 10 minutes then air-dried overnight. Slides were washed three times in cold 1× PBS. Primary antibodies were diluted together in 7% nonfat dry milk (1:200 for entactin, 1:10 for laminin α2), applied to the sections, and incubated overnight at 4°C in a humidified dish. Slides were then washed successively for 5, 7, and 10 minutes with cold 1× PBS. Secondary antibodies were diluted together at 1:00 (Texas red α-rabbit for lam-α2 and fluorescein isothiocyanate α-rat for entactin) in the blocking agent and applied for 4 hours at 4°C. After PBS washes, Vectashield (Vector Laboratories, App Imaging, Santa Clara, CA) anti-fade mounting media was applied and sections were coverslipped, sealed, and imaged. Images were collected using a Cytovision Ultra image analysis system (Applied Imaging, Inc.) interfaced with an Olympus BH-2 fluorescence microscope.
For the remaining laminin chain-specific antibodies, slides were brought to room temperature, then postfixed in cold (−20°C) acetone for 15 minutes, and air-dried overnight at 5°C. The specimens were rehydrated by three successive washes in PBS for 10 minutes each at room temperature, then denatured by immersing them in 0.1% sodium dodecyl sulfate at 37°C for 45 minutes. This step greatly enhanced immunoreactivity, presumably by exposing the masked laminin epitopes (B. Patton, personal communication). The specificity of the laminin chain-specific antibodies have been established in previous publications. 24,25 The anti-laminin α1 (8B3) and β1 (5A2) antibodies were mouse monoclonals, and a gift from D. Abrahamson (Department of Anatomy and Cell Biology, University of Kansas Medicine Center, Kansas City, KS). 26 Anti-laminin α2 chain-specific rabbit antibodies were a gift from Dr. Peter Yurchenco (Robert Wood Johnson Medical School, Piscataway, New Jersey). 27 The anti-laminin α3 chain-specific antibodies were a gift from Dr. Bob Burgeson (anti laminin-5 rabbit antiserum No. 4101) and described previously. 28,29 The anti-laminin α4 (C0877) and α5 (8948) rabbit antisera were provided by Jeff Miner (Washington University School of Medicine Department of Nephrology, St Louis, MO). 24 The anti-laminin β2 (Gpb1) chain-specific guinea pig antisera were a gift from Joshua Sanes (Washington University School of Medicine Department of Anatomy and Neurobiology, St. Louis, MO). 31 Anti-laminin β3 antibodies were provided by Yoshi Yamada (National Institute of Dental and Craniofacial Research, Bethesda, MD). 31 A rat monoclonal antibody for the laminin γ1 chain was purchased from Chemicon (Temecula, CA). All secondary reagents were purchased from Vector Laboratories.
For fibronectin immunostaining, the antibody was purchased from Sigma (St. Louis, MO; catalogue no. F3648). The same procedure was used as used for the laminin α2 chain.
Construction, Purification, and Activity of the Murine TGFβR:Fc
The extracellular domain of the murine TGF-β type II receptor was amplified from a murine lung cDNA library (Clontech, Palo Alto, CA) by PCR, and engineered to contain a 5′ NotI, and a 3′ SalI restriction site. The Fc region of murine IgG2a was amplified by PCR from a murine hybridoma, and engineered to contain a 5′ SalI restriction site and a 3′ NotI restriction site. The receptor and Fc fragments were purified, digested with the appropriate restriction enzymes, and ligated into the expression vector pEAG347, which contains tandem SV40, and adenovirus major late promoters; the SV40 late polyA termination signal; an ampicillin resistance gene; and a pSV2 dihydrofolate reductase-derived selection marker. The resulting construct (pAA002) was transformed into competent JM109, and plasmids were selected for the correct orientation of the receptor-Fc fusion gene. Proper sequence and alignment was confirmed by DNA sequencing.
pAA002 was transfected in Chinese hamster ovary cells (CHO DUKX-B1) by electroporation. After selection in 200 nmol/L methotrexate, single clones were selected and screened for the expression of mTGFβR:Fc. The clone with the highest titer was picked for expansion to 20 L fermentors, and the expressed protein was purified over protein A-Sepharose (Pharmacia, Piscataway, NJ), under sterile, endotoxin-free conditions. The protein is >95% pure as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and contains <1 U endotoxin per mg protein.
Activity of the mTGFβR:Fc was assessed in the mink lung epithelial cell assay as described. 32-34 Briefly, Mv1Lu cells (ATCC CCL-64) were maintained in minimal essential medium supplemented with 100 U/ml penicillin, 100 Tg/ml streptomycin, 10% fetal bovine serum, and 4 mmol/L l-glutamine. To test, serial dilutions of TGFβR:Fc were incubated with 0.1 ng/ml TGF-β1, 0.5 ng/ml TGF-β2, and 0.05 ng/ml TGF-β3 (R&D Systems, Minneapolis, MN) for 1 hour in assay medium (minimal essential medium supplemented with 100 U/ml of penicillin, 100 Tg/ml of streptomycin, and 10% fetal bovine serum) in a 96-well microtiter tissue culture plate. Mv1Lu cells were resuspended in assay medium and added to the assay plate at a concentration of 6,000 cells per well. The cells were incubated at 37°C for 48 hours and pulsed with [3H]thymidine (70 to 86 Ci/mmol; Amersham) for an additional 6 hours. DNA synthesis, which reflects cell proliferation, was determined by measuring incorporation of [3H]thymidine. As reported for similar TGF-β receptor antagonists, 32,33 mTGFβR:Fc blocks TGF-β1 (IC50 = 1 nmol/L), and TGF-β3 (IC50 = 1 nmol/L), but not TGF-β2-mediated inhibition of Mv1Lu cell proliferation.
