

Abstract
The pathogenesis of keloid remains poorly understood. As no effective therapy for keloid is as yet available, an insight into its pathogenesis may lead to novel approaches. Apoptosis has been found to mediate the decrease in cellularity during the transition between granulation tissue and scar. Here, we report that in contrast to hypertrophic scar-derived and normal skin-derived fibroblasts, keloid-derived fibroblasts are significantly resistant to both Fas-mediated and staurosporine-induced apoptosis. The caspases-3, -8, and -9 were not activated indicating that the block in the apoptotic pathway in keloid is upstream of the caspases. There were no significant differences in the level of expression of Fas, Bcl-2, and Bax between the three groups but addition of transforming growth factor (TGF)-β1 significantly inhibited Fas-mediated apoptosis in hypertrophic scar-derived and normal skin-derived fibroblasts and neutralization of autocrine TGF-β1 with anti-TGF-β1 antibody abrogated the resistance of keloid-derived fibroblasts. Anti-apoptotic activity was not observed with TGF-β2. This is the first study linking refractory Fas-mediated apoptosis to cellular phenotype in keloids and indicating a pivotal role for the anti-apoptotic effect of TGF-β1 in this resistance. Hence, it becomes important to treat keloids as a separate entity different from hypertrophic scars and enhancement of Fas-sensitivity could be a promising therapeutic target.
A keloid is a unique human dermal fibroproliferative disorder that occurs after trauma, inflammation, burns, surgery, and possibly spontaneously. Although it is not fatal, it is a major cosmetic problem and symptoms like itching and pain can significantly disturb the patient’s quality of life. The incidence is highest among the Black population, which has been estimated at 4 to 6% 1 but is also not uncommon among Hispanics and Orientals. It is often addressed as a benign dermal tumor as it spreads to invade normal skin beyond the boundaries of the original wound and does not regress spontaneously. Recurrence is common after surgical excision, which often exacerbates the condition. 2 Hypertrophic scars, on the other hand, are raised scars that remain within the boundaries of the original wound, frequently regress spontaneously and recurrence is rare after surgical excision. Because no effective therapy for keloid is as yet available, an insight into its pathogenesis may lead to novel approaches.
Keloids form when the normal wound-healing process is dysregulated and the evolving scar remains in the proliferative phase of healing 2-4 but the mechanism is still unclear. Keloid-derived fibroblasts (KFs) demonstrate a reduced growth-factor requirement in vitro, 5,6 a unique sensitivity to transforming growth factor (TGF)-β, 7-11 coupled with an increased production of TGF-β1 and 2, 12 which results in increased proliferation and collagen production, and have a greater proliferative capacity than hypertrophic scar-derived fibroblasts (HFs) and normal skin-derived fibroblasts (NFs). 13-15 Furthermore, it has recently been demonstrated that there is p53 gene mutations in keloids. 16,17
Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. 18 The cell-surface Fas receptor (Fas), also termed Apo-1 or CD95, is a member of the tumor necrosis factor and nerve growth factor family of receptors. 19,20 Fas is widely distributed in skin components 21 and it has been shown that Fas receptor stimulation can induce apoptosis or proliferation in human dermal fibroblasts, 22-24 depending on the magnitude of Fas aggregation. 22 We hereby performed a comparative study on the apoptotic properties of human dermal fibroblasts derived from keloids, hypertrophic scars, and normal skin specimens in response to Fas ligation with anti-Fas antibody (clone CH-11). Our results show that a dysregulation in the Fas mediated apoptosis that normally occurs during the process of wound healing may be an important mechanism by which keloids arise and that TGF-β1 is an important factor responsible for this resistance.
Materials and Methods
Cell Culture
We prepared primary fibroblast cultures from fresh keloid and hypertrophic scar tissues obtained at the time of surgical excision after informed consent. Only typical, clinically clear-cut cases of keloid and hypertrophic scar were included in this study. Age-, gender-, and site-matched normal skin specimens were obtained at the time of other unrelated operations. Third generation cells from 15 keloid, nine hypertrophic scar, and nine normal skin specimens were used. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Inc., Gaithersburg, MD) supplemented with 20% and 10% (v/v) heat-inactivated fetal bovine serum for primary culture and subsequent cultures, respectively, and 5 mg/ml l-glutamine in an atmosphere of 5% CO2.
Induction of Apoptosis
Cells (10,000 cells per well) were plated in 24-well plates in DMEM supplemented with 10% fetal bovine serum. After 24 hours, the medium was changed to serum-free medium and incubated for 48 hours after which induction of apoptosis was performed with 0, 0.1, or 1 μg/ml of anti-human Fas antibody (clone CH-11; Medical & Biological Laboratories, Nagoya, Japan) and 0 or 10 nmol/L of staurosporine (Sigma Chemical Co., St. Louis, MO) dissolved in dimethyl sulfoxide.
Detection of Apoptosis
Cell viability was determined by staining with Trypan blue and counting the live/dead cells using a Neubauer hemocytometer (Kayagaki Irikha Kougyou Co. Ltd., Tokyo, Japan). For the assessment of nuclear morphology, cells were stained with 5 μg/ml of Hoechst 33342 dye (Sigma Chemical Co.) and counting of nuclear condensation or fragmentation-positive or -negative cells was done using an inverted fluorescence microscope (IX-70; Olympus, Tokyo, Japan).
Flow Cytometry
Fluorescence-activated cell sorting analysis was done using standard protocols. Cells were detached from culture plates with 0.02% ethylenediaminetetraacetic acid (Boehringer Mannheim, Indianapolis, IN). Detached cells (1 million cells) were washed two times with phosphate-buffered saline containing 1% bovine serum albumin and 0.02% NaN3 and incubated with anti-Fas antibody (Transduction Laboratories, Lexington, KY) for 1 hour on ice. Cells were washed three times and incubated with fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Sigma Chemical Co.) for another hour on ice. After three washes, cells were analyzed on a Becton-Dickinson FACScan (Mountain View, CA). Controls lacking primary antibody or both primary and secondary antibodies were analyzed with each series.
Western Blot Analysis
Equal numbers of cells were lysed with a buffer containing 1% Nonidet P-40, 0.1% sodium deoxycholate, 150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH7.5, 1 mmol/L phenylmethyl sulfonyl fluoride, 0.2 U/ml aprotinin, 10 mmol/L Na4P2O 7, 10 mmol/L NaF, 4 mmol/L ethylenediaminetetraacetic acid, and 2 mmol/L Na3VO4. The protein content was measured by the Bradford assay using Bio-Rad protein assay reagent (Bio-Rad Laboratories, Richmond, CA). Cell lysates (20 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to an Immobilon polyvinylidene difluoride membrane (Millipore, Bedford, MA). After blocking with 5% milk and overnight incubation at 4°C in the presence of anti-human Fas monoclonal antibody (Transduction Laboratories), anti-Bcl-2 monoclonal antibody (Pharmingen, San Diego, CA), anti-Bcl-xL polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), or anti-Bax monoclonal antibody (Medical & Biological Laboratories), the membrane was further processed using horseradish peroxidase-conjugated secondary antibody and a chemiluminescence system (Amersham, Arlington Heights, IL).
Caspase Activation
Induction of apoptosis with anti-Fas antibody or staurosporine was performed as described above. Cells including the detached cells were lysed with a buffer containing 20 mmol/L Tris-HCl, pH 7.2, 1% Triton X-100, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L N-ethylmaleimide, and a cocktail of proteinase inhibitors (phenylmethyl sulfonyl fluoride, chymopapain, aprotinin, leupeptin, and pepstatin). Cell lysates (40 μg) were analyzed using 10 to 14% polyacrylamide gels and Western blotting was done using monoclonal anti-caspase-8 and polyclonal rabbit anti-caspase-9 antibody (Medical & Biological Laboratories), monoclonal anti-caspase-3, and monoclonal anti-caspase-7 (Transduction Laboratories) or monoclonal anti-gelsolin antibody (GS2C4, Sigma).
Treatment with TGF-β1 and TGF-β2
Cells were plated in 24-well plates and treated with recombinant human TGF-β1 (Genzyme/Techne, Cambridge, MA) or recombinant human TGF-β2 (Austral Biologicals, San Ramon, CA) at a dose of 0, 5, or 10 ng/ml, 6 hours before induction of apoptosis with anti-Fas antibody or staurosporine. Hoechst staining and counting were done at 24, 48, and 72 hours. As controls, recombinant human epidermal growth factor (Earth Chemical Co., Hyougo, Japan) and recombinant human platelet-derived growth factor (Genzyme) at a dose of 20 ng/ml were used.
Neutralization of TGF-β1 and TGF-β2 Bioactivity
KFs were plated in 24-well plates and treated with 50 μg/ml of monoclonal anti-human TGF-β1 antibody or 0.1 μg/ml of anti-TGF-β2 neutralizing antibody (concentration for maximal inhibition of the cytokine activity according to the antibody concentration versus percent neutralization curve on the product data sheet; R & D Systems, Minneapolis, MN) at the time of induction of apoptosis with anti-Fas antibody or staurosporine and analyzed at 24, 48, and 72 hours.
Statistical Analysis
The data were analyzed using analysis of variance (ANOVA) followed by Scheffé’s post hoc analysis.
Results
KFs Are Refractory to Fas-Mediated Apoptosis
Third generation fibroblasts derived from keloid, hypertrophic scar, and normal skin of patients by explant method were treated with anti-Fas antibody to induce apoptosis. To create pro-apoptotic stress, serum was withdrawn from the cultures for 48 hours before stimulation with anti-Fas antibody. We observed blebbing, cell rounding, and shrinking, characteristic of apoptotic cells in the anti-Fas-treated cells in NFs and HFs, which was absent in the untreated controls and significantly few in the anti-Fas treated KFs (Figure 1 ▶ ; a, c, and e). To assess cell viability after anti-Fas stimulation, Trypan blue exclusion test was performed and we found that in contrast to 85 and 80.5% survival of KFs when treated with 0.1 and 1 μg/ml of anti-Fas antibody, respectively, for 72 hours, only 49.3 and 35.2% of NFs and 66 and 52.9% of HFs survived (Figure 2, a and b) ▶ . The decrease in cell viability was dependent on both antibody concentration and time of exposure (Figure 2, a and b) ▶ . In addition, anti-Fas treated cells were stained with the fluorescent chromatin dye Hoechst 33342 to assess nuclear morphology. Cells were scored as apoptotic when they showed nuclear chromatin condensation or fragmentation (Figure 1 ▶ ; b, d, and f). Consistent with the results of Trypan blue exclusion test, cell mortality in the KF group was significantly lower than in the NF and HF groups (Figure 2, c and d) ▶ .
Figure 1.

KFs are refractory to Fas-mediated apoptosis. Cells were grown in DMEM and 10% fetal bovine serum and after 24 hours, starved in serum-free medium for 48 hours followed by stimulation with 1 μg/ml of anti-Fas antibody (clone: CH-11; MBL, Japan) for 48 hours. a and b: NFs. c and d: HFs. e and f: KFs. Cellular morphological changes of apoptosis analyzed by phase contrast microscopy. Original magnification, ×200 (a, c, and e). The same fields as that of (a, c, and e) examined by Hoechst 33342 staining (b, d, and f). Note the staining of many condensed nuclei indicative of apoptosis (b and d) that corresponds to the cell rounding seen in (a and c), respectively, and their relative absence in the KFs (e and f).
Figure 2.

KFs are refractory to Fas-mediated apoptosis. Cells were stimulated with anti-Fas antibody as described. Viability assay by trypan blue exclusion (a and b) and percentage of nuclear condensation assessed by Hoechst staining (c and d) after stimulation with 0.1 and 1 μg/ml of anti-Fas, respectively, for 24, 48, and 72 hours. *, P < 0.01 compared with NFs and HFs (ANOVA with Scheffé’s post hoc tests). Values shown are the mean and SD of 15 independent experiments.
Expression of Fas, Bcl-2, Bcl-xL, and Bax
Fas oligomerization in dermal fibroblasts may initiate dual signaling programs, either proliferation or apoptosis, and the chosen outcome may depend on the magnitude of Fas aggregation, ie, the quantity of Fas receptor expression. 23 To determine whether there is any correlation between the level of expression of Fas cell surface receptor and sensitivity to apoptosis, we analyzed the level of cell surface Fas receptor expression by flow cytometry but no significant differences between the groups were observed (Figure 3a) ▶ . Immunoblot analysis of the whole cellular lysates confirmed similar levels of Fas protein expression in the three groups (Figure 3b) ▶ .
Figure 3.

Expression of Fas, Bcl-2, and Bax. a: Flow cytometric analysis of cell surface Fas expression on fibroblasts derived from normal skin (NF), hypertrophic scar (HF), and keloid (KF). Cells were incubated with fluorescein isothiocyanate-conjugated goat anti-mouse IgG alone for control (open curve) or with monoclonal anti-Fas antibody followed by incubation with fluorescein isothiocyanate-conjugated goat anti-mouse IgG (filled curve). b: Representative Western blots depicting expression of Fas, Bcl-2, and Bax. To ensure equal loading, each blot was probed for the presence of actin. Here, Fas and Bcl-2 were probed on the same membrane. N4, N7, H1, H3, K6, K7, and K9, are each fibroblasts derived from normal skin, hypertrophic scar, and keloid of different patients.
Members of the Bcl-2 family of proteins are regulators of the apoptotic signaling, and have been shown to be dysregulated in diverse pathological conditions associated with resistance to apoptosis. 25-28 Therefore, we checked the level of expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL and the pro-apoptotic protein Bax by immunoblotting. No significant differences were seen in the level of expression of Bcl-2 and Bax (Figure 3b) ▶ . Expression of Bcl-xL was not detected in any of the three groups (data not shown). Again, we analyzed the level of expression of Bcl-2, Bax, and Bcl-xL by immunoblotting after apoptotic stimuli (anti-Fas antibody, staurosporine) in a time-course manner (24 hours, 48 hours, 72 hours) but there were no significant differences and Bcl-xL was not detected (data not shown).
KFs Are Refractory to Staurosporine-Induced Apoptosis
To explore the resistance of KFs further, we treated cells with the protein kinase inhibitor staurosporine (10 nmol/L). We found that KFs are also least affected by staurosporine toxicity (Figure 4, a and b) ▶ . The mean viability of the KF group was 88.04% and that of the NF and HF groups were 42.1 and 20.6%, respectively, at 72 hours (Figure 4a) ▶ . Similarly, the mean percentage of cells with nuclear condensation or fragmentation even at 72 hours was only 11.03% in the KF group, whereas it was 38.8 and 59.3% in the NF and HF groups, respectively (Figure 4b) ▶ . Interestingly, in contrast to the results of anti-Fas treatment, we observed that HFs are more susceptible to staurosporine toxicity than NF.
Figure 4.
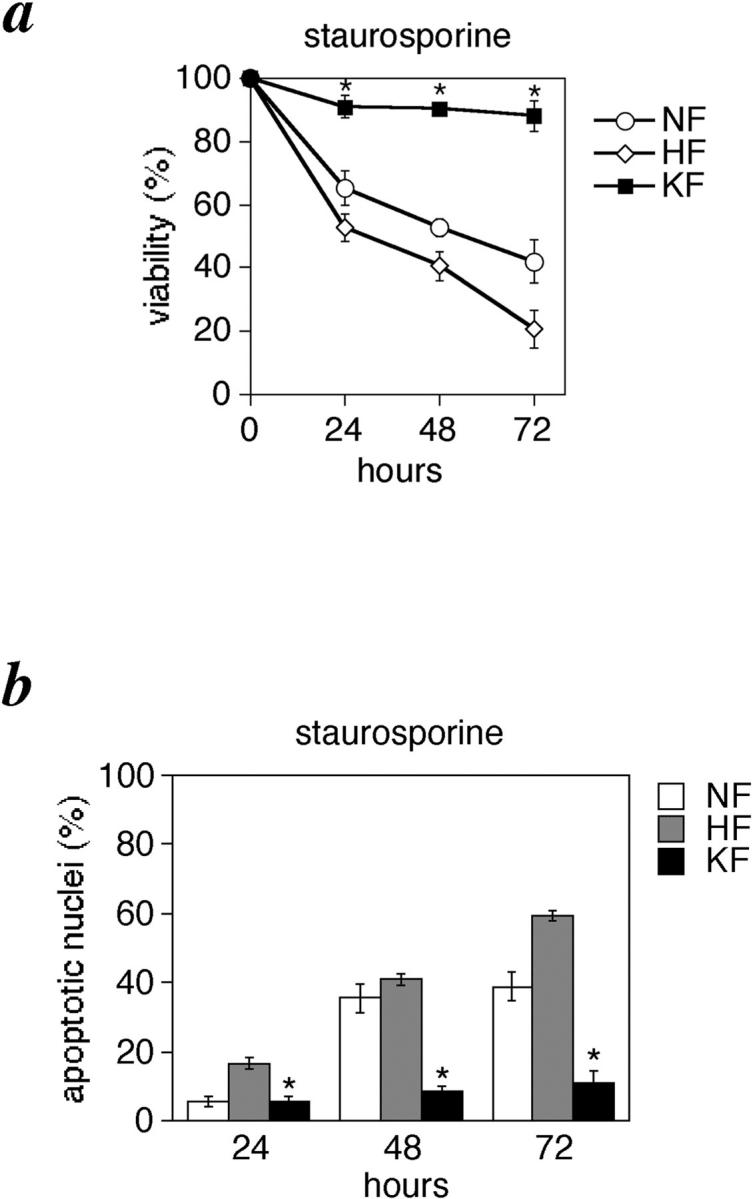
KFs are refractory to staurosporine-induced apoptosis. Cells were grown in DMEM and 10% fetal bovine serum and after 24 hours they were treated with staurosporine (10 nmol/L). Viability assay by trypan blue exclusion (a), and percentage of nuclear condensation assessed by Hoechst staining (b) at 24, 48, and 72 hours of treatment. *, P < 0.01 compared with NFs and HFs (ANOVA with Scheffé’s post hoc tests). Values shown are mean and SD of 15 independent experiments.
Caspases-3, -8, and -9 Are Not Activated in KFs
Several lines of evidence indicate that caspases are the key players in the induction and progression of apoptosis. Caspase activation correlates with the onset of apoptosis and caspase inhibition attenuates apoptosis. 29-32 The expression of Fas or of Fas ligand (FasL) can regulate Fas-mediated apoptosis, but it has been reported that differences in cell susceptibilities to Fas-mediated apoptosis can also be controlled by the regulation of signaling cascades, because not all Fas-positive cell types undergo apoptosis similarly after stimulation of Fas. 33,34 On analyzing the activation of caspases by immunoblotting, we observed that caspase-3 was activated in the NFs and HFs on stimulation with anti-Fas or staurosporine, whereas it was not in the KFs (Figure 5) ▶ . Gelsolin has been identified as a substrate for caspase-3 and expression of the cleavage product in multiple cell types caused the cells to round-up, detach from the plate, and undergo nuclear fragmentation. 35 To confirm the activation of caspase-3, we analyzed the cleavage of gelsolin by immunoblotting. The cleavage product of gelsolin was clearly observed in case of NFs and HFs, which increased in amount with the time of exposure to the apoptotic stimulus accordingly (Figure 5) ▶ . Caspase-7, although expressed in NFs, HFs, and KFs, was not activated, either with anti-Fas or with staurosporine (data not shown). In NFs and HFs, caspase-8 was activated on stimulation with anti-Fas whereas it was not when treated with staurosporine (Figure 5) ▶ . However, in KFs, caspase-8 was not activated on either stimulation with anti-Fas or staurosporine (Figure 5) ▶ . Caspase-9 was activated in HFs when treated with both anti-Fas and staurosporine but in NFs, it was activated only on stimulation with staurosporine (Figure 5) ▶ . In KFs, stimulation with neither anti-Fas nor staurosporine caused caspase-9 activation (Figure 5) ▶ . In summary, all caspases tested were not activated in KFs after apoptotic stimuli.
Figure 5.

Caspases are not activated in KFs. After induction of apoptosis with anti-Fas or staurosporine for 24, 48, and 72 hours as described in Materials and Methods, cells were lysed. Cell lysates (40 μg) were loaded onto 10 to 14% polyacrylamide gels and analyzed by Western blotting using monoclonal anti-caspase-3 (top), monoclonal anti-caspase-8 (second row), polyclonal anti-caspase-9 (third row), or monoclonal anti-gelsolin antibody (bottom).
TGF-β1 Inhibits Fas-Mediated Apoptosis
Keloid fibroblasts have been shown to produce TGF-β 36-38 and there is evidence suggesting that TGF-β1 can inhibit apoptosis in certain types of cells 39-47 through various mechanisms such as up-regulation of Bcl-xL, 41 down-regulation of c-myc, 42 and through the mitogen-activated protein kinase pathway. 43 To explore this, we treated cells with TGF-β1 at a dose of 5 ng/ml or 10 ng/ml 6 hours before the apoptotic stimuli. Both the doses significantly inhibited anti-Fas antibody-induced apoptosis in the NFs and HFs (Figure 6) ▶ . No significant increase in the resistance of the KFs was observed with the addition of exogenous TGF-β1 (Figure 6) ▶ . On the other hand, TGF-β1 failed to inhibit staurosporine-induced apoptosis in NFs and HFs and addition of exogenous TGF-β1 had no effect on the degree of resistance to staurosporine-induced apoptosis in KF (data not shown).
Figure 6.

TGF-β1 inhibits Fas-mediated apoptosis. Cells were treated with recombinant human TGF-β1 or recombinant human TGF-β2 at a dose of 5 and 10 ng/ml, recombinant human epidermal growth factor or recombinant human platelet-derived growth factor at a dose of 20 ng/ml 6 hours before stimulation with anti-Fas, treated with anti-Fas only or in the absence of both (control) and the percentage of nuclear condensation analyzed by Hoechst staining. *, p < 0.01 compared with the untreated controls and the TGF-β2-, epidermal growth factor-, platelet-derived growth factor-treated groups (ANOVA with Scheffé’s post hoc tests). Values shown are mean and SD of six independent experiments.
Enhanced expression of TGF-β2 12 has been reported in KFs and there is also evidence indicating that TGF-β2 activates proliferative scar fibroblasts. 11,48 To determine whether TGF-β2 has a similar anti-apoptotic function as that of TGF-β1, we treated cells with TGF-β2 but no significant inhibition of anti-Fas antibody-induced apoptosis (Figure 6) ▶ and staurosporine-induced apoptosis (data not shown) were observed. Inhibition of apoptosis was also not observed in the case of epidermal growth factor and platelet-derived growth factor that were used as controls (Figure 6) ▶ .
Neutralizing Antibody to TGF-β1 Abrogates the Resistance of KFs
To determine whether TGF-β1 is essential for making KFs resistant to apoptosis, we treated KFs with a neutralizing antibody directed against TGF-β1. This made the KFs significantly sensitive to both anti-Fas- and staurosporine-induced apoptosis (Figure 7a) ▶ . On stimulation with anti-Fas, the percentage of apoptotic cells at 72 hours was 9.66% in the untreated group, whereas it was 78.23% in the TGF-β1-neutralizing antibody-treated group (Figure 7a) ▶ .
Figure 7.
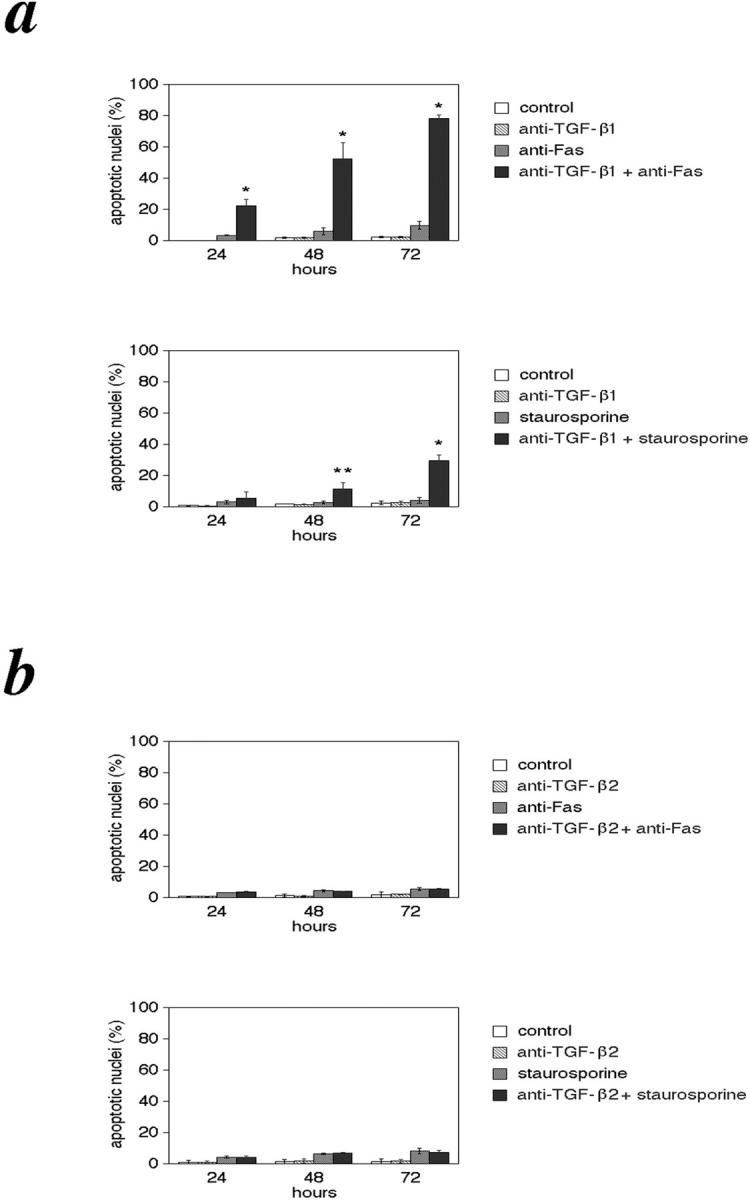
Neutralizing antibody to TGF-β1 abrogates the resistance of KFs. KFs were treated with monoclonal anti-human TGF-β1 antibody (a) or monoclonal anti-human TGF-β2 antibody (b) alone or together with anti-Fas or staurosporine as described in Materials and Methods and analyzed by Hoechst staining. Control indicates cells not treated with the above mentioned agents. *, P < 0.01; **, P < 0.05 compared with the untreated controls and the anti-TGF-β2 antibody-treated groups (ANOVA with Scheffé’s post hoc tests). Values shown are mean and SD of six independent experiments.
Similarly, we treated KFs with an anti-TGF-β2 neutralizing antibody. Consistent with the finding that the addition of TGF-β2 could not inhibit anti-Fas and staurosporine-induced apoptosis in HFs and NFs, neutralization of autocrine TGF-β2 did not abrogate the resistance of KFs (Figure 7b) ▶ .
Treatment with anti-TGF-β1 antibody or anti-TGF-β2 antibody alone without anti-Fas or staurosporine did not cause cell death. These results confirm an anti-apoptotic role of TGF-β1 in the resistance of KFs to apoptosis.
Discussion
In normal wound healing, the scar evolves from granulation tissue rich in cells and as the wound becomes epithelialized, there is a sharp increase in the number of apoptotic cells, suggesting that apoptosis is the mechanism responsible for the evolution of granulation tissue into a normal scar. 18 Fibroblasts and keratinocytes both come into close contact with Fas ligand-bearing lymphoid cells that infiltrate at the site of injury. Hence, apoptosis during wound remodeling is possibly mediated by Fas/FasL interaction. Here, we have demonstrated that KFs are refractory to apoptosis. This is consistent with the in vivo finding that keloid lesions were found to have lower rates of apoptosis than the normal controls 16 and p53 gene mutations were observed in keloid. 16,17 This indicates that keloid forms as a result of an abnormal wound-healing process with a prolonged proliferative phase because of an apoptosis resistant phenotype which in turn allows a state of continued production of excessive collagen.
It has been shown that low-level Fas expression signals proliferation whereas high-level Fas expression signals apoptosis in human fibroblasts. 23 Furthermore, Bcl-2 and Bcl-xL are reported to counteract apoptotic signaling whereas Bax can generate pro-apoptotic signals. 25-28 The balance between the anti- and pro-apoptotic members of the Bcl-2 family has been shown to be involved in the outcome of cell death or survival. However, there is also evidence suggesting that levels of Fas and Bcl-2 family members are not necessarily the cause of biological responsiveness 49 and consistent with this, there was no difference in the level of expression of Fas, Bcl-2, and Bax in NFs, HFs, and KFs.
Another possible explanation for resistance to apoptosis is caspase inactivation. 50 Fas receptor stimulation provokes the recruitment of the adapter protein FADD and activation of the initiator caspase-8 which in turn activates downstream executioner caspases such as caspase-3 and -7. 51 In nonreceptor cell death, such as those induced by staurosporine, ceramide, chemotherapeutic agents, or DNA damage, mitochondria plays a central role in apoptotic signaling: cytochrome c released from mitochondria binds to APAF-1 and in the presence of ATP or dATP, activates pro-caspase-9 which in turn activates the executioner caspases. 52,53 Consistent with this, in NFs and HFs, caspase-8 was activated on stimulation with anti-Fas but not with staurosporine and caspase-9 was activated when stimulated with staurosporine. Both anti-Fas antibody and staurosporine failed to activate caspases in KFs. This indicates that the inhibition of apoptosis seen in KFs is occurring upstream of caspase activation.
Our data provide evidence for a pivotal role for TGF-β1 in the resistance of KFs to apoptosis. TGF-β1 and -β2 proteins were found to be at higher levels in keloid fibroblast cultures compared with normal human dermal fibroblast cultures. 12 This autocrine TGF-β signaling may be contributing to the keloid phenotype. It has been extensively reported that TGF-β can stimulate the production of excessive collagen deposited in the dermis of keloids. 8,10,54 Here we show that neutralization of autocrine TGF-β1 can revert the apoptosis resistant state of KFs, providing evidence for the first time that TGF-β1 protects KFs from apoptosis, which can explain the prolonged proliferative phase of wound healing seen in keloid scars. Treatment of cells with TGF-β1 significantly inhibited anti-Fas antibody-induced apoptosis in the NFs and HFs but a similar effect could not be obtained in the case of staurosporine-induced apoptosis. One possibility is that the effect of TGF-β1 is mediated through a protein kinase C pathway and staurosporine is a potent protein kinase C inhibitor. 55 The fact that KFs are also resistant to staurosporine-induced apoptosis indicates that a higher sensitivity to TGF-β1 observed in KFs is the key factor although other protective factors besides TGF-β1 may exist.
The other pro-fibrotic TGF-β isoform involved in keloid is TGF-β2. There are evidences regarding the role of TGF-β2 in cell growth, proliferation and extracellular matrix production but this study shows that TGF-β2 does not contribute to the resistance of KFs to apoptosis. TGF-β isoforms represent structurally similar, yet functionally diverse growth factors which can regulate different aspects of cell behavior 56 and they differ in their binding affinity for TGF-β receptors. 57
Inadequate Fas-mediated apoptosis attributed to an exaggerated response to TGF-β1 and autocrine production of TGF-β, leading to increased proliferation of fibroblasts and continued excessive production of collagen may be the key mechanism of keloid formation. Until now, keloid and hypertrophic scars have been considered as different degrees of the same pathology. 58 Our data indicate that KFs display a distinctive phenotype from NFs and HFs and thus should be treated as a separate entity. Hypertrophic scar fibroblasts probably represent a heightened state of normal fibroblasts, as a result of factors such as infection, wound tension, and other stimulating external factors. This study opens the door to a new therapeutic strategy and indicates that agents that overcome blocked apoptotic signaling processes and potentiate endogenous apoptotic signaling mechanisms may be promising targets for the treatment of keloids.
Footnotes
Address reprint requests to Prof. Dr.Tsuneki Sugihara, Department of Plastic and Reconstructive Surgery, Hokkaido University School of Medicine, Kita-15, Nishi-7, Kita-ku, Sapporo 060-8638, Japan. E-mail: t-sugiha@med.hokudai.ac.jp.
Supported by research grants from the Ministry of Education, Science and Culture of Japan.
References
- 1.Alhady S: Keloids in various races. Plast Reconstr Surg 1969, 44:564-566 [DOI] [PubMed] [Google Scholar]
- 2.Murray JC, Pollack SV, Pinnelli SR: Keloids: a review. J Am Acad Dermatol 1981, 4:461-470 [DOI] [PubMed] [Google Scholar]
- 3.Ladin DA, Garner WL, Smith DJ: Excessive scarring as a consequence of healing. Wound Rep Reg 1995, 3:6-14 [DOI] [PubMed] [Google Scholar]
- 4.Nicoletis C, Bazin S, LeLous M: Clinical and biochemical features of normal, defective and pathologic scars. Clin Plast Surg 1977, 4:347-359 [PubMed] [Google Scholar]
- 5.Russell SB, Trupin KM, Rodriguez-Eaton S, Russell JD, Trupin JS: Reduced growth-factor requirement of keloid-derived fibroblasts may account for tumor growth. Proc Natl Acad Sci USA 1988, 85:587-591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch RJ, Goode RL, Simpson GT: Serum-free keloid fibroblast cell culture: an in vitro model for the study of aberrant wound healing. Plast Reconstr Surg 1997, 99:1094-1098 [DOI] [PubMed] [Google Scholar]
- 7.Babu M, Diegelmann R, Oliver N: Keloid fibroblasts exhibit an altered response to TGF-β. Invest Dermatol 1992, 99:650-655 [DOI] [PubMed] [Google Scholar]
- 8.Younai S, Nichter LS, Wellisz T, Reinisch J, Nimni ME, Tuan TL: Modulation of collagen synthesis by transforming growth factor-beta in keloid and hypertrophic scar fibroblasts. Ann Plast Surg 1994, 33:148-151 [DOI] [PubMed] [Google Scholar]
- 9.Kikuchi K, Kadono T, Takehara K: Effects of various growth factors and histamine on cultured keloid fibroblasts. Dermatology 1995, 190:4-8 [DOI] [PubMed] [Google Scholar]
- 10.Bettinger DA, Yager DR, Diegelmann RF, Cohen IK: The effect of TGF-β on keloid fibroblast proliferation and collagen synthesis. Plast Reconstr Surg 1996, 98:827-833 [DOI] [PubMed] [Google Scholar]
- 11.Polo M, Smith PD, Kim YJ, Wang X, Ko F, Robson MC: Effect of TGF-β on proliferative scar fibroblast cell kinetics. Ann Plast Surg 1999, 43:185-190 [PubMed] [Google Scholar]
- 12.Lee TY, Chin GS, Kim WJ, Chau D, Gittes GK, Longaker MT: Expression of transforming growth factor beta 1, 2, 3 proteins in keloids. Ann Plast Surg 1999, 43:179-184 [PubMed] [Google Scholar]
- 13.Nakaoka H, Miyauchi S, Miki Y: Proliferating activity of dermal fibroblasts in keloids and hypertrophic scars. Acta Derm Venereol 1995, 75:102-104 [DOI] [PubMed] [Google Scholar]
- 14.Calderon M, Lawrence WT, Banes AJ: Increased proliferation in keloid fibroblasts wounded in vitro. J Surg Res 1996, 61:343-347 [DOI] [PubMed] [Google Scholar]
- 15.Ghazizadeh M, Miyata N, Sasaki Y, Arai K, Aihara K: Silver-stained nucleolar organizer regions in hypertrophic and keloid scars. Am J Dermatopathol 1997, 19:468-472 [DOI] [PubMed] [Google Scholar]
- 16.Ladin DA, Hou Z, Patel D, McPhail M, Olson JC, Saed GM, Fivenson DP: p53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Rep Reg 1998, 6:28-37 [DOI] [PubMed] [Google Scholar]
- 17.Saed GM, Ladin D, Olson J, Han X, Hou Z, Fivenson D: Analysis of p53 gene mutations in keloids using polymerase chain reaction-based single-strand conformational polymorphism and DNA sequencing. Arch Dermatol 1998, 134:963-967 [DOI] [PubMed] [Google Scholar]
- 18.Desmouliere A, Redard M, Darby I, Gabbiani G: Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995, 146:56-66 [PMC free article] [PubMed] [Google Scholar]
- 19.Nagata S, Golstein P: The Fas death factor. Science 1995, 267:1449-1456 [DOI] [PubMed] [Google Scholar]
- 20.Schulzeosthoff K, Ferrari D, Los M, Wesselbourg S, Peter ME: Apoptosis signaling by death receptors. Eur J Biochem 1998, 254:439-459 [DOI] [PubMed] [Google Scholar]
- 21.Oishi M, Maeda K, Sugiyama S: Distribution of apoptosis-mediating Fas antigen in human skin and effects of anti-Fas monoclonal antibody on human epidermal keratinocyte and squamous cