

Abstract
The endothelial cell-derived endothelin-1 (ET-1) is a potent mitogen for endothelial cells, vascular smooth muscle cells, and tumor cells. In this study, we analyzed the role of ET-1 on human umbilical vein endothelial cell (HUVEC) phenotype related to different stages of angiogenesis. ET-1 promoted HUVEC proliferation, migration, and invasion in a dose-dependent manner. The ETB receptor (ETBR) antagonist, BQ 788, blocked the angiogenic effects induced by ET-1, whereas the ETAR antagonist was less effective. ET-1 stimulated matrix metalloproteinase-2 mRNA expression and metalloproteinase-2 production, as determined by reverse transcriptase-polymerase chain reaction and gelatin zymography. Furthermore ET-1 was able to enhance HUVEC differentiation into cord vascular-like structures on Matrigel. When tested in combination with vascular endothelial growth factor (VEGF), ET-1 enhanced VEGF-induced angiogenic-related effects on endothelial cells in vitro. Finally, using the Matrigel plug neovascularization assay in vivo, ET-1 in combination with VEGF stimulated an angiogenic response comparable to that elicited by basic fibroblast growth factor. These findings demonstrated that ET-1 induces angiogenic responses in cultured endothelial cells through ETBR and that stimulates neovascularization in vivo in concert with VEGF. ET-1 and its receptors acting as angiogenic regulators might represent new targets for anti-angiogenic therapy.
Angiogenesis, the sprouting of new capillaries from pre-existing blood vessels, is a multistep process that involves migration and proliferation of endothelial cells, remodeling of the extracellular matrix, and functional maturation of the newly assembled vessels. 1 Several angiogenic growth factors have been characterized so far, including members of vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) families. 2,3 Tissue hypoxia represents a physiological stimulus for angiogenesis, enhancing the production of several autocrine angiogenic mediators produced by endothelial cells. 4,5 Endothelin-1 (ET-1), one such mediator, is produced by endothelial cells, by vascular smooth muscle cells, and its concentration is elevated in many tumors. 6 ET-1 acts through two distinct subtypes of G protein-coupled receptors, ETA and ETB, expressed in a wide variety of tissues. 7 Because ET-1 stimulates proliferation and migration of endothelial cells, 8-11 and is a potent mitogen for vascular smooth muscle and tumor cells, 12,13 it has been suggested that this peptide could stimulate angiogenesis. We have previously demonstrated that the expression of ET-1 is significantly increased in the majority of ovarian carcinomas when compared with normal ovarian tissues. In these tumor cells ET-1 acts as an autocrine growth factor selectively through ETAR, as demonstrated by the inhibitory proliferative effects induced by a specific ETAR antagonist. 14-16 Moreover the presence of ET-1 correlates with tumor vascularity and malignancy in well-vascularized brain tumors, 17 in colorectal cancer, 18 and the ET-1 binding sites have also been characterized in the vessels of pulmonary tumors. 19
The aim of our study was to investigate the role of ET-1 in the angiogenic process by evaluating the effect of ET-1 at the different stages of neovascularization including proliferation, migration, invasion, protease production, and morphogenesis of human umbilical vein endothelial cells (HUVECs). Furthermore, in the present study, we evaluated the receptor subtype mediating the angiogenic effects of ET-1 on endothelial cells and investigated whether specific antagonists inhibit these ET-1-induced responses. There is increasing evidence that the nature of the response elicited by a specific angiogenic factor is contextual, ie, depends on the presence or absence of other regulatory molecules in the pericellular environment of the responding cell. 20 Because ET-1, predominantly through ETAR, stimulates the synthesis of VEGF in vascular smooth muscle cells and the VEGF-mediated migration and proliferation of endothelial cells, 21 we investigated whether ET-1 would be able to induce an angiogenic phenotype in endothelial cells in association with VEGF. Finally, using the model of in vivo angiogenesis in the pellet of reconstituted basement membrane (Matrigel), we evaluated the ability of ET-1 to stimulate the formation of new vessels. The results clearly demonstrate that ET-1 is able to induce angiogenic responses in cultured endothelial cells through the ETB receptor and stimulates neovascularization in vivo in concert with VEGF.
Materials and Methods
Cells
Human endothelial cells were isolated from human umbilical vein as previously described 22 and maintained in M199 supplemented with 10% fetal calf serum (FCS), 10% newborn calf serum, 0.1 mg/ml endothelial growth supplement (crude extract from bovine brain), 0.1 mg/ml heparin, and 20 mmol/L HEPES. All culture reagents were from Gibco (Paisley, Scotland).
Thymidine Incorporation Assay
Cells were seeded in 96-well plates at 80% confluence (1 × 104cells/well) and incubated in 0.5% FCS medium for 24 hours. Mitogenic stimuli (ET-1, Peninsula Laboratories, Belmont, CA; VEGF165, R&D Systems, Minneapolis, MN) were then added, and after 18 hours 1 μCi of [methyl]-[3H] thymidine (6.7 Ci/mmol; DuPont, New England Nuclear Research Products, Wilmington, DE) was added to each well. ET receptor antagonists (Peninsula Laboratories) were incubated 15 minutes before the addition of ET-1. The antagonists had no effect on basal cellular proliferation. The effects of ET-1 were compared with the basal control condition of 0.5% FCS medium. Six hours later the culture media were removed and the cells were washed three times with phosphate-buffered saline, treated with 10% trichloroacetic acid for 15 minutes, washed twice with 100% ethanol, and solubilized in 0.4 N sodium hydroxide. The cell-associated radioactivity was then determined by liquid scintillation counting. Responses to all agents were assayed in sextuplicate and results were expressed as means of three separate experiments.
Chemoinvasion and Chemotaxis Assay
Chemotaxis was conducted in a 48-well modified Boyden chamber (NeuroProbe, Pleasanton, CA) as previously described. 23 Eight micrometer pore size polyvinylpyrrolidone-free polycarbonate Nucleopore filters (Costar, New York, NY) were coated with gelatin by immersing them overnight in a solution of 100 μg/ml gelatin in 0.1% acetic acid and then dried. The filter separated the attractants from the upper part of the chamber in which HUVECs were added. For chemoinvasion the filter was coated with an even layer of Matrigel (0.5 mg/ml; Becton Dickinson, Milan, Italy), as previously described. 24 HUVECs were harvested in trypsin/ethylenediaminetetraacetic acid solution, collected by centrifugation, and resuspended in Dulbecco’s modified Eagle’s medium supplemented with 0.1% bovine serum albumin. The lower compartment of the chamber was filled with chemoattractants or inhibitors (27 μl/well). Cells (5 × 10 5 cells/ml) were placed in the upper compartment (55 μl/well). ET receptor antagonists (Peninsula Laboratories) were previously added to the cells and preincubated for 15 minutes at 37°C and had no effect on basal cellular migration. After 4 hours (chemotaxis) or 6 hours (chemoinvasion) of incubation at 37°C, filters were stained with Diff-Quick (Merz-Dade, Dudingen, Switzerland) and the migrated cells in 10 high-power fields were counted. Each experimental point was analyzed in triplicate.
Gelatin Zymography
For analysis by zymography, HUVECs at 80% confluence were cultured for 24 hours with M199 with 0.5% FCS and then stimuli were added for an additional 24 hours. The supernatants were collected, concentrated, and electrophoresed (40 μg of protein) for analysis in 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels co-polymerized with 1 mg/ml gelatin. The gels were then washed for 30 minutes at 22°C in 2.5% Triton X-100 to remove sodium dodecyl sulfate and then incubated in 50 mmol/L Tris, pH 7.6, 1 μmol/L ZnCl2, 5 mmol/L CaCl2 for 18 hours at 37°C. After incubation the gels were stained with 0.1% Coomassie blue. Enzyme-digested regions were identified as white bands on a blue background and quantified by computerized image analysis of the band. Molecular sizes were determined from the mobility using gelatinase zymography standards (Bio-Rad Laboratories, Richmond, CA).
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) Analysis
Total RNA was isolated by the guanidium thiocyanate-phenol-chloroform extraction method. The RT-PCR was performed using a GeneAmp RNA PCR kit (Perkin-Elmer Corp., Norway, CT). Briefly, 1 μg of mRNA was used for cDNA synthesis using 50 μmol/L oligo d(T)16 as reverse transcriptase primer at 42°C for 15 minutes. The cDNA were amplified in a final reaction volume of 100 μl containing 0.15 μmol/L oligonucleotide primers. Sequence of the primer set used for metalloproteinase (MMP)-2 was 5′-TTTGGACTGCCCCAGACAGG-3′ and 5′-GCTGCGGCCAGTATCAGTGC-3′ (518-bp fragment). 25 All 5′ primers covered splice junctions, thus evaluating the amplification of genomic DNA. In all experiments, two control reactions, one that did not contain any RNA and the other containing mRNA but no reverse transcriptase, were included. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Gibco) was used as an internal control. The semiquantitative analysis was performed essentially as described by Rieckmann et al. 26 Ten μl of the amplified products were analyzed on a 1.5% agarose gel containing ethidium bromide (Bio-Rad) and photographed. Densitometric scanning was performed with a Mustek MFS-6000CX apparatus, and data were analyzed with Phoretix 1D software and normalized to those of GAPDH. The mRNA values are expressed as relative units calculated according to the following formula: density of the MMP-2 amplification product/density of the GAPDH amplification product × 100. To compare the results of different experiments, optimal cycle conditions for linear amplification were determined by a semiquantitative assay of the amplified products at different cycles. Thirty-six cycle products (94°C for 1 minute, 54°C for 1 minute, and 72°C for 1 minute), which were within the linear logarithmic phase of the amplification curve, were chosen for comparative analysis.
In Vitro Angiogenesis
Morphogenesis assay on Matrigel was performed as previously described. 27 HUVECs cultured for 24 hours in M199 with 0.5% FCS were then plated at 3.5 × 10 5 cells/well in 24-well plates precoated with 250 μl of Matrigel (10.7 mg/ml; Becton Dickinson) in M199 with 0.5% FCS in the absence or in the presence of ET-1 (10 nmol/L) or VEGF (1 ng/ml). After 24 hours of incubation in a 5% CO2-humidified atmosphere at 37°C, the cell three-dimensional organization was examined under an inverted photomicroscope. Each treatment was performed in triplicate wells.
In Vivo Angiogenic Assay
The method described by Passaniti et al 28 has been used with minor modification. 29 Briefly, 0.5 ml Matrigel (10.7 mg/ml) alone or containing angiogenic stimuli was injected subcutaneously into C57BL/6N male mice (Charles River, Calco, Como, Italy). After 7 days animals were sacrificed and the gels were recovered. The angiogenic response was evaluated by macroscopic analysis and by measurement of the hemoglobin content. Hemoglobin was mechanically extracted by mincing the pellet. The hemoglobin content was measured using Drabkin reagent kit 525 (Sigma Chemical Co., St Louis, MO) and the final concentration of hemoglobin was calculated from a standard calibration curve.
Statistical Analysis
All statistical analysis were performed by the Inplot software system (GraphPad Software Inc., San Diego, CA).
Results
Proliferation of Endothelial Cells Induced by ET-1
RT-PCR analysis, using HUVEC total cellular RNA and specific ET-1 and its receptor primers, demonstrated the presence of endogenous transcripts of the appropriate molecular size, thereby confirming that HUVECs express mRNA for ET-1 and for the ETB receptor as the major population, whereas the expression of ETA mRNA was weaker (data not shown).
ET-1 induced a concentration-dependent proliferation of HUVEC with a maximal effect of ET-1 at 10 nmol/L (50% increase over 0.5% FCS medium, used as control) (Figure 1) ▶ . The maximal proliferative effect induced by 10 nmol/L of ET-1 was equipotent with that elicited by 1 ng/ml VEGF. Additive proliferative effects were observed when adding ET-1 (10 nmol/L) to VEGF (1 ng/ml) as demonstrated by a significant (P < 0.001) increase in DNA synthesis. To elucidate the subtype of the ET receptor involved in HUVEC proliferation, we co-incubated endothelial cells with ET-1 and the ETAR antagonist, BQ 123, and the ETBR antagonist, BQ 788. The stimulatory action of 10 nmol/L ET-1 was completely blocked in the presence of 1 μmol/L BQ 788. In the same experiment, the addition of the ETAR antagonist (BQ 123 1 μmol/L) caused only a slight decrease in [3H]thymidine incorporation by HUVECs. Endothelial cell proliferation induced by bFGF was not inhibited by either the addition of 1 μmol/L BQ 788 or BQ 123, indicating that the inhibitory effect induced by the ETBR and ETAR antagonist was specific and was not because of cytotoxicity (data not shown). Taken together, these data confirm that mitogenic signaling by ET-1 is mediated mainly by the ETBR subtype.
Figure 1.
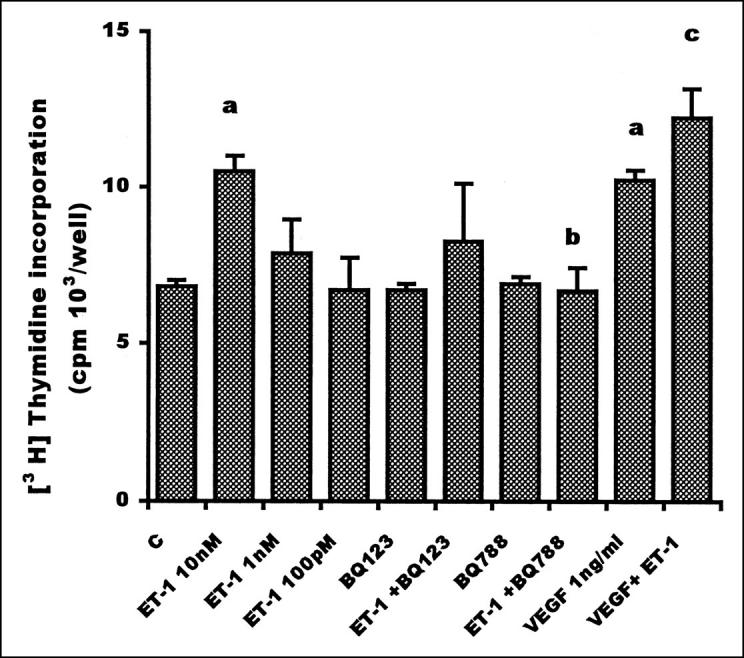
Proliferative effect of ET-1 and VEGF on HUVEC. Endothelial cells (1 × 10 4 cells/ml) were plated in 0.5% FCS medium. Different doses of ET-1 were added to endothelial cells. ET receptor antagonists (1 μmol/L) were incubated 15 minutes before the addition of ET-1 (10 nmol/L). VEGF (1 ng/ml) was added in the absence or presence of ET-1 (10 nmol/L). In all cases, [3H]thymidine incorporation was analyzed 24 hours after the addition of agonists. Data are means of results from three experiments, each performed in sextuplicate. Bars, SD. a: P ≤ 0.001 compared to control. b: P ≤ 0.001 compared to ET-1 10 nmol/L. c:P ≤ 0.01 compared to ET-1 and VEGF.
Endothelial Cell Migration Induced by ET-1
To test whether ET-1 could affect endothelial cell motility, HUVECs were incubated in a Boyden chamber with ET-1 for 4 hours. ET-1 induced a dose-dependent increase in HUVEC migration, as shown in Table 1 ▶ . A significant (P ≤ 0.02) 50% increase in the number of migrated cells was detected with ET-1 (10 nmol/L), similar to that observed in the presence of VEGF (1 ng/ml). When endothelial cells were treated with ET-1 in the presence of VEGF, additive chemotactic effects were observed, as shown in Figure 2 ▶ . To elucidate the type of ET receptor involved in the stimulation of HUVEC migration, experiments were performed with an antagonist of the ETA receptor and agonist and antagonist of ETB receptors. Ten nmol/L of selective ETB agonists, ET-3 and S6c, were able to significantly (P ≤ 0.02) enhance endothelial cell migration. In the same experiment, a selective ETBR antagonist, BQ 788, strongly inhibited the stimulatory action of ET-1, whereas the addition of BQ 123 only induced a partial decrease. Taken together, these data indicate that ET-1 induces endothelial cell migration and that chemotactic signaling is mediated mainly by the ETB receptor subtype.
Table 1.
Dose-Dependent Effect of ET-1 on the Migration and on the Invasion of Matrix by HUVEC
| Chemotaxis No. of cells | Chemoinvasion No. of cells | |
|---|---|---|
| Control | 16 ± 2.6 | 34 ± 1.4 |
| ET-1 100 nmol/L | 28 ± 3.2 | 56 ± 1.4* |
| ET-1 10 nmol/L | 31 ± 6.5 | 50 ± 1.9 |
| ET-1 1 nmol/L | 26 ± 3.0 | 40 ± 1.7 |
| ET-1 100 pmol/L | 22 ± 2.8 | 35 ± 1.3 |
Results are expressed as the mean ± SD.
*P≤0.001 compared to control
Figure 2.

Effect of ET-1 and VEGF on HUVEC migration. Endothelial cells (5 × 10 5 cells/ml) pretreated for 15 minutes with 1 μmol/L of the ETAR (BQ 123) or ETBR antagonist (BQ 788) were placed in the upper compartment of a 48-well Boyden chamber. VEGF (1 ng/ml), ET-1 (10 nmol/L), and ETBR agonists (ET-3 and S6c, 10 nmol/L) were added in lower wells. Cells migrated through the filter were counted after 4 hours. Data are expressed as the number of migrated cells in 10 high-power fields and are the means of results from three experiments each performed in triplicate. Bars, SD. a: P ≤ 0.02 compared to control. b: P ≤ 0.001 compared to control. c: P ≤ 0.02 compared to ET-1. d: P ≤ 0.01 compared to VEGF.