Results
Loss of α1β1 Integrin Slows Renal Disease Progression, Attenuates Expansion of the Mesangial Matrix, and Rescues Podocyte Foot Process Architecture in Alport Mice
The collagen α3(IV)-deficient mouse developed in this laboratory 14 was crossed with the integrin α1-deficient mouse 19 to produce an animal null at both alleles. Comparative analysis of renal disease pathogenesis of Alport mice versus these double-knockout mice revealed a markedly slower rate of renal disease in the double knockouts. Both the time of onset and the rate of progression of proteinuria was delayed relative to the Alport mice (Figure 1A ▶ and Table 1 ▶ ), suggesting improved function of the glomerular filter. The data in Table 1 ▶ represent successive analysis of urinary albumin in three sets of experimental animals, and illustrate that differences in onset and progression of proteinuria in the Alport mouse versus the double knockout are highly reproducible. Microhematuria was also assessed, and did not vary significantly among the experimental groups. The mean age of end-stage renal failure (as defined in the Methods section), based on analysis of 10 experimental sets (10 controls and 10 Alport, 10 integrin α1 null mice, and 10 double-knockout animals) was extended from 8.5 ± 0.5 weeks in the Alport mice to 14.5 ± 0.9 weeks in the double knockouts.
Figure 1.

Proteinuria in experimental mice. Proteinuria was measured by gel electrophoresis of urine samples taken at weekly intervals from an individual set of experimental animals. The age of the mice (in weeks) at the time of sample collection is indicated above each lane. Group A(1) is the profile for an untreated 129 Sv/J Alport mouse; Group A(2) is that for a double-knockout mouse; Group B(1) is the extended profile for a double-knockout mouse; Group B(2) is a littermate double knockout treated with mTGFβR:Fc. Mr, molecular weight standards.
Table 1.
Inactivating α1β1 Integrin and TGF-β1 Results in Additive Improvement in Slowing Both the Rate of Onset and Progression of Albuminuria in the Alport Mouse System
| Age (Wks) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Control | − | − | − | − | − | − | − | − | − | − |
| − | − | − | − | − | − | − | − | − | − | |
| − | − | − | − | − | − | − | − | − | − | |
| Control Sol Rec | − | − | − | − | − | − | − | − | − | − |
| − | − | − | − | − | − | − | − | − | − | |
| − | − | − | − | − | − | − | − | − | − | |
| Alport | − | − | ± | + | + | ++ | +++ | +++ | Renal failure | |
| − | − | ± | + | + | ++ | +++ | +++ | |||
| − | − | ± | + | + | ++ | +++ | +++ | |||
| DKO | − | − | − | − | ± | + | + | ++ | ++ | ++ |
| − | − | − | − | ± | + | + | + | ++ | ++ | |
| − | − | − | − | − | + | + | + | ++ | ++ | |
| DKO Sol Rec | − | − | − | − | − | − | ± | ± | ± | + |
| − | − | − | − | − | − | ± | ± | ± | + | |
| − | − | − | − | − | − | − | ± | ± | ± | |
Semiquantitative assessment of proteinuria was performed using Albustrix (Bayer Corporation, Elkhart, IN) at the indicated times. Each symbol represents an individual urine analysis. The data represents successive analysis of three individual animals for each of the indicated experimental groups. Approximate albumin concentrations: (−) = negative; (±) = trace; (+) = about 0.3 mg/ml; (++) = about 1 mg/ml; (+++) = greater than 3 mg/ml. DKO = double knockout; SolRec = animals treated with mTGFβR:Fc.
Transmission electron microscopic analysis (TEM) of the glomerular capillary loops revealed attenuated GBM damage in 7-week-old double-knockout versus age-matched Alport mice (compare focal thickening of the GBM in Figure 2G ▶ with D), thus the absence of α1 integrin appears to have provided a protective effect on GBM damage in the Alport mouse model. The morphology of the podocyte foot processes was normal in the double knockout as compared with the Alport mouse both by transmission electron microscopy (Figure 2 ▶ ; A, D, and G) and scanning electron microscopy (Figure 2 ▶ ; B, E, and H). Effacement of the podocyte foot processes, as observed in the Alport mouse (Figure 2, D and E) ▶ , obliterates the slit diaphragms, potentially blocking the glomerular filter.
Figure 2.
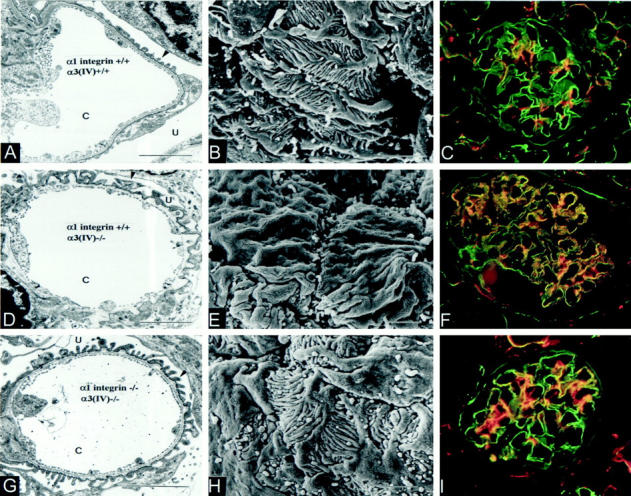
Ultrastructural damage to the glomerular capillary loop and glomerular distribution of the laminin α2 chain in control and double-mutant mice. Renal cortex from normal (A–C), Alport (D–F), and double-knockout (G–I) mice was harvested at 7 weeks. Tissue was analyzed by transmission electron microscopy (A, D, and G), scanning electron microscopy (B, E, and H), or dual immunofluorescence using antibodies specific for entactin (green), and the laminin α2 chain (red) (C, F, and I). Arrows denote foot processes. C, capillary lumen; U, urinary space. Scale bars, 1.5 μm for transmission electron microscopic photographs and 2 μm for scanning electron microscopy photographs. Immunofluorescence panels illustrate sections of whole glomeruli at ×400 magnification.
One underlying mechanism driving effacement of the foot processes is thought to be the loss of focal contact adhesion between the podocytes and the GBM. 35 Thus, we performed a comparative examination of the molecular composition of the GBM in normal mice and Alport mice. Earlier studies have shown that type IV collagen and fibronectin, which localize primarily to the mesangial matrix in control mice, are abundant in the GBM of Alport mice. 14,15 A comprehensive analysis of the known laminin chains in normal versus Alport mice has not yet been performed. We used antibodies specific for the different laminin chains to examine the GBM composition of normal versus Alport mice. The results in Figure 3 ▶ illustrate a dramatic shift in laminin α2 (Figure 3, C ▶ versus D) and laminin β1 (Figure 3, G ▶ versus H) chain expression from the mesangial matrix in control mice to the GBM in Alport mice. No changes in the glomerular localization of the α5, β2, or γ1 chains were observed in normal versus Alport mice. Consistent with earlier reports, 36 the α3, α4, and β3 chains are not expressed in the renal glomerulus. By co-localization inference, these data suggest that either laminin-2 (a heterotrimer comprised of the α2, β1, and γ1 chains) or a combination of laminin-2 and laminin-4 (comprised of the α2, β2, and γ1 chains), whereas they are completely absent from the GBM of normal mice, are abundant in the GBM of Alport mice.
Figure 3.
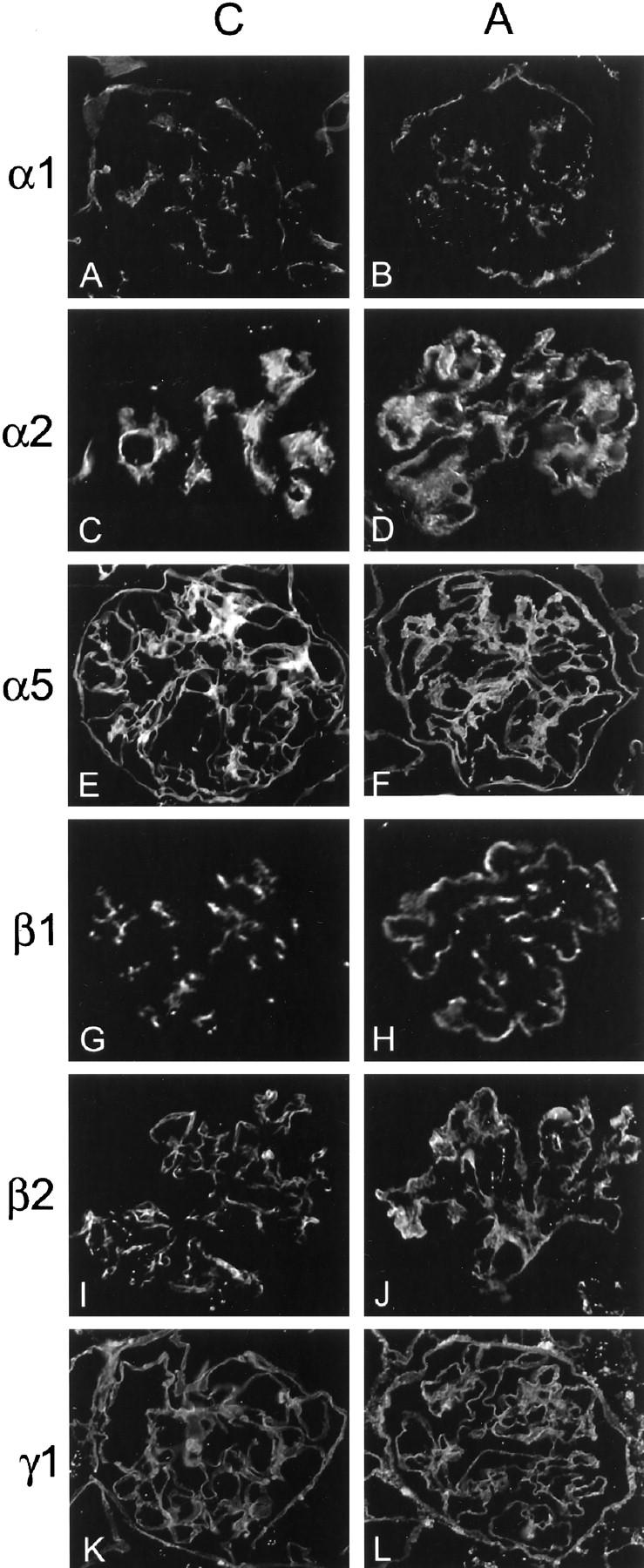
Analysis of the laminin chain composition of the GBM in normal versus Alport mice. Antibodies specific for the indicated laminin chains were used to immunostain glomeruli from 7-week-old normal mice and Alport littermates. The laminin chain specificity of the antibody used is indicated to the left of each set of panels. C, control; A, Alport. Based on co-localization, either laminin-2 (α1β1γ1) or both laminin-2 and laminin-4 (α1β2γ1) are present in the GBM of the Alport mouse, but not in the GBM of the control.
In Figure 2, C, F, and I ▶ , the GBM is stained in green, and laminin α2 chain is stained in red to illustrate that the laminin α2 chain, which normally localizes specifically to the glomerular mesangium (Figure 2C) ▶ localized heavily in the GBM of Alport mice (Figure 2F) ▶ . In double-knockout mice, the laminin α2-chain localization was similar to that in normal mice, however some laminin α2 staining is observed in the GBM (Figure 2I) ▶ . Similar results were obtained using antisera specific for the β1-laminin chain (data not shown). The changes in laminin composition of the GBM in normal versus Alport mice may be causally linked to effacement of the podocyte foot processes.
As α1β1 integrin plays a key role in cell adhesion, spreading, and migration in other systems, 19,37,38 it seemed plausible that expansion of the mesangial matrix, an early marker of Alport glomerular pathogenesis, might be attenuated in the double-knockout mice. In Figure 4 ▶ a fibronectin immunostain was used to illustrate that this is indeed the case. This abundant mesangial matrix marker reveals extensive expansion of the mesangial matrix in Alport versus normal mice at 7 weeks of age (Figure 4, A ▶ versus B). Age matched double-knockout mice, however, do not exhibit noticeable expansion of the glomerular mesangium (Figure 4C) ▶ . By 12 weeks of age, however, the mesangial matrix in double-knockout mice is expanded (data not shown), illustrating that the absence of α1 integrin attenuates, but does not inhibit expansion of the mesangial matrix.
Figure 4.

Absence of integrin α1β1 is associated with attenuated expansion of the mesangial matrix. The glomerular mesangium from 7-week-old control (A), Alport (B), and double-knockout (C) mice was stained using antibodies specific for fibronectin.
Blocking TGF-β1 Inhibits Focal GBM Thickening, but Not Podocyte Foot Process Effacement in Alport Mice
Recently we described induction of TGF-β1 mRNA as well as fibronectin, laminin β1, and collagen α1(IV) mRNAs in the podocytes of the Alport mouse. 23 We further evaluated the role of TGF-β1 on renal disease progression in the Alport mouse by using a newly designed soluble receptor inhibitor. A number of groups, including ours, have reported the development of soluble TGF-β type II receptors as in vitro and in vivo antagonists of TGF-β. 32,33,39 As reported for these similar TGF-β receptor antagonists, mouse TGFβR:Fc blocks TGF-β1 (IC50 = 1 nmol/L), and TGF-β3 (IC50 = 1 nmol/L), but not TGF-β2-mediated inhibition of mink lung tumor (Mv1Lu) cell proliferation (Figure 5) ▶ . We noted no inflammation or immune infiltration in normal mice chronically administered effective doses of this inhibitor (data not shown).
Figure 5.

The soluble TGF-β type II receptor (mTGFβR:Fc) inhibits TGF-β1 and TGF-β3 activities, but not TGF-β2 activity. The biological activity of mTGFβR:Fc was tested in the mink lung cell (Mv1Lu) proliferation assay. MvL1 cells were pre-incubated with either 0.1 ng/ml TGF-β1, 0.5 ng/ml TGF-β2, or 0.05 ng/ml TGF-β3 for 1 hour, then transferred to wells containing the indicated concentrations of the mTGFβR:Fc. Cells were incubated for 48 hours, pulsed with [3H]thymidine for 6 hours, and [3H]thymidine incorporation into the DNA measured by liquid scintillation spectrometry (y axis).
Alport mice were injected twice weekly through the tail vein with 25 μg of mTGFβR:Fc starting at 3 weeks of age and kidneys were harvested at 7 weeks of age. Transmission electron microscopy illustrates that treatment with the inhibitor prevents irregular thickening of the GBM, but does not prevent podocyte foot process effacement (Figure 6, A–C) ▶ . Scanning electron microscopy reveals the podocyte foot processes are effaced in Alport animals treated with mTGFβR:Fc (Figure 6F) ▶ . Laminin α2 is present in the GBM of Alport mice treated with mTGFβR:Fc (Figure 6G) ▶ . Alport mice treated with mTGFβR:Fc die of renal failure at approximately the same time as untreated Alport mice, presumably because of effacement of the podocyte foot processes which results in massive proteinuria (data not shown).
Figure 6.
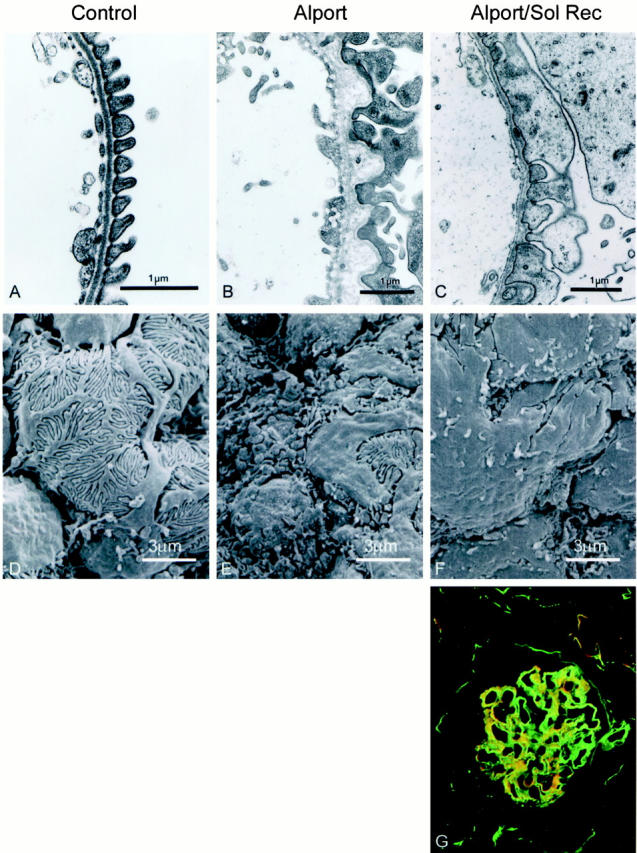
TGF-β1 inhibition prevents GBM thickening, but not podocyte foot process effacement in the Alport mouse. Renal cortex from 7-week-old normal mice (A and D), Alport mice (B and D), and Alport mice treated with mTGFβR:Fc (C and E) was analyzed by both transmission electron microscopy (A, B, and C) and scanning electron microscopy (D, E, and F). Glomerular capillary loops and podocyte surface architecture are shown. F: Dual immunofluorescence immunostain (laminin α2 in red and entactin, a marker for GBM, in green) of a glomerulus from a 7-week-old Alport mouse treated with mTGFβR:Fc.
Blocking TGF-β1 and Integrin α1β1 in Alport Mice Restores Both GBM and Podocyte Architecture
The results shown in Figures 2 and 6 ▶ ▶ suggest α1β1 integrin and TGF-β1 play distinct roles in Alport glomerular pathogenesis. If so, blocking TGF-β1 in Alport mice null for α1β1 integrin should prevent both podocyte foot process effacement and GBM thickening. To test this hypothesis, mTGFβR:Fc was administered to double-knockout mice starting at 4 weeks of age, and the kidneys were analyzed at 10 weeks of age. The treated animals were compared directly with untreated double-knockout littermates. Data shown in Table 1 ▶ and Figure 1B ▶ illustrates that both the time of onset and progression of proteinuria are markedly attenuated in double knockouts treated with mTGFβR:Fc relative to untreated double-knockout mice. Double-knockout animals treated with mTGFβR:Fc die at 18 to 24 weeks of age. As shown in Figure 7 ▶ , the GBM of the double-knockout mice at 10 weeks of age contains many areas of focal thickening, indicating progression of the glomerular pathology relative to 7-week-old double knockouts (compare Figures 7B and 2G ▶ ▶ ). The podocyte foot processes have normal morphology, except for areas where the GBM is focally thickened. In these regions, the foot processes appear fused. In the double knockouts treated with the soluble receptor, both basement membrane thickening and effacement of the podocyte foot processes are virtually absent. Most of the glomeruli in the treated double knockouts reflect that illustrated in Figure 7C ▶ , where some irregularities in the glomerular basement membrane are visible. Approximately 20% of the glomeruli, however, are morphologically indistinguishable from those of control animals (Figure 7D) ▶ . By comparison, out of >50 glomeruli examined by transmission electron microscopy, none of the glomeruli in 3-week-old Alport mice are morphologically normal. Laminin α2 in the untreated 10-week-old double knockouts shows considerable staining in the GBM (Figure 7E) ▶ , however the staining is focally deposited rather than distributed throughout the full length of the GBM, as observed in the 7-week-old Alport mice (compare Figure 7E ▶ with 2F). This is consistent with the focal thickening observed on transmission electron microscopy micrographs (Figure 7B) ▶ . In 10-week-old double knockouts treated with mTGFβR:Fc, laminin α2 staining is restricted to the glomerular mesangium (Figure 7F) ▶ , identical to that observed in normal animals (Figure 2C) ▶ . Thus, blocking α1β1 integrin and TGF-β1 is synergistic in slowing the rate of both onset and progression of glomerular pathology in the Alport mouse.
Figure 7.
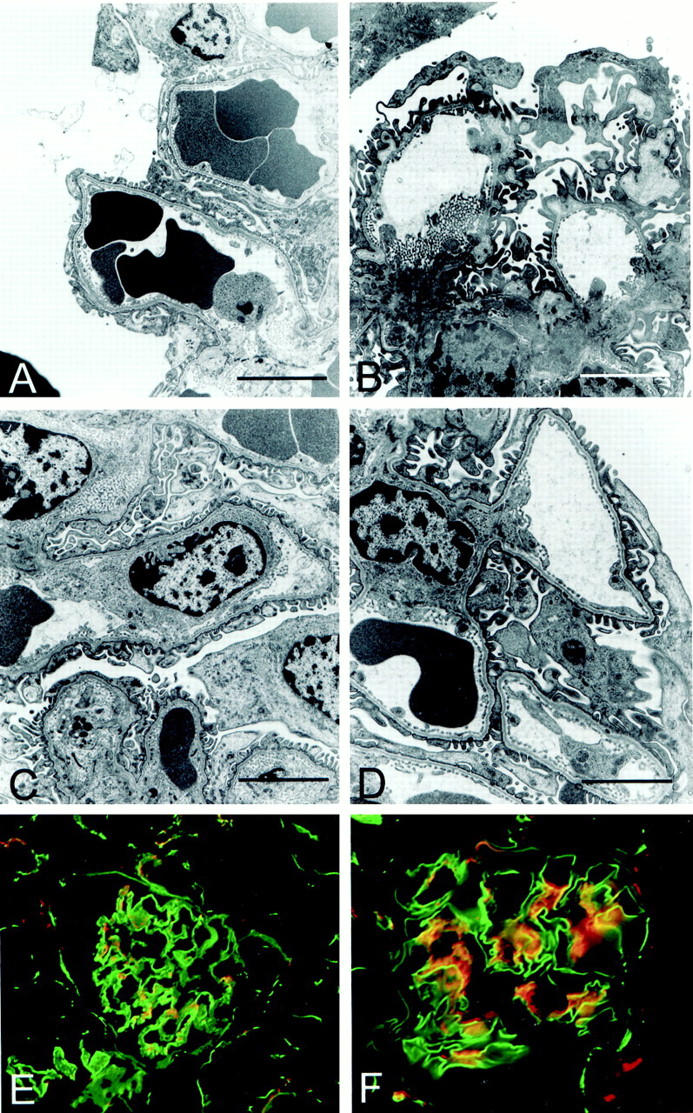
TGF-β1 inhibition primarily restores both GBM and podocyte architecture in double-knockout mice. Renal cortex from normal mice (A), double-knockout mice (B), and double-knockout mice treated with mTGFβR:Fc (C and D) was harvested at 10 weeks of age and analyzed by transmission electron microscopy. Fields represent the GBM architecture of typical glomerular capillary loops. E and F: Dual immunofluorescence staining using antibodies specific for entactin (green) and the laminin α2 chain (red). E: Glomerulus from a 10-week-old untreated double-knockout mouse. F: Glomerulus from a 10-week-old double-knockout mouse treated with mTGFβR:Fc. Scale bars, 3 μm.
Discussion
The delayed onset and progressive nature of Alport renal pathogenesis suggests the existence of a molecular triggering mechanism which initiates an imbalance in GBM homeostasis. The age of onset for Alport renal disease, although usually in the juvenile, can be quite variable, suggesting that the metabolic pathways affecting onset and progression are modulated by differences in the genetic background between individuals. Thus, identification of the specific metabolic imbalances might provide targets for therapies that delay onset, progression, or both.
Herein we describe that at least two pathways play distinct contributory roles in the glomerular pathology of Alport syndrome. The first involves integrin α1β1, which is the most prevalent integrin found on the surface of mesangial cells. 17,18 Our data illustrates this integrin is involved in the mechanisms of both podocyte foot process effacement and mesangial matrix expansion. Its effect on podocyte effacement is likely mediated via GBM deposition of laminin isoforms not native to the GBM. The second pathway involves TGF-β1, which we show is driving focal thickening of the GBM, a hallmark for diagnosis of Alport GBM pathogenesis. When both TGF-β1 and α1β1 integrin are removed from Alport mice, synergistic improvement in renal morphology and function are observed, indicating that these two pathways work in distinct ways to drive renal disease progression in Alport syndrome.
Integrin α1β1 and Alport Renal Disease
A major indicator of Alport glomerular pathogenesis is expansion of the mesangial matrix. 2 How or whether changes in the glomerular mesangium have anything to do with observed GBM damage in Alport syndrome, however, is not clear. Integrin α1β1 has been shown to be important for gel contraction of mesangial cells in vitro. 21 It is well documented that mesangial cells depend on β1 integrins for adhesion and migration, 40 and α1β1 integrin has been directly implicated in both adhesion and spreading of chondrocytes. 38 Thus, the absence of α1β1 in the double knockouts might affect the migration of cytoplasmic processes that interface the mesangium with the glomerular capillary loops. 41,42 These contact points provide direct access for mesangial enzymes and proteins to be deposited into the GBM. Focal degradation by matrix metalloproteinase derived from the glomerular mesangium with concomitant deposition of mesangial matrix proteins might explain why focal thickening and splitting are observed in Alport GBM disease. Indeed, the proteins that accumulate in the GBM as a function of Alport renal disease pathogenesis include many matrix proteins normally synthesized by mesangial cells, 14,23 and mesangial cells are known to produce both matrix metalloproteinase (MMP)-2, MMP-9 and the tissue inhibitors of metalloproteinases-I, -II, and -III. 43,44 The mRNAs encoding both the integrin α1 and β1 subunits are induced by TGF-β1, 45 as are mesangial expression of laminins, fibronectin, and collagen α1(IV) and α2(IV). An alternative explanation for the appearance of typically mesangial proteins in the GBM is gene activation in the podocytes. Activation in podocytes of the genes encoding collagen α1(IV), fibronectin, and the laminin β1chain, all typically mesangial matrix proteins, has been demonstrated in Alport mice. 23 Thus, an integrin α1β1 regulated paracrine factor may exist that can activate these genes in podocytes.
Absence of α1β1 integrin results in an inhibition of laminin α2 and β1 chain deposition in the GBM. This report is the first to illustrate GBM deposition of these laminin chains in GBM pathogenesis. The importance of this observation lies with its likely association with effacement of the podocyte foot processes. Known laminin heterotrimers that contain the α2 chain include laminin-2 (α2β1γ1) and laminin-4 (α2β2γ1). 46 Normal mature GBM contains only laminin-11 (α5β2γ1). 24 Recent studies used a recombinant soluble α3β1 integrin receptor to illustrate that the integrin heterodimer binds specifically to laminin heterotrimers that contain the α5 chain, but not those that contain the α1 or α2 chains. 47 It is widely believed that adhesion of the podocyte foot processes to the GBM requires α3β1 integrin. 35 Immunogold localization studies place α3β1 integrin at the interface between the podocytes and the basement membrane. 48 An α3 integrin knockout has been produced, and dies at birth from acute renal failure, with complete effacement of the podocyte foot processes. 49 Integrin α1β1 and α2β1 bind to the amino terminus of the laminin α1 chain and the laminin α2 chain, 50,51 creating precedent for laminin α chains as specific ligands for integrins. Thus, deposition of the laminin heterotrimers containing the α2 chain might mask binding of α3β1 integrin to its preferred ligand, laminin-11. Such masking might lead to progressive loss of focal contact adhesion, resulting in foot process effacement. Because laminin α2 is found only in the mesangial matrix of normal mice and humans, how it gets into the GBM in Alport syndrome is an important unanswered question. Clearly α1β1 integrin plays an key role in this process.
Role of TGF-β1 in Alport Glomerular Pathogenesis
TGF-β1 is strongly implicated as a molecular driver in the rat model for acute mesangial proliferative glomerulonephritis. 52 The induced disease can be suppressed by injecting the animals with neutralizing antibodies against TGF-β1 or antisense oligonucleotides complimentary to the TGF-β1 mRNA. 53,54 More recently, this work has been extended to include gene therapy by transduction with an expression vector expressing a soluble TGF-β type II receptor/Fc fusion peptide, much like the soluble receptor used in our studies. 39 TGF-β1 has been implicated in a number of glomerular diseases, 54,55 including Alport syndrome. 23
In this report we show that by inhibiting TGF-β1 we can inhibit thickening of the GBM. We do not, however, inhibit effacement of the podocyte foot processes. Laminin α2 is present in the GBM of 7-week-old Alport mice treated with mTGFβR:Fc, suggesting that the events resulting in deposition of laminin α2 containing isoforms in the GBM are not mediated by TGF-β1. The animals die at about the same time as untreated animals, however why they die is not clear. They do develop acute massive proteinuria, so the cause of death may be hypoalbuminemia and hypovolemia. This is very different from the results obtained with experimental glomerulonephritis, where inhibition of TGF-β1 was sufficient to restore normal glomerular function. 22,53 Thus, although TGF-β1 might be induced in a variety of glomerular disease, it is likely that its contributory role may be unique for each.
Combined, the observations reported herein support a potential therapeutic paradigm for Alport renal disease by treatment with inhibitors for both integrin α1β1 and TGF-β1.
Acknowledgments
We thank John (Skip) Kennedy for his help in the preparation of figures for publication; Kelly Peterson for the genetic typing of animals; and Peter Yurchenco, Jeffery Miner, Joshua Sanes, Yoshi Yamada, Bob Burgeson, and Richard Hynes for the generous gifts of antibodies.
Footnotes
Address reprint requests to Dominic Cosgrove, Ph.D., Department of Genetics, Boys Town National Research Hospital, 555 No. 30th Street, Omaha, NE 68131. E-mail: cosgrove@boystown.org.
Supported by National Institutes of Health Grants R01 DK55000 (to D. C.) from the National Institute of Diabetes and Digestive and Kidney Diseases, P01 DC01813 (to D. C.) from the National Institute on Deafness and Other Communication Disorders, and by R01 DK51711 (to R. K.); the 1998 Carl Gottschalk award (to R. K.); the 1998 NKF Murray Award (to R. K.); and funds from Beth Israel Deaconess Medical Center.
References
- 1.Flinter F: Alport syndrome. J Med Genet 1992, 34:326-330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregory MC, Terreros DA, Barker DF, Fain PN, Denison JC, Atkin CL: Alport syndrome—clinical phenotypes, incidence, and pathology. Contrib Nephrol 1996, 117:1-28 [DOI] [PubMed] [Google Scholar]
- 3.Barker DE, Hostikka SL, Zhou J, Show LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K: Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 1990, 248:1224-1227 [DOI] [PubMed] [Google Scholar]
- 4.Lemmink HH, Mochizuki T, van den Heuvel LPWJ, Schroder CH, Barrientos A, Monnens LAH, van Oost BA, Brunner HG, Reeders ST, Smeets HJM: Mutations in the type IV collagen α3 (COLCOL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet 1994, 3:1269-1273 [DOI] [PubMed] [Google Scholar]
- 5.Mochizuki T, Lemmink HH, Mariyama M, Antignac C, Gubler MC, Pirson Y, Verellen-Dumoulin C, Chan B, Schroder CH, Smeets HJ, Reeders ST: Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 1994, 8:77-82 [DOI] [PubMed] [Google Scholar]
- 6.Kashtan CE, Kim Y: Distribution of the α1 and α2 chains of collagen IV and of collagens V and VI in Alport syndrome. Kidney Int 1992, 42:115-126 [DOI] [PubMed] [Google Scholar]
- 7.Gubler M-C, Knebelmann B, Beziau A, Broyer M, Pirson Y, Haddoum F, Kleppel MM, Antignac C: Autosomal recessive Alport syndrome: immunohistochemical study of type IV collagen chain distribution. Kidney Int 1995, 47:1142-1147 [DOI] [PubMed] [Google Scholar]
- 8.Mazzucco G, Barsotti P, Muda AO, Fortunato M, Mihatsch M, Torri-Tarelli L, Renieri A, Faraggiana T, De Marchi M, Monga G: Ultrastructural and immunohistochemical findings in Alport’s syndrome: a study of 108 patients from 97 Italian families with particular emphasis on COL4A5 gene mutation correlations. J Am Soc Nephrol 1998, 9:1023-1031 [DOI] [PubMed] [Google Scholar]
- 9.Desjardins M, Bendayan M: Ontogenesis of glomerular basement membrane: structural and functional properties. J Cell Biol 1991, 113:689-700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleppel MM, Fan WW, Cheong HI, Michael AF: Evidence for separate networks of classical and novel basement membrane collagen. J Biol Chem 1992, 267:4137-4142 [PubMed] [Google Scholar]
- 11.Gunwar S, Ballester F, Noelken ME, Ninomiya Y, Hudson B: Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of α, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem 1998, 273:8767-8775 [DOI] [PubMed] [Google Scholar]
- 12.Miner JH, Sanes JR: Collagen IV α3, α4, and α5 chains in rodent basal lamina: sequence, distribution, association with laminins, and developmental switches. J Cell Biol 1994, 127:879-891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalluri R, Shield CF, III, Todd P, Hudson BG, Nielson EG: Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest 1997, 99:2470-2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, Samuelson GC: Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev 1996, 10:2981-2992 [DOI] [PubMed] [Google Scholar]
- 15.Miner JH, Sanes JR: Molecular and functional defects in kidneys of mice lacking collagen α3(IV): implications for Alport syndrome. J Cell Biol 1996, 135:1403-1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu W, Phillips CL, Killen PD, Hlaing WR, Elder FF, Miner JH, Overbeek PA, Meisler MH: Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model for Alport syndrome. Genomics 1999, 15:113-124 [DOI] [PubMed] [Google Scholar]
- 17.Patey N, Halbwachs-Mecarelli D, Droz D, LeSavre P, Noel LH: Distribution of integrin subunits in normal human kidney. Cell Adhes Commun 1994, 2:159-167 [DOI] [PubMed] [Google Scholar]
- 18.Sterk LM, deMelker AA, Kramer D, Kuikman I, Chand A, Claessen N, Weening JJ, Sonnenberg A: Glomerular extracellular matrix components and integrins. Cell Adhes Commun 1998, 5:177-192 [DOI] [PubMed] [Google Scholar]
- 19.Gardner H, Kreidberg J, Koteliansky V, Jaenisch R: Deletion of integrin α1 by homologous recombination permits normal murine development, but gives rise to a specific deficit in cell adhesion. Dev Biol 1996, 175:301-313 [DOI] [PubMed] [Google Scholar]
- 20.Pozzi A, Wary KK, Giancotti FG, Gardner HA: Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol 1999, 142:587-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kagami S, Kondo S, Loster K, Reutter W, Kuhara T, Yatsumoto K, Kuroda Y: α1β1 integrin-mediated collagen matrix remodeling by rat mesangial cells is differentially regulated by transforming growth factor-β and platelet-derived growth factor-BB. J Am Soc Nephrol 1999, 10:779-789 [DOI] [PubMed] [Google Scholar]
- 22.Border WA, Ruoslahti E: Transforming growth factor-β in disease: the dark side of tissue repair. J Clin Invest 1992, 90:1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayers R, Kalluri R, Rodgers KD, Shield CF, III, Meehan DT, Cosgrove D: Role for TGF-β in Alport renal disease progression. Kidney Int 1999, 56:1662-1673 [DOI] [PubMed] [Google Scholar]
- 24.Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkens NA, Copeland NG, Sanes JR: The laminin α chains: expression, developmental transitions, and chromosomal locations of α1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel α3 isoform. J Cell Biol 1997, 137:685-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patton BL, Miner JH, Chiu AY, Sanes JH: Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol 1997, 139:1507-1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abrahamson DR, Irwin MH, St John PL, Perry EW, Accavitti MA, Heck LW, Couchman JR: Selective immunoreactivities of kidney basement membranes to monoclonal antibodies against laminin: localization of the end of the long arm and the short arms to discrete microdomains. Cell Biol 1989, 109:2477-2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E: Merosin and laminin in myogenesis: specific requirement for merosin in myotube stability and survival. J Cell Biol 1996, 134:1483-1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rousselle P, Lundstrum GP, Keene DR, Burgeson RE: Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J Cell Biol 1991, 114:567-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marinkovich MP, Lunstrum GP, Keene DR, Burgeson RE: The dermal-epidermal junction of human skin contains a novel laminin variant. J Cell Biol 1992, 119:695-703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanes JR, Engvall E, Butkowski R, Hunter DD: Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J Cell Biol 1990, 111:1685-1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Utani A, Kopp JB, Kozak CA, Matsuki Y, Amizuka N, Sugiyama S, Yamada Y: Mouse kalinin B1 (laminin β3 chain): cloning and tissue distribution. Lab Invest 1995, 72:300-310 [PubMed] [Google Scholar]
- 32.Tsang ML-S, Zhou L, Zheng BL, Wenker J, Fransen G, Humphrey J, Smith JM, O’Connor-McCourt M, Lucas R, Weatherbee JA: Characterization of recombinant soluble human transforming growth factor-β Type II (rhTGF-bsRII). Cytokine 1995, 7:389-397 [DOI] [PubMed] [Google Scholar]
- 33.O’Connor-McCourt M, Segarini P, Grothe S, Tsang ML-S, Weatherbee JA: Analysis of the interaction between two TGF-β binding proteins and three TGF-β isoforms using surface plasmon resonance. Ann NY Acad Sci 1995, 766:300-302 [DOI] [PubMed] [Google Scholar]
- 34.Smith JD, Bryant SR, Couper LL, Vary CPH, Gotwals PJ, Koteliansky VE, Lindner V: Soluble transforming growth factor-β type II receptor inhibits negative remodeling, fibroblast transdifferentiation, and intimal lesion formation but not endothelial regrowth. Circ Res 1999, 84:1212-1222 [DOI] [PubMed] [Google Scholar]
- 35.Smoyer WE, Mundel P: Regulation of podocyte structure during the development of nephrotic syndrome. J Mol Med 1998, 76:172-183 [DOI] [PubMed] [Google Scholar]
- 36.Miner JH: Renal basement membrane components. Kidney Int 1999, 56:2016-2024 [DOI] [PubMed] [Google Scholar]
- 37.Carver W, Molano I, Reaves TA, Borg TK, Terracio L: Role of the α1 β1 integrin complex in collagen gel contraction in vitro by fibroblasts. J Cell Physiol 1995, 165:425-437 [DOI] [PubMed] [Google Scholar]
- 38.Makihira S, Yan W, Ohno S, Kawamoto T, Fujimoto K, Okimura A, Yoshida E, Noshiro M, Hamada T, Kato Y: Enhancement of cell adhesion and spreading by a cartilage-specific noncollagenous protein, cartilage matrix protein (CMP/Matrilin-1), via integrin alpha1beta1. J Biol Chem 1999, 274:11417-11423 [DOI] [PubMed] [Google Scholar]
- 39.Isaka Y, Akagi Y, Ando Y, Tsujie M, Sudo T, Ohno N, Border W, Noble NA, Kaneda Y, Hori M, Imai E: Gene therapy by transforming growth factor-β receptor-IgG Fc chimera suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int 1999, 55:465-475 [DOI] [PubMed] [Google Scholar]
- 40.Prols F, Hartner A, Schlockman HO, Stersel RB: Mesangial cells and their adhesive properties. Exp Nephrol 1999, 7:137-146 [DOI] [PubMed] [Google Scholar]
- 41.Sakai T, Kriz W: The structural relationship between mesangial cells and basement membrane of the renal glomerulus. Anat Embryol 1987, 176:373-386 [DOI] [PubMed] [Google Scholar]
- 42.Kriz W, Elger M, Lemley KV, Sakai T: Mesangial cell-glomerular basement membrane connections counteract glomerular capillary and mesangium expansion. Am J Nephrol 1990, 10(Suppl 1):4-13 [DOI] [PubMed] [Google Scholar]
- 43.Anderson SS, Wu K, Nagase H, Stettler-Stevenson WG, Kim Y, Tsilibary EC: Effect of matrix glycation on expression of type IV collagen, MMP-2, MMP-9, and TIMP-1 by human mesangial cells. Cell Adhes Commun 1996, 2:89-101 [DOI] [PubMed] [Google Scholar]
- 44.Mozes MM, Bottinger EP, Jacot TA, Kopp JB: Renal expression of fibrotic matrix proteins and of transforming growth factor-β (TGF-β) isoforms in TGF-β transgenic mice. J Am Soc Nephrol 1999, 10:271-280 [DOI] [PubMed] [Google Scholar]
- 45.Kagami S, Kuhara T, Yatsumoto K, Okada K, Loster K, Reutter W, Kuroda Y: Transforming growth factor β (TGF-β) stimulates the expression of β1 integrins and adhesion by rat mesangial cells. Exp Cell Res 1996, 229:1-6 [DOI] [PubMed] [Google Scholar]
- 46.Engvall E, Earwicker D, Haaparanta T, Rouslahti E, Sanes JR: Distribution and isolation of four laminin variants: tissue restricted distribution of heterotrimers assembled from five different subunits. Cell Regul 1990, 1:731-740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eble JA, Wucherpfennig KW, Gauthier L, Dersch P, Krukonis E, Isberg RR, Hemler ME: Recombinant soluble human α3β1 integrin: purification, processing, regulation, and specific binding to laminin-5 and invasion in a mutually exclusive manner. Biochemistry 1998, 37:10945-10955 [DOI] [PubMed] [Google Scholar]
- 48.Regoli M, Bendayan M: Alterations in the expression of the α3β1 integrin in certain membrane domains of the glomerular epithelial cells (podocytes) in diabetes mellitus. Diabetologia 1997, 40:15-22 [DOI] [PubMed] [Google Scholar]
- 49.Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R: α3 β1 integrin has a crucial role in kidney and lung organogenesis. Development 1996, 122:3537-3547 [DOI] [PubMed] [Google Scholar]
- 50.Colognato H, MacCarrick M, O’Rear JJ, Yurchenco PD: The laminin α2-chain short arm mediates cell adhesion through both the α1β1 and α2β1 integrins. J Biol Chem 1997, 272:29330-29336 [DOI] [PubMed] [Google Scholar]
- 51.Ettner N, Gohring W, Sasaki T, Mann K, Timpl R: The N-terminal globular domains of the laminin α1 chain binds to α1β1 and α2β1 integrins and to the heparin sulfate-containing domains of perlecan. FEBS Lett 1998, 430:217-222 [DOI] [PubMed] [Google Scholar]
- 52.W.A., Okuda S, Languino LR, Sporn MB, Ruoslahti E: Suppression of experimental glomerulonephritis by antiserum against transforming growth factor β1 Nature (London) 1990, 346:371–374 [DOI] [PubMed]
- 53.Agaki Y, Isaka Y, Arai M, Kaneko T, Takenaka M, Moriyama T, Kaneda Y, Ando A, Orita Y, Kamada T, Ueda N, Imai E: Inhibition of TGF-β1 expression by antisense oligonucleotides: suppressed extracellular matrix accumulation in experimental border glomerulonephritis. Kidney Int 1996, 50:148-155 [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto T, Noble NA, Miller DE, Border WA: Sustained expression of TGF-β1 underlies development of progressive kidney fibrosis. Kidney Int 1994, 45:916-927 [DOI] [PubMed] [Google Scholar]
- 55.Yang C-W, Vlassara H, Striker GE, Striker LJ: Administration of AGEs in vivo induces genes implicated in diabetic glomerulosclerosis. Kidney Int 1995, 47:S53-S58 [PubMed] [Google Scholar]