

Abstract
Microdissection techniques allow a cell-type or even cell-specific mRNA analysis within complex tissues. Furthermore, valid mRNA quantitation can be performed by real-time reverse transcriptase-polymerase chain reaction from a few isolated cells obtained from cryosections. For a more precise access to many cell types, this technique has to be complemented by a cell-type-specific immunostaining. To evaluate its effect on mRNA quantitation, we analyzed alveolar macrophages (AMs) from control rat lungs and those undergoing stimulation with lipopolysaccharide and interferon-γ nebulization. Whereas AMs from the left lung were directly harvested for mRNA extraction by bronchoalveolar lavage, tissue sections of the right lung were stained with an optimized immunofluorescence protocol detecting AMs. Fifteen AM profiles per sample were picked by laser-assisted sampling technique. Normalizing to a standard gene, nitric oxide synthase II (NOSII) and tumor necrosis factor (TNF)-α mRNA were quantified by real-time reverse transcriptase-polymerase chain reaction. In stimulated lungs, the percentage of picked samples positive for NOSII or TNF-α mRNA increased significantly. Moreover, a marked increase in the ratio of target gene mRNA to standard gene mRNA was noted for both NOSII and TNF-α in picked AMs from stimulated lungs, which matched very well the increase detected in the lavaged AMs undergoing direct RNA extraction. Thus, when using an optimized protocol for immunofluorescence, this approach may be reliably combined with laser-assisted cell picking and real-time mRNA quantitation in a few immunohistochemically characterized cell profiles within complex tissues.
Tissue microdissection has become one of the key tools in molecular biomedicine and modern pathology. In combination with various downstream applications, this technique provides the possibility of cell-type or even cell-specific investigation of DNA, RNA, and proteins. 1 For reliable mRNA analysis in microdissected samples of complex tissues, several preconditions have to be fulfilled: 1) cells of interest have to be detected unambiguously. Thus, in many cases, an immunostaining procedure is inevitably required. 2) Microdissection and isolation of single cell profiles must be performed in a precise manner, without destruction of the relevant mRNA and without contamination by adjacent tissues. 3) Next to qualitative mRNA detection by reverse transcriptase-polymerase chain reaction (RT-PCR), a sensitive and reliable mRNA quantitation has to be established when transcriptional regulatory events are targeted with the number of mRNA copies increasing or decreasing.
Using an ultraviolet laser microdissection system, mRNA analysis was shown to be possible even in a few cell profiles originating from formalin-fixed routine material 2 or cryosections. 3 Additionally, this technique allows identification of distinct splicing variants within a few cell profiles. 4-6 Moreover, our group as well as other workers have demonstrated a reliable mRNA quantitation in microdissected samples from hemalaun- or hemalaun/eosin-stained tissue sections. 7,8 Immunostaining and microdissection for mRNA analysis were found to be possible for several hundred cell profiles, 9 for single dispersed, but intact cells, 10 and for single cell profiles further processed by an RNA amplification step. 11 However, we observed a decrease in efficiency rates for mRNA detection (percentage of samples positive for the respective mRNA) when routine immunostaining was applied. 12 In an attempt to optimize the technique with respect to mRNA recovery, we showed that short-term formalin fixation, utmost reduction of antibody incubation times, application of immunofluorescence, and digestion with proteinase K forwarded the best results when applied to oligocellular clusters.
In the present study, we combined immunolabeling, microdissection, and picking of a few stained cell profiles and mRNA quantitation by real-time RT-PCR. 13,14 In ex vivo ventilated and perfused rat lungs, aerosolization of lipopolysaccharide (LPS) and interferon-γ (IFN-γ) was used to up-regulate alveolar macrophage (AM) nitric oxide synthase II (NOSII) and tumor necrosis factor-α (TNF-α) mRNA levels. AMs were then obtained by conventional bronchoalveolar lavage. Alternatively, they were stained within tissue sections of the lung by use of the indirect immunofluorescence technique, and ∼15 cell profiles per sample were isolated for mRNA quantitation. Relative quantitation was performed by normalizing NOSII and TNF-α mRNA to mRNA of the standard gene porphobilinogen deaminase (PBGD). Thus, by comparing the data obtained by combined immunofluorescence and cell picking to those from nonstained AMs recovered by a standard procedure, validation of the technique was obtained.
Materials and Methods
Lung Preparation
Male CD rats (Sprague-Dawley, 60 to 70 days old, 350 to 400 g body weight; Charles River, Sulzfeld, Germany) were deeply anesthetized with phenobarbital-Na. Lungs were isolated, ventilated, and ex vivo-perfused with Krebs-Henseleit buffer in a closed perfusion circuit as previously described. 15 Measurement of perfusion pressure, ventilation pressure, and organ weight was performed continuously. The lungs had a homogeneous white appearance without signs of hemostasis or edema formation and the pulmonary as well as ventilation pressures were in the normal ranges. They were isogravimetric during a steady-state period of 30 minutes. For stimulation, an ultrasonic nebulizer was used afterward to aerosolize 75 μg of LPS and 1,000 U of IFN-γ in a volume of 5 ml into the afferent limb of the ventilator circuit for 10 minutes. Experiments were then continued under standard conditions for 6 hours. Control lung did not undergo LPS/IFN-γ challenge. Certain differences in the extent of mRNA up-regulation were seen among the stimulated lungs and could most probably be because of individual variability of the organs or smallest and nonsusceptible alterations of the experimental system.
For fixation, lungs were perfused with 4.5% formaldehyde solution (Roti-Histofix; Roth, Karlsruhe, Germany) for 15 minutes, followed by a rinsing step with Krebs-Henseleit buffer to remove the residual formalin. Finally, the left lung was lavaged, while the right lung was instilled with Tissue-Tek (Sakura Finetek, Zoeterwoude, The Netherlands) and snap-frozen in liquid nitrogen.
Bronchoalveolar Lavage
As detailed by Fink and colleagues, 7 six aliquots of 3 ml of saline were instilled into the left lung and re-aspirated immediately. Cells were centrifuged at 300 × g, washed, and counted in a hemocytometer chamber. For in vitro stimulation, AM aliquots were transferred to plastic dishes and suspended in RPMI 1640 (Life Technologies, Paisley, Scotland) supplemented with 2% rat serum. Next, they were incubated at 37°C with 5% CO2. For mRNA extraction, AMs were directly transferred into 300 μl of lysis buffer (Dynal, Oslo, Norway).
In Vitro Stimulation
AMs were allowed to adhere for 2 hours. Afterward, medium was replaced by medium supplemented with 10 μg of LPS and 1,000 U IFN-γ. Cells were harvested after the respective incubation times at 37°C, 5% CO2, by removing the medium, lysed in 300 μl of lysis buffer (Dynal) and transferred into 1.5 ml reaction tubes.
mRNA Extraction
Using the Dynabeads mRNA direct kit (Dynal), cell lysate was applied to magnetic separation. The mRNA was caught by attachment to oligo-dT fragments that are coupled to supermagnetic glass particles. Per sample, 100-μg beats were used. Isolated mRNA was finally solved in 2 × 10 μl of diethyl pyrocarbonate-treated H2O.
Immunofluorescence Labeling
Cryostat sections (∼8 μm) were mounted on poly-l-lysine- (Sigma Aldrich, Deisenhofen, Germany) covered glass slides and stored in acetone for 1 to 3 minutes. Afterward, the murine primary monoclonal antibody ED-1 (specific for rat macrophage/monocyte, 1:25; Serotec via Biozol Diagnostika, Eching, Germany) was incubated for 5 to 8 minutes at room temperature, and after shortly washing in buffer, incubation with fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin (1:100; Organon Teknika, Durham, NC) at room temperature was performed for 5 to 8 minutes. Finally, sections were washed and immersed in 70 to 90% ethanol until picking.
Immunoalkaline-Phosphatase Staining
Immunohistochemistry was performed by the alkaline-phosphatase monoclonal anti-alkaline phosphatase (APAAP) technique, slightly modified from the method of Cordell et al. 16 Cryostat sections (8 μm) were mounted on poly-l-lysine-covered glass slides. To unmask the epitopes, sections were placed in a solution of citric acid (2 μmol/L) and sodium citrate (9.1 μmol/L). They were heated three times for 5 minutes in a microwave oven (600 W; Bosch, Stuttgart, Germany) and cooled down to room temperature for 15 minutes. After storage in acetone for 10 minutes, they were incubated with the primary antibodies rabbit polyclonal anti-NOSII (N-20, 1:250; Santa Cruz Biotechnology, Heidelberg, Germany) or rabbit polyclonal anti-TNF-α (ICC-TNF-9B, 1:250; Innogenetics, Heiden, Germany) for 20 minutes. In a second incubation step for 20 minutes, monoclonal mouse anti-rabbit immunoglobulin (1:500; Dako Diagnostika, Hamburg, Germany) was applied, followed by incubation with rabbit anti-mouse immunoglobulin (rabbit-“link,” 1:40; Dako) and finally mouse-APAAP complex (1:50; Dako), each at room temperature for 20 minutes. Second and third antibodies were supplemented with pooled rat serum (1:750; Sigma). Between these steps, samples were washed twice in Tris-buffered saline (pH 7.5). Alkaline-phosphatase substrate reaction was performed at pH 9.0 with new fuchsin (100 μg/ml) and levamisole (400 μg/ml) for 25 minutes at room temperature. Finally, sections were counterstained with hemalaun for 45 seconds.
Laser-Assisted Cell Picking, Proteinase K Digestion, and RT
Cell picking was performed as described in detail recently. 5,12 Using the UV-laser Robot Microbeam (P.A.L.M., Bernried, Germany), a mercury vapor lamp was coupled to the epifluorescence illumination path. After immunofluorescence staining, AMs were selected by immunofluorescence microscopy. Every single AM localization was stored by the stage position, so that a precise return was possible. Afterward, bright-field illumination was switched on. The AMs were then identified by nearly closing the condenser. Adjacent tissue was removed by UV-laser photolysis under visual control. Finally, the AM profiles were isolated by a sterile syringe needle and transferred into a reaction tube containing 10 μl of first-strand-buffer. Samples with 15 AM profiles each were snap-frozen in liquid nitrogen and stored for further preparation. After a short thawing period, proteinase K (1 μl, 1 mg/ml; Sigma) was added to the samples and incubated for 1 hour at 53°C. Proteinase K as well as RNA were denaturated for 5 minutes at 95°C, and samples were stored on ice for another 5 minutes. To create identical conditions for picked samples and extracted mRNA (10 μl), they underwent denaturation and after cDNA synthesis in parallel using the same composition of the reagents. cDNA synthesis was performed using 2 μl MgCl2 (25 mmol/L), 2 μl GeneAmp 10× PCR buffer II (100 mmol/L Tris-HCl, pH 8.3, 500 mmol/L KCl), 1 μl dNTP (10 mmol/L each), 1 μl random hexamers (50 μmol/L), 0.5 μl RNase inhibitor (10 U), and 1 μl murine leukemia virus RT (50 U). Except for dNTP (Eurobio, Raunheim, Germany), all reagents were purchased from PE Biosystems (Weiterstadt, Germany). Samples were incubated at 20°C for 10 minutes and at 43°C for 60 minutes. Finally, the reaction was stopped by exposure to 99°C for 5 minutes.
Relative mRNA Quantitation
Real-time PCR is based on the 5′ nuclease activity of Taq polymerase for fragmentation of a dual-labeled fluorogenic hybridization probe. 13,14 Using the Sequence Detection System 7700 (PE Biosystems), it was performed as recently described in detail by Fink and colleagues. 7 For relative quantitation, the target gene was normalized to an internal standard gene. Therefore, PBGD mRNA was used. 5 This kind of quantitation is calculated by the following equation:
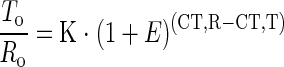 |
where To is the initial number of target gene mRNA copies, Ro is the initial number of standard gene mRNA copies, E is the efficiency of amplification, CT,T is the threshold cycle of target gene, CT,R is the threshold cycle of standard gene, and K is constant.
Using the mentioned primer/probe sets (Table 1) ▶ , pilot experiments showed that amplification efficiencies of PBGD, TNF-α, and NOSII mRNA were noted to be approximately equal and amounted to 0.95 ± 0.02.
Table 1.