Endothelial Cell Invasion Induced by ET-1
To form new blood vessels, endothelial cells have to migrate and cross basement membranes. The invasive capacity of HUVECs in response to ET-1 was investigated by measuring the invasion of a Matrigel layer in a Boyden chamber assay. Addition of ET-1 induced a dose-dependent increase in HUVEC invasion through Matrigel, as shown in Table 1 ▶ . Maximal stimulation, corresponding to a 50% increase in the number of migrated cells, was obtained at 100 nmol/L ET-1. Furthermore, ET-1 (100 nmol/L) potentiated the VEGF-induced invasive effects (1 ng/ml). Addition of BQ 788 significantly (P ≤ 0.001) affected endothelial cell invasion, whereas BQ 123 did not induce a significant inhibition (Figure 3) ▶ indicating that this response to ET-1 is mediated by the ETB receptor. Phenanthroline, used as a reference inhibitor of invasiveness, completely blocked the invasive capacity of HUVECs (data not shown). These data demonstrate, for the first time, that ET-1 is able to induce endothelial cell invasion through ETBR and to cooperate with VEGF inducing the invasion through the basement membrane.
Figure 3.

Effect of ET-1 and VEGF on HUVEC invasion. HUVEC (5 × 10 5 cells/ml) were seeded on Matrigel layer in a Boyden chamber assay. Endothelial cells were preincubated for 15 minutes with ETA (BQ 123) or ETB (BQ 788) receptor antagonists (1 μmol/L) before the addition of 100 nmol/L ET-1. VEGF (1 ng/ml) in the absence or presence of ET-1 (100 nmol/L) was added in the lower wells. Cells migrating through the filter were counted after 6 hours. Data are expressed as the number of migrated cells in 10 high-power fields and are means of results from three experiments each performed in triplicate. Bars, SD. a: P ≤ 0.001compared to control. b: P ≤ 0.001 compared to ET-1. c: P ≤ 0.01 compared to VEGF.
ET-1 Enhances Gelatinolytic Activity Released by Endothelial Cells
To invade through the basement membrane, endothelial cells must degrade various constituents of the interstitial stroma and basement membrane. MMP-2 is a major extracellular matrix proteolytic enzyme and it is secreted when endothelial sprouting takes place, thus enhancing endothelial cell migration across the matrix. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis gelatin zymography of conditioned media of HUVECs not exposed to angiogenic stimuli, demonstrated a characteristic gelatinase activity corresponding to the 72-kd form of proMMP-2. When the cells were grown in the medium supplemented with different concentrations of ET-1 for 24 hours, the secretion of MMP-2 was increased in a dose-dependent manner (data not shown). A low level of the MMP-9 was also detected. Soft laser scanning of the band, in fact, showed a 2.5-fold increase in the conditioned media of HUVECs treated with ET-1 (100 nmol/L), equipotent to that induced by VEGF (1 ng/ml) when compared with nonstimulated cultures (Figure 4) ▶ . 30 An ET-1 concentration of 100 nmol/L is saturating in terms of MMP-2 production in HUVECs, as observed in a dose-response experiment (not shown). The addition of VEGF to ET-1 was not able to induce a further increase in the MMP-2 zymogram induced by ET-1 alone (data not shown). To determine whether the observed effects of ET-1 and VEGF on the MMP-2 production were transcriptionally regulated, we established a sensitive RT-PCR analysis to detect mRNA transcript for the MMP-2 gene. The RT-PCR amplified cDNA fragments for MMP-2 were detectable in all samples as a single band at the expected size (Figure 4B) ▶ . Primers for the amplification of the GAPDH gene were used as control. Densitometric analysis of these bands and comparison with the intensity of the bands of the GAPDH expression, indicated an up-regulation of MMP-2 mRNA after 4 hours of stimulation with ET-1 (100 nmol/L, Figure 4C ▶ ). These results correlated closely with the protein expression observed by zymography. Hence, RT-PCR analysis showed that MMP-2 mRNA levels correlated well with the secreted protein levels, suggesting that MMP-2 up-regulation induced by ET-1 and VEGF was at the transcriptional level.
Figure 4.


Effect of ET-1 and VEGF on MMP-2 production in HUVEC. Endothelial cells were cultured for 24 hours in 0.5% FCS in the absence or in presence of 100 nmol/L ET-1 or 1 ng/ml VEGF. A: After incubation, the conditioned medium of control (lane 1), 100 nmol/L ET-1 (lane 2), and 1 ng/ml VEGF (lane 3) -treated cells was analyzed by gelatin-zymography, showing that HUVEC released the 72-kd form of MMP-2 into their supernatants. B: MMP-2 mRNA detected by RT-PCR in HUVEC. PCR products for MMP-2 and GAPDH of control (lane 1), 100 nmol/L ET-1 (lane 2), and 1 ng/ml VEGF (lane 3) were shown as visualized by ethidium bromide. C: The relative expression of MMP-2 mRNA (open bars) or protein (hatched bars) in HUVEC. Protein production was determined by densitometric scanning of zymography of cells treated with ET-1 or VEGF. mRNA levels were determined by densitometric scanning of ethidium-bromide bands normalized to those of GAPDH. Data are the mean ± SD of three independent experiments and are expressed as the percent of control. **, P < 0.001 compared to control.
ET-1 Induces in Vitro Morphological Differentiation
To examine whether ET-1 induces morphogenetic changes resembling capillary like-structure tube formation, HUVECs were plated on Matrigel. HUVECs in the presence of 0.5% FCS exhibited small round shapes, isolated cells, and did not spread (Figure 5A) ▶ . Treatment with different doses of ET-1 for 24 hours resulted in morphological changes (data not shown). At the optimal dose of 10 nmol/L of ET-1, the cells became elongated, forming thin cords of interconnecting cells (Figure 5B) ▶ . Similar effects were also observed with 1 ng/ml VEGF (Figure 5C) ▶ . 31 Co-addition of ET-1 and VEGF resulted in a marked increase in morphogenesis in vitro, compared to that observed with ET-1 and VEGF alone, inducing HUVEC elongation and branching to form a network of capillary-like structures (Figure 5D) ▶ . These data demonstrate that ET-1, like VEGF, is able to mediate morphogenetic effects and that it cooperates with VEGF to enhance HUVEC differentiation into vascular structures, which would be necessary in vivo for the sprouting of endothelial cells and tube formation.
Figure 5.
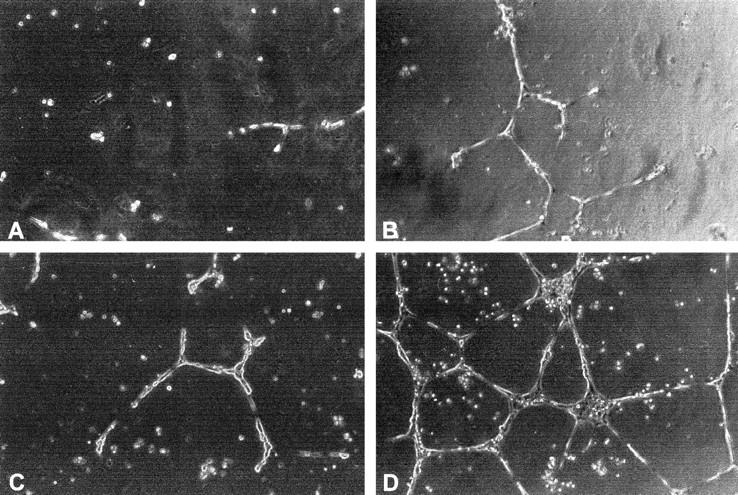
Morphogenic activity of ET-1 and VEGF. HUVEC (3.5 × 10 5 cells/well) were seeded onto Matrigel-precoated wells and cultured in low-serum conditions (0.5% FCS) in absence (control) (A) or in presence of 10 nmol/L ET-1 (B), 1ng/ml VEGF (C), or ET-1 (10 nmol/L) + VEGF (1ng/ml) (D). Photographs were taken 24 hours later. 0riginal magnification, ×200.
ET-1 Enhances VEGF-Induced in Vivo Angiogenesis
The effect of ET-1 on angiogenesis in vivo was tested using an experimental model in which angiogenesis was induced by factors embedded in a pellet of Matrigel. 29 The gels were then injected subcutaneously in mice and after 7 days, the degree of vascularization was evaluated. Quantitation of angiogenesis by hemoglobin content showed that the addition of ET-1 (100 nmol/L) or VEGF (up to 300 ng/ml) to the Matrigel did not induce an increased angiogenic response compared to the negative control (Matrigel plugs containing vehicle). At variance, ET-1 added with VEGF to the pellets caused a significant increase of the angiogenic response compared to ET-1 or VEGF alone. The angiogenic response of Matrigel neovascularization was comparable to that elicited by the prototypic angiogenic factor bFGF (300 ng/ml) used as positive control (Table 2) ▶ . These results were confirmed by histological analysis of Matrigel pellets, demonstrating that the addition of both ET-1 and VEGF to the Matrigel resulted in the induction of cellularity and in the formation of cords, tubules, and several blood-filled channels containing red blood cells (Figure 6B) ▶ , whereas Matrigel pellets without angiogenic stimuli presented only few infiltrating single elongated cells (Figure 6A) ▶ .
Table 2.
Angiogenesis in Matrigel Implanted in Vivo
| Factors | ET-1 (nmol/L) | n | Hemoglobin content of the pellet (g/dl) mean ± SD |
|---|---|---|---|
| - | − | 8 | 0.027 ± 0.017 |
| bFGF | - | 10 | 0.057 ± 0.010* |
| VEGF | - | 9 | 0.028 ± 0.024 |
| ET | 100 nmol/L | 9 | 0.027 ± 0.013 |
| ET+VEGF | 100 nmol/L | 10 | 0.062 ± 0.014*†‡ |
Angiogenesis response, measured as hemoglobin content in the Matrigel plug, was evaluated 7 days after implantation.
*P ≤ 0.05 compared to negative control.
†P ≤ 0.02 compared to VEGF.
‡P ≤ 0.001 compared to ET-1 100 nmol/L.
Figure 6.

Histological analysis of the angiogenic response induced by ET-1 and VEGF in the Matrigel pellets. A: The histology of Matrigel pellets containing vehicle presented only few infiltrating single cells. B: The addition of both ET-1 (100 nmol/L) and VEGF (300 ng/ml) to the Matrigel resulted in the induction of cellularity and in the formation of cords, tubules, and blood-containing vessels. Original magnification, ×300; counterstained with hematoxylin.
Discussion
The process of angiogenesis is the outcome of an imbalance between positive and negative angiogenic factors. 32 However, the relative expression of the individual angiogenesis factors that contribute to this process, has to be investigated more thoroughly. In the present study we examined the role of a putative angiogenic factor, ie, ET-1, an autocrine regulator of endothelial cells, in the process of neovascularization.
ET-1 is a potent mitogen for vascular smooth muscle cells as well as for endothelial cells and is produced by neoplastic cells. 8,12,13 Several investigators have provided functional evidence of the expression of the ETB receptor in HUVECs, by amplifying the ETBR numbers through the use of phosphoramidon, demonstrating the presence of 100% ETBR in HUVEC. 33 Thus, HUVEC can actively produce and secrete ET-1 and simultaneously express ETB receptor (Kd = 17 pmol/L) as the major population, indicating an autocrine role for synthesized ET-1. Moreover ET-1, through a positive autocrine feed-back action, increases its own synthesis in HUVECs acting via ETBR. 34,35
Previous observations demonstrated that ET-1 and ET-3, acting through ETB receptors, have a dose-dependent stimulatory proliferative and migratory effect on endothelial cells isolated from bovine adrenal capillaries and HUVECs. 8-11 ET-1 has also been shown to accelerate endothelial wound healing via the ETB receptor. 9 These findings suggested that ET-1 may exert an angiogenic activity, 36 as also seen in the case of well-vascularized brain tumors such as gliomas or astrocytomas in which the expression of ET-1 correlates with tumor vascularity and malignancy. 17
We have demonstrated that ET-1 induces a proangiogenic phenotype in human endothelial cells. This phenotype includes both early (ie, increase in cell proliferation and migration and MMP-2 production) and late angiogenic events (differentiation into vascular cords). During the formation of new blood vessels, endothelial cells are stimulated to migrate, proliferate, and invade surrounding tissue to form capillaries. ET-1 and VEGF, the two growth factors that we studied, both induced these angiogenic effects, and by acting in concert have a potent additive effect on different stages of the angiogenesis process. The ability of ET-1 to induce a significant increase of MMP-2, released by endothelial cells, may represent an important step in ET-1-mediated vascularization, because MMP-2, a major extracellular matrix proteolytic enzyme, is secreted when endothelial sprouting takes place, thus enhancing endothelial cell migration across the extracellular matrix. 37 Endothelial cells exposed to ET-1 migrate throughout the Matrigel surface and align to form vascular cord-like structures, indicating that ET-1 is able to stimulate morphogenesis in cultured endothelium. The most striking effect was seen in combination with VEGF: when added simultaneously, VEGF and ET-1 induced a marked increase in the tube formation in cultured endothelial cells.
Overall, our observations demonstrate that ET-1 stimulates vasoproliferative processes and at maximally effective concentrations its effects were additive to that of VEGF. The mechanisms underlying the cooperation between ET-1 and VEGF are not well defined. In our study, the predominately additive actions of ET-1 and VEGF on angiogenic events at saturating agonist concentrations reflect the independence between their individual signaling mechanisms.
Recent studies, exploring a potential interaction between VEGF and ET-1 in vascular cells, evidenced that VEGF enhanced ET-1 mRNA expression and ET-1 secretion in endothelial cells. 38 Similarly, in vascular smooth muscle cells, ET-1, predominantly through ETAR, enhances VEGF mRNA expression and VEGF secretion and stimulated the VEGF-induced endothelial cell proliferation and invasion. 21,39 This indicated that VEGF and ET-1 have reciprocal stimulatory interactions, resulting in concomitant proliferation of endothelial and vascular smooth muscle cells. In pathological conditions such as cancer, VEGF and ET-1 may be up-regulated by various stimuli including hypoxia, growth factors, and inflammatory cytokines. 39 Because hypoxia constitutes a potent stimulus for VEGF and ET-1 production, it is reasonable to hypothesize that in tumor tissues, acute or chronic hypoxia may stimulate VEGF production through both a direct and indirect effect, the latter involving ET-1 secretion as well. 40 Furthermore, because ET-1 functions as an antiapoptotic factor for endothelial cells and vascular smooth muscle cells, 41,42 this peptide may also contribute to endothelial cell integrity acting as a survival factor for newly formed blood vessels.
Previous observations had shown that ET-1 significantly increased angiogenesis evaluated by blood vessel growth in the rat aortic ring assay, and that erythropoietin-induced neovascularization is partially because of the enhanced autocrine release of ET-1 induced by erythropoietin, because the stimulation of angiogenesis by erythropoietin had been blunted by the ET-1 antibody. 43,44 On the contrary, other data show that ET-1 is unable to stimulate blood vessel growth in the chick embryo chorioallantoic membrane 45 and in a rat sponge model. 46 Having shown that ET-1 cooperates with VEGF in the induction of an angiogenic phenotype in HUVECs in several in vitro assays, it was important to investigate the in vivo relevance of these properties. In this study, ET-1 in association with VEGF has a clear angiogenic activity in the Matrigel in vivo assay, comparable to that promoted by bFGF, demonstrating that ET-1 enhances formation of new vessels in vivo.
These results demonstrated that ET-1 like VEGF, induces angiogenic responses, including endothelial cell protease production, migration, and invasion, via a direct effect on the endothelial cells, and by acting in concert, these two factors have an additive effect on the induction of angiogenesis in vitro and in vivo, suggesting that the interactions between VEGF and ET-1 play an important role in the control of angiogenesis.
In tumors, such as in ovarian carcinoma, ET-1 and VEGF apparently play a complementary and coordinated role during neovascularization and malignant ascite formation. 47 In these cells, we demonstrated that ET-1 stimulated VEGF production through ETAR in a manner equipotent to hypoxia, a recognized potent stimulus of VEGF production. Moreover, we demonstrated that elevated levels of ET-1 released by ovarian carcinoma cells in ascitic fluids and in culture media were primarily responsible for endothelial cell migration. The significant inhibition of migration obtained co-incubating endothelial cells with ETBR antagonists and with antibody to VEGF strongly indicated that ET-1, together with VEGF, could modulate angiogenesis.
Altogether, these results provide evidence of the role of ET-1 in neovascularization via a mechanism requiring the activation of the ETB receptor on endothelial cells. Thereafter, the tumor promoting effect of ET-1 could be mediated through direct angiogenic effects on endothelial cells and in part through the VEGF stimulation in the existing tumor.
Because angiogenesis is not only controlled by the presence of VEGF, but may be mediated by several angiogenic factors, we identified ET-1 and its receptors as angiogenic regulators that could represent novel targets for anti-angiogenic therapies. If an angiogenic response is sustained by both VEGF and ET-1, neutralization of one of these two factors may greatly decrease that response by suppressing the contextual effect. New therapeutic strategies using specific antagonists for ET receptor, 48 which have no genotoxic effects, provide an additional approach for the treatment of tumors characterized by active angiogenesis, and of other angiogenesis-dependent diseases.
Acknowledgments
We wish to dedicate this work to our colleague and friend, Raffaele Tecce, of the Regina Elena Cancer Institute, who died last summer. His loss ends a long time of friendly and precious collaboration that we will miss. We thank Marco Varmi for excellent technical assistance, Dr. Roberta Ceruti for histopathological analysis, Dr. Pier Giorgio Natali for critical reading and discussion of this manuscript, and Paula Franke for the formal revision of the manuscript.
Footnotes
Address reprint requests to Dr. Anna Bagnato, Molecular Pathology Laboratory, Regina Elena Cancer Institute, Via delle Messi d’Oro 156, 00158 Rome, Italy. E-mail: bagnato@ifo.it.
Supported by grants from the Associazione Italiana Ricerca sul Cancro and Fondazione Italiana per la Ricerca sul Cancro. D. S. and L. R. are recipients of fellowships from Fondazione Italiana Ricerca sul Cancro.
References
- 1.Folkman J, Shing Y: Angiogenesis. J Biol Chem 1992, 267:10931-10934 [PubMed] [Google Scholar]
- 2.Ferrara N, Davis-Smith T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-23 [DOI] [PubMed] [Google Scholar]
- 3.Bikfalvi A, Klein S, Pintucci G, Rifkin DB: Biological roles of fibroblast growth factor-2. Endocr Rev 1997, 18:26-45 [DOI] [PubMed] [Google Scholar]
- 4.Kourembanas S, Maraden PA, Mcquillan LP, Faller DV: Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest 1991, 88:1054-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodi I, Bishopric NM, Discher DJ, Wu X, Webster KA: Cell-specificity and signaling pathway of endothelin-1 gene regulation by hypoxia. Cardiovasc Res 1995, 30:975-980 [DOI] [PubMed] [Google Scholar]
- 6.Levin ER: Endothelins. N Engl J Med 1995, 333:356-363 [DOI] [PubMed] [Google Scholar]
- 7.Rubanji GM, Polokoff MA: Endothelins: molecular biology, biochemistry, pharmacology, physiology and pathophysiology. Pharmacol Rev 1994, 46:325-414 [PubMed] [Google Scholar]
- 8.Morbidelli L, Orlando C, Maggi CA, Ledda F, Ziche M: Proliferation and migration of endothelial cells is promoted by endothelins via activation of ETB receptors. Am J Physiol 1995, 269:4686-4695 [DOI] [PubMed] [Google Scholar]
- 9.Wren AD, Hiley CR, Fan TD: Endothelin-3 mediated proliferation in wounded human umbilical vein endothelial cells. Biochem Biophys Res Commun 1993, 196:369-375 [DOI] [PubMed] [Google Scholar]
- 10.Noiri E, Hu Y, Bahou WF, Keese CR, Giaever I, Goligorsky MS: Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. J Biol Chem 1997, 272:1747-1752 [DOI] [PubMed] [Google Scholar]
- 11.Goligorsky MS, Budzikowski AS, Tsukahara H, Noiri E: Co-operation between endothelin and nitric oxide in promoting endothelial cell migration and angiogenesis. Clin Exp Pharmacol Physiol 1999, 26:269-271 [DOI] [PubMed] [Google Scholar]
- 12.Alberts GF, Peifley KA, Johns A, Kleha JF, Winkles JA: Constitutive endothelin-1 overexpression promotes smooth muscle cell proliferation via an external autocrine loop. J Biol Chem 1994, 269:10112-10118 [PubMed] [Google Scholar]
- 13.Bagnato A, Catt KJ: Endothelins as autocrine regulators of tumor cell growth. Trends Endocrinol Metab 1998, 9:378-383 [DOI] [PubMed] [Google Scholar]
- 14.Bagnato A, Tecce R, Moretti C, Di Castro V, Spergel DJ, Catt KJ: Autocrine actions of endothelin-1 as a growth factor in human ovarian carcinoma cells. Clin Cancer Res 1995, 1:1059-1066 [PubMed] [Google Scholar]
- 15.Bagnato A, Salani D, Di Castro V, Wu-Wong JR, Tecce R, Nicotra MR, Venuti A, Natali PG: Expression of endothelin-1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res 1999, 59:1-8 [PubMed] [Google Scholar]
- 16.Bagnato A, Tecce R, Di Castro V, Catt KJ: Activation of mitogenic signaling by endothelin-1 in ovarian carcinoma cells. Cancer Res 1997, 57:1306-1311 [PubMed] [Google Scholar]
- 17.Stiles JD, Ostrow PT, Balos LL, Greenberg SJ, Plunkett R, Grand W, Heffner RR: Correlation of endothelin-1 and transforming growth factor β1 with malignancy and vascularity in human gliomas. J Neuropathol Exp Neurol 1997, 56:435-439 [DOI] [PubMed] [Google Scholar]
- 18.Shankar A, Loizidou M, Aliev G, Fredericks S, Holt D, Boulos PB, Burnstock G, Taylor I: Raised endothelin-1 levels in patients with colorectal liver metastases. Br J Surg 1998, 85:502-506 [DOI] [PubMed] [Google Scholar]
- 19.Zaho Y, Spingall D, Hamid P, Levene M, Polak J: Localization and characterization of endothelin-1 receptor binding in the blood vessels of human pulmonary tumors. J Cardiovasc Pharmacol 1995, 26(Suppl 3):9341-9345 [PubMed] [Google Scholar]
- 20.Sporn MB, Roberts AB: Peptide growth factors are multifunctional. Nature 1988, 332:217-219 [DOI] [PubMed] [Google Scholar]
- 21.Pedram A, Rasandi M, Hu RM, Levin ER: Vasoactive peptides modulate vascular endothelial cell growth factor production and endothelial cell proliferation and invasion. J Biol Chem 1997, 272:17097-17103 [DOI] [PubMed] [Google Scholar]
- 22.Jaffe E, Nachman R, Becker C, Mimick C: Culture of human endothelial cells derived from umbilical veins: identification of morphologic and immunologic criteria. J Clin Invest 1973, 52:2745-2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taraboletti G, Roberts D, Liotta LA, Giavazzi R: Platelet thrombospondin modulates endothelial cell adhesion, motility and growth: a potential angiogenesis regulatory factor. J Cell Biol 1990, 111:765-772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albini A, Iwamato Y, Kleinman HK, Martin GW, Aaronson SA, Korlowski JM, McEwan RN: A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res 1987, 47:3239-3245 [PubMed] [Google Scholar]
- 25.Puyraimond A, Weitzman JB, Babole E, Menashi S: Examining the relationship between the gelatinolytic balance and the invasive capacity of endothelial cells. J Cell Sci 1999, 112:1283-1290 [DOI] [PubMed] [Google Scholar]
- 26.Rieckmann P, Albrecht M, Ehrenreich H, Weber T, Michel U: Semiquantitative analysis of cytokine gene expression in blood and cerebrospinal fluid cells by reverse transcriptase polymerase chain reaction. Res Exp Med 1995, 195:17-29 [DOI] [PubMed] [Google Scholar]
- 27.Kubota Y, Kleinman HK, Martin GR, Lawley TJ: Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J Cell Biol 1988, 107:1589-1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Passaniti A, Taylor RM, Pili R, Guo Y, Long PV, Haney A, Pauly RR, Grant DS, Martin GR: A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab Invest 1992, 67:519-528 [PubMed] [Google Scholar]
- 29.Belotti D, Vergani V, Drudis T, Borsotti P, Pitelli MR, Viale G, Giavazzi R, Taraboletti G: The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin Cancer Res 1996, 2:1843-1849 [PubMed] [Google Scholar]
- 30.Zucher S, Mirza H, Conner CE, Lorenz AF, Drews MH, Bahou WF, Jesty J: Vascular endothelial growth factor induces tissue factor and matrix metalloproteinase production in endothelial cells: conversion of prothrombin to thrombin results in progelatinase A activation and cell proliferation. Int J Cancer 1998, 75:780-786 [DOI] [PubMed] [Google Scholar]
- 31.Bikfalvi A, Sauzeau C, Moukodizi H, Maclouf J, Busso N, Bryckaert M, Plouet J, Tobelem G: Interaction of vasculotropin/vascular endothelial cell growth factor with human umbilical vein endothelial cell: binding, internalization, degradation, and biological effects. J Cell Physiol 1991, 149:50-59 [DOI] [PubMed] [Google Scholar]
- 32.D’Amore PA, Thomson RW: Mechanisms of angiogenesis. Annu Rev Physiol 1987, 49:453-464 [DOI] [PubMed] [Google Scholar]
- 33.Flynn MA, Haleen SJ, Welch KM, Cheng XM, Reynolds EE: Endothelin B receptors on human endothelial and smooth-muscle cells show equivalent binding pharmacology. J Cardiovasc Pharmacol 1998, 32:106-116 [DOI] [PubMed] [Google Scholar]
- 34.Fujitani Y, Oda K, Takimoto M, Inui T, Okada T, Urade Y: Autocrine receptors for endothelins in the primary culture of endothelial cells of human umbilical vein. FEBS Lett 1992, 298:79-83 [DOI] [PubMed] [Google Scholar]
- 35.Saijonmaa O, Nyman T, Fyyyhrquist F: Endothelin-1 stimulates its own synthesis in human endothelial cells. Biochem Biophys Res Commun 1992, 188:286-291 [DOI] [PubMed] [Google Scholar]
- 36.Dawas K, Loizidou M, Shankar A, Ali H, Taylor I: Angiogenesis in cancer: the role of endothelin-1. Ann R Coll Surg Engl 1999, 81:306-310 [PMC free article] [PubMed] [Google Scholar]
- 37.Mignatti P, Rifkin DB: Biology and biochemistry of proteinase in tumor invasion. Physiol Rev 1993, 73:161-195 [DOI] [PubMed] [Google Scholar]
- 38.Matsuura A, Yamochi W, Hirata K, Kawashima S, Yokoama M: Stimulatory interaction between vascular endothelial growth factor and endothelin-1 on each gene expression. Hypertension 1998, 32:89-95 [DOI] [PubMed] [Google Scholar]
- 39.Kourembanas S, Maraden PA, Mcquillan LP, Faller DV: Hypoxia induces endothelin gene expression and secretion in cultured endothelium. J Clin Invest 1991, 88:1054-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okuda Y, Tsurumaru K, Suzuki S, Miyauchi T, Asano M, Hong Y, Sone H, Fujita R, Mizutani M, Kawakami Y, Nakajima T, Soma M, Matsuo K, Suzuki H, Yamashita K: Hypoxia and endothelin-1 induce VEGF production in human vascular smooth muscle cells. Life Sci 1998, 6:477-484 [DOI] [PubMed] [Google Scholar]
- 41.Shichiri M, Kato H, Marumo F, Hirata Y: Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension 1997, 30:1198-1203 [DOI] [PubMed] [Google Scholar]
- 42.Wu-Wong JR, Chiou WJ, Dickinson R, Opgenorth TJ: Endothelin attenuates apoptosis in human smooth muscle cells. Biochem J 1997, 328:733-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlini RG, Dusso AS, Obialo CI, Alvarez UM, Rothstein M: Recombinant human erythropoietin (rHuEpo) increases endothelin-1 release by endothelial cells. Kidney Int 1993, 43:1010-1014 [DOI] [PubMed] [Google Scholar]
- 44.Carlini RG, Reyes AA, Rothstein M: Recombinant human erythropoietin stimulates angiogenesis in vitro. Kidney Int 1995, 47:740-745 [DOI] [PubMed] [Google Scholar]
- 45.Ribatti D, Presta M, Vacca A, Ria R, Guiliani R, Dell’Era P, Nico B, Roncali L, Dammacco F: Human erythropoietin induces a pro-angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Blood 1999, 93:2627-2639 [PubMed] [Google Scholar]
- 46.Hu D, Hiley RC, Fau TD: Comparative studies of the angiogenic activity of vasoactive intestinal peptide, endothelins-1 and -3 and angiotensin II in a rat sponge model. Br J Pharmacol 1996, 117:545-551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salani D, Di Castro V, Nicotra MR, Rosanò L, Tecce R, Venuti A, Nateli PG, Bagnato A: Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol 2000, 157:1537-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lahav R, Heffner G, Patterson PH: An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci USA 1999, 96:11496-11500 [DOI] [PMC free article] [PubMed] [Google Scholar]