Sequences, Amplicon Sizes, Exon Localization in the Genes, and Position of Primers and Probes
| Primer name | Nucleotide sequence | Exon localization | Position |
|---|---|---|---|
| PBGD amplicon size: 135 bp | |||
| PBGD-forward | 5′CAAGGTTTTCAGCATCGCTACCA3′ | e4 | 140–118† |
| PBGD-reverse | 5′ATGTCCGGTAACGGCGGC3′ | e1 | 1–18* |
| PBGD-probe | 5′CCAGCTGACTCTTCCGGGTGCCCAC3′ | e4–e3 | 91–67* |
| TNF-α amplicon size: 173 bp | |||
| TNFα-forward | 5′GGTGATCGGTCCCAACAAGGA3′ | e1 | 162–182 |
| TNFα-reverse | 5′CACGCTGGCTCAGCCACTC3′ | e4 | 334–316 |
| TNFα-probe | 5′TGGCCCAGACCCTCACACTCAGATCA3′ | e2–e3 | 221–246 |
| NOSII amplicon size: 177 bp | |||
| NOSII-forward | 5′CTCGCTGCATCGGCAGGAT3′ | e6 | 604–622 |
| NOSII-reverse | 5′AGTCATGCTTCCCATCGCTCC3′ | e8 | 780–760 |
| NOSII-probe | 5′CCTGCAGGTCTTCGATGCCCGGA3′ | e6–e7 | 635–657 |
Intron-spanning primers were selected so that cDNA amplicon was much shorter than genomic DNA amplicon. Each hybridization probe also spanned an intron to prevent annealing to genomic DNA. Excluding falsification by amplification of possible pseudogene sequences, all primer sets were shown to detect no genomic DNA with cDNA sequence. The primer/probe-sets work under identical PCR cycling conditions to obtain simultaneous amplification of PBGD, TNF-α, and NOSII in the same run. Sequences were taken from GeneBank, Accession Y17155* (reverse + probe) and X06827† (forward) for PBGD, X66539 for TNF-α, and D14051 for NOSII.
After RT, every picked sample as well as samples with extracted mRNA were divided into two aliquots of 8 μl for target gene and standard gene analysis.
Twenty-five μl of Universal Master Mix (PE Biosystems), oligonucleotide primers (final concentration, 900 nmol/L), and hybridization probe (final concentration, 200 nmol/L) were added to an end volume of 50 μl. Cycling conditions were 95°C for 6 minutes, followed by 55 cycles of 95°C for 20 seconds, 61°C for 30 seconds, and 73°C for 30 seconds.
Results
To establish the most advantageous conditions for the experiments in intact lungs, we first evaluated the time course of NOSII mRNA induction in AMs by in vitro stimulation. After 2 hours of adhesion to plastic dishes, LPS and IFN-γ were added. Maximal up-regulation of NOSII occurred after 6 hours (Figure 1) ▶ . In consequence, the isolated rat lungs were ventilated and perfused for this time span.
Figure 1.

Relative NOSII mRNA expression in AMs after in vitro stimulation. After 2 hours of adhesion, time was set at zero, and AMs were incubated with LPS/IFN-γ for the given time periods. Five independent experiments each were performed and data are given as mean ± SEM. K, constant.
Next, we determined the efficiency rates of PBGD and NOSII mRNA in formalin-fixed stimulated lung tissue (Figure 2) ▶ . Within hemalaun-stained tissue sections of these lungs, PBGD mRNA was detected in ∼70% of the samples containing 15 cell profiles or cell clusters with 15 nuclei. Although this PBGD efficiency rate displayed virtually no variation between different cells in the lung, NOSII mRNA was found in <10% of the samples from bronchial epithelium and endothelium of both pulmonary arteries and veins. Highest NOSII mRNA recovery in picked lung cells was obtained for AMs (37.5%). In samples from alveolar septae (including some monocytes/macrophages), the NOSII efficiency rate was 18.8%. According to these results, immunohistochemical staining of lung tissue for NOSII detected predominantly AMs (Figure 3H) ▶ .
Figure 2.

Efficiency rates of PBGD and NOSII mRNA detection after picking from formalin-fixed and hemalaun-stained lung sections. Cell clusters with 15 nuclei each or 15 single cell profiles were isolated from the given structures.
Figure 3.

Technique of immunodetection and cell picking. A–F: Laser-assisted cell picking of AMs after immunofluorescence labeling in formalin-fixed lungs. Indirect immunofluorescence was performed within 14 minutes, using anti-ED-1 and fluorescein isothiocyanate-coupled rabbit anti-mouse immunoglobulin. Original magnifications, ×40. A: An ED-1-immunolabeled AM is selected. B: Switching to bright-field microscopy, AM is identifiable in the given position next to the septum. C: Adjacent septum is removed by UV-laser. D: A sterile syringe needle is approximated. E: The AM profile adheres to the needle and is lifted. F: To prove the isolation process, switch-back to fluorescence microscopy is performed, demonstrating the fluorescent AM profile adherent to the needle. G and H: APAAP immunohistochemical staining of NOSII in formalin-fixed control and stimulated lung tissue. AMs are marked by arrows. Original magnifications, ×20. G: Control lung. H: LPS/IFN-γ stimulated lung.
To evaluate the efficiency rates of cell picking after immunofluorescence labeling, two stimulated lungs and one control lung, fixed by formalin perfusion, were prepared. While each left lung was lavaged, each right lung was processed for tissue sectioning of various regions. After short-term ED-1 immunofluorescence labeling, ∼15 AM profiles per sample were picked (Figure 3, A–F) ▶ , and relative quantitation of NOSII and TNF-α mRNA was obtained by real-time RT-PCR.
Overall, 214 samples were analyzed from which 121 were positive for PBGD mRNA (=56.5%; Table 2 ▶ ). The mean threshold cycle (CT) for PBGD mRNA varied only negligibly, thus confirming the presence of similar mRNA amounts within all samples.
Table 2.
Efficiency of Housekeeping and Target Gene mRNA Detection in Lungs Undergoing Immunofluorescence Labeling
| PBGD-positive picked samples | TNF-α-positive picked samples | NOSII-positive picked samples | PBGD threshold cycle CT ± SEM | |
|---|---|---|---|---|
| Control lung | 42 /61 | 5 /28 | 0 /33 | 37.02 ± 0.40 |
| Stimulated lung 1 | 37 /68 | 17 /47 | 8 /21 | 37.18 ± 0.22 |
| Stimulated lung 2 | 42 /85 | 17 /35 | 16 /50 | 37.24 ± 0.21 |
Per sample 15 AM profiles were picked, and the number of samples positive for the investigated mRNA is given in relation to the total number of samples (= efficiency rate). The threshold cycles (CT) for PBGD mRNA varied only slightly, thus confirming the presence of similar amounts of mRNA within the samples.
In the control lung, TNF-α mRNA was detected in 17.8% of the samples, whereas NOSII mRNA was totally undetectable. In contrast, 41.5% of the samples from the stimulated lungs were found to express TNF-α mRNA and 33.8% NOSII mRNA, respectively. Thus, efficiency rates of samples positive for TNF-α and NOSII mRNA differed significantly between control and stimulated lungs (NOSII, P < 0.001; TNF-α, P = 0.019; Person’s chi-square test).
As a basis for the relative mRNA quantitation, the mean threshold cycle for PBGD mRNA was calculated for each lung. The corresponding threshold cycles of the target gene samples were substracted from mean PBGD CT (ΔCT = CTPBGD − CTtarget gene; given as mean ± SEM). Thus, normalization to the nonregulated standard gene PBGD was achieved. For lavaged and picked AMs, these ΔCT values are shown in Figure 4 ▶ (top). Calculating a PCR efficiency of 0.95 ± 0.02 for the three investigated genes, the values of relative mRNA expression were obtained by the formula K·1.95ΔCT (K = constant). These values are given in Figure 4 ▶ (bottom).
Figure 4.

NOSII and TNF-α mRNA quantitation—comparison of picked and lavaged cells. Top: For relative mRNA quantitation, ΔCT (difference of standard gene mean threshold cycle CT and threshold cycle of the target gene) is calculated. Values of picked cells are compared to those of lavaged AMs. Values of lavage represent the mean of two PCR samples. Bottom: Using the ΔCT values, relative mRNA expression is calculated by the formula K·(1 + E)ΔCT with E = 0.95 (K, constant). Again, picked AM profiles are compared to lavaged AMs in control and stimulated lungs. Mean and SEM are given. Statistical comparison of picked samples was performed for control versus stimulated lungs by the Wilcoxon test and resulted in P < 0.001 for both NOSII and TNF-α values.
Relative NOSII mRNA Expression in Lavaged and Picked Immunostained AMs
Not only the recovery of positive NOSII samples was shown to differ significantly between control and stimulated lungs, the value of relative NOSII mRNA expression in the positive samples also increased significantly from control to stimulated lungs (P < 0.001). In the control lung, NOSII mRNA was not detected in either lavaged or in picked samples. In stimulated lung case 1, 0.92 copies NOSII mRNA per copy PBGD mRNA was found for lavaged AMs, and picked AM samples exhibited 0.56 ± 0.28 copies NOSII mRNA per copy PBGD mRNA (Figure 4 ▶ , bottom). Relative NOSII expression in lavaged AMs of stimulated lung case 2 amounted to 0.42 copies per copy PBGD mRNA, corresponding to 0.39 ± 0.09 copies in picked AMs.
Relative TNF-α mRNA Expression in Lavaged and Picked Immunostained AMs
Relative TNF-α mRNA expression in the control lung amounted to 0.17 copies per copy PBGD for lavaged AMs, and the picked AMs expressed 0.32 ± 0.05 copies TNF-α mRNA. In stimulated lung case 1, TNF-α expression in lavage was found to be 4.68 copies per copy PBGD mRNA, in the second stimulated lung 9.49. Corresponding expression in picked samples amounted to 1.70 ± 0.67 copies and 2.13 ± 0.62 copies, respectively. Again, apart from the significant increase in the percentage of positive TNF-α mRNA samples in stimulated lungs, the value of relative TNF-α mRNA expression differed significantly between the stimulated lungs and the control (P < 0.001).
To confirm the results of mRNA quantitation, immunohistochemical staining for NOSII and TNF-α was performed in the control lung and stimulated lungs. Within the stimulated lungs, NOSII was exclusively stained in AMs. In the control lung, no staining was detectable (Figure 3, G and H) ▶ . Additionally, immunohistochemical staining for TNF-α was performed. Again, only AMs of the stimulated lungs were labeled, corresponding closely to the NOSII staining (data not shown).
Discussion
Immunolabeling is a precondition for many cell types to be precisely detected within complex tissues. To extend and complement the technique of isolating few isotypic cell profiles from tissue sections for further qualitative and quantitative mRNA analysis, immunospecific staining had to be introduced. Thus, we transferred our optimized protocol for immunofluorescence staining and cell picking 12 to real-time RT-PCR quantitation of target genes in intact lungs. 7 By focusing on AMs, which can be recovered in parallel by a conventional technique (bronchoalveolar lavage) not demanding immunostaining, and by cell picking, we investigated the effect of the immunodetection procedure on mRNA efficiency rates. Further we sought for the validity of mRNA quantitation in cells undergoing the entire protocol of immunostaining and laser-assisted cell picking.
Because long-term immersion of tissue in aqueous media is deleterious for RNA preservation, 9 short-term indirect immunofluorescence proved to provide the best mRNA efficiency rates. 12 Using ED-1 antibody to stain AMs, a specific detection was obtained within 15 minutes. Unfortunately, the additionally used anti-NOSII and anti-TNF-α antibodies did not detect their specific epitopes in formalin-fixed tissue. Pretreatment with citrate buffer and microwave did indeed unmask these epitopes and allowed specific staining. This, however, was shown to be disadvantageous for nucleic acids and amplification. 17 In consequence, the suitability and shortest possible incubation time for staining had to be tested for each primary antibody. Finally, the influence of the fluorochrome conjugated to the secondary antibody on the real-time PCR technology, based on fluorescence detection, must be taken into consideration. In our experience, however, the combination of fluorescein isothiocyanate-coupled secondary antibody and 6-carboxy fluorescein or VIC-labeled PCR probes has never resulted in interference with analysis or deviation of background measurement.
Although hemalaun staining of formalin-fixed tissue resulted in an efficiency rate of ∼70% for PBGD in picked AMs, this was decreased to a mean of 56.5% after immunofluorescence labeling. These rates correspond well with our previous data. 12 The loss might be because of exogenous RNase activity introduced by the applied antibodies or also to endogenous degradation mechanisms and elution, despite the mild fixation. Additionally, the stimulation procedure itself apparently influenced the efficiency rate of PBGD mRNA detection in picked AMs (51.6% in stimulated as compared to 68.9% in control lungs); the underlying reasons are currently still unknown. Nevertheless, the mean CT for PBGD mRNA was substantially decreased when compared to our previous study (37.15 versus 41.13), underlining the beneficial effect of the newly introduced optimizing steps.
Concerning the preferable type of tissue fixation, the best results in RT-PCR amplification products were obtained from cryosections fixed by a precipitating fixative like ethanol 5,18 or methacarn 19 in combination with a short routine staining (eg, hemalaun, hematoxylin and eosin). Unfortunately, in picked samples from organs fixed by ethanol, the RNA efficiency rate after the immunostaining procedure decreased considerably when compared to samples from organs fixed by the cross-linking fixative formalin. 12
The mRNA analysis in picked AMs differs in many respects from mRNA extracted from lavaged AMs. Whereas lavaged cells are intact with integral cell membranes, picked cell profiles are sectioned and thus lose a considerable portion of their cytoplasm and protecting membrane. In addition, they have to pass the immunostaining procedure, storage in alcohol, lasering, and picking. Nevertheless, these few picked immunostained cell profiles are sufficient to reliably detect the up-regulation of both NOSII and TNF-α in response to inflammatory challenge of the intact lungs. This was demonstrated first by a significant increase in the percentage of both NOSII- and TNF-α-positive samples as compared to samples from control lungs. Second, it was shown by a manifold increase in target gene mRNA copies (normalized to PBGD mRNA) within the positive picked samples from the stimulated lungs. This matches well the increase in the target mRNA/PBGD mRNA ratio of the lavaged AMs serving as reference. Two aspects have to be addressed in further detail:
First, in the control lung, 17% of picked samples were positive for TNF-α mRNA. However, the TNF-α mRNA expression in these positive samples was stronger than in the lavaged AMs. This clearly suggests that the analysis of (a large number of) lavaged cells averages the expression of this cell population without considering individual differences, whereas such individual differences between the cells are revealed by the cell picking approach.
Second, concerning the quantitation of NOSII expression, the data obtained from the picked AMs reflected closely those from the lavaged AMs. A somewhat lower target gene mRNA/PBGD mRNA ratio was, however, obtained for TNF-α in picked AMs as compared to lavaged AMs from stimulated lungs. This might be because of the fact that TNF-α mRNA is relatively unstable and short-lived, as compared to PBGD mRNA, which may be assumed to be particularly relevant for the longer work-up procedures for immunostaining at room temperature. In contrast, NOSII mRNA is known to possess a long half-life (1 to 2 hours), which moreover increases by LPS/IFN-γ stimulation. 20,21 In addition, nitric oxide is known to destabilize TNF-α mRNA by reducing its half-life. 22 This type of reasoning again demonstrates the necessity for an utmost short staining protocol.
In conclusion, when using an optimized protocol for immunofluorescence, laser-assisted cell picking can be effectively combined with real-time quantitative RT-PCR from a few immunodetected cells. The results obtained from lavaged AMs as reference confirmed the quantitative data obtained by cell picking. Thus, the combination with immunodetection is a substantial extension of the current technology allowing detection of pathophysiological gene regulation in a cell-specific manner within intact tissue structures.
Acknowledgments
We thank M. M. Stein for skillful technical assistance, G. Jurat for photographic arrangement, and Dr. R. Snipes for critical reading of the manuscript.
Footnotes
Address reprint requests to Dr. Ludger Fink, Institut für Pathologie, Universität Giessen, Langhansstrasse 10, D 35392 Giessen, Germany. E-mail: ludger.fink@patho.med.uni-giessen.de.
Supported by the Deutsche Forschungsgemeinschaft (SFB 547 “Cardiopulmonary vascular system,” Project Z1).
References
- 1.Sirivatanauksorn Y, Drury R, Crnogorac-Jurcevic T, Sirivatanauksorn V, Lemoine NR: Laser-assisted microdissection: applications in molecular pathology. J Pathol 1999, 189:150-154 [DOI] [PubMed] [Google Scholar]
- 2.Schütze K, Lahr G: Identification of expressed genes by laser-mediated manipulation of single cells. Nat Biotech 1998, 16:737-742 [DOI] [PubMed] [Google Scholar]
- 3.Hölschermann H, Bohle RM, Zeller H, Schmidt H, Stahl U, Fink L, Grimm H, Tillmanns H, Haberbosch W: In situ detection of tissue factor within coronary intima in rat cardiac allograft vasculopathy. Am J Pathol 1999, 154:211-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kummer W, Fink L, Dvorakova M, Haberberger R, Bohle RM: Rat cardiac neurons express the non-coding R-exon (exon 1) of the cholinergic gene locus. NeuroReport 1998, 9:2209-2212 [DOI] [PubMed] [Google Scholar]
- 5.Fink L, Stahl U, Ermert L, Kummer W, Seeger W, Bohle RM: Rat porphobilinogen deaminase: a pseudogene-free internal standard for laser-assisted cell picking. BioTechniques 1999, 26:510-516 [DOI] [PubMed] [Google Scholar]
- 6.Pauls K, Fink L, Franke FE: Angiotensin-converting enzyme (CD143) in neoplastic germ cells. Lab Invest 1999, 79:1425-1435 [PubMed] [Google Scholar]
- 7.Fink L, Seeger W, Ermert L, Hänze J, Stahl U, Grimminger F, Kummer W, Bohle RM: Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med 1998, 4:1329-1333 [DOI] [PubMed] [Google Scholar]
- 8.Nagasawa Y, Takenaka M, Matsuoka Y, Imai E, Hori M: Quantitation of mRNA expression in glomeruli using laser-manipulated microdissection and laser pressure catapulting. Kidney Int 2000, 57:717-723 [DOI] [PubMed] [Google Scholar]
- 9.Fend F, Emmert-Buck M, Chuaqui R, Cole K, Lee J, Liotta LA, Raffeld M: Immuno-LCM: laser capture microdissection of immunostained frozen sections for mRNA analysis. Am J Pathol 1999, 154:61-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin L, Thompson CA, Qian X, Kuecker SJ, Kulig E, Lloyd RV: Analysis of anterior pituitary hormone mRNA expression in immunophenotypically characterized single cells after laser capture microdissection. Lab Invest 1999, 79:511-512 [PubMed] [Google Scholar]
- 11.Crino PB, Trojanowski JQ, Dichter MA, Eberwine J: Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc Natl Acad Sci USA 1996, 93:14152-14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fink L, Kinfe T, Stein MM, Ermert L, Hänze J, Seeger W, Kummer W, Bohle RM: Immunostaining and cell picking for mRNA analysis. Lab Invest 2000, 80:327-333 [DOI] [PubMed] [Google Scholar]
- 13.Heid CA, Stevens J, Livak KJ, Williams PM: Real time quantitative PCR. Genome Res 1996, 6:986-994 [DOI] [PubMed] [Google Scholar]
- 14.Gibson UEM, Heid CA, Williams PM: A novel method for real time quantitative RT-PCR. Genome Res 1996, 6:995-1001 [DOI] [PubMed] [Google Scholar]
- 15.Ermert L, Ermert M, Merkle M, Goppelt-Struebe M, Duncker HR, Grimminger F, Seeger W: Rat pulmonary cyclooxygenase-2 expression in response to endotoxin challenge: differential regulation in the various types of cells in the lung. Am J Pathol 2000, 156:1275-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordell JL, Falini B, Erber WN, Gosh AK, Abdulaziz Z, MacDonald S, Pulford KAF, Stein H, Mason DY: Immunoenzymatic labeling of monoclonal antibodies using immune complexes of alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes). J Histochem Cytochem 1984, 32:219-229 [DOI] [PubMed] [Google Scholar]
- 17.Murase T, Inagaki H, Eimoto T: Influence of histochemical and immuno-histochemical stains on polymerase chain reaction. Mod Pathol 2000, 13:147-151 [DOI] [PubMed] [Google Scholar]
- 18.Goldsworthy SM, Stockton PS, Trempus CS, Foley JF, Maronpot RR: Effects of fixation on RNA extraction and amplification from laser capture microdissected tissue. Mol Carcinog 1999, 25:86-91 [PubMed] [Google Scholar]
- 19.Shibutani M, Uneyama C, Miyazaki K, Toyoda K, Hirosa M: Methacarn fixation: a novel tool for analysis of gene expressions in paraffin-embedded tissue specimens. Lab Invest 2000, 80:199-208 [DOI] [PubMed] [Google Scholar]
- 20.Weisz A, Oguchi S, Citatiello L, Esumi H: Dual mechanism for the control of inducible-type NO synthase gene expression in macrophages during activation by interferon-gamma and bacterial lipopolysaccharide. Transcriptional and post-transcriptional regulation. J Biol Chem 1994, 269:8324-8333 [PubMed] [Google Scholar]
- 21.Kunz D, Muhl H, Walker G, Pfeilschifter J: Two distinct signaling pathways trigger the expression of inducible nitric oxide synthase in rat renal mesangial cells. Proc Natl Acad Sci USA 1994, 91:5387-5391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha B, Eigler A, Baumann KH, Greten TF, Moeller J, Endres S: Nitric oxide downregulates tumour necrosis factor in mRNA in RAW 264.7 cells. Res Immunol 1998, 149:139-150 [DOI] [PubMed] [Google Scholar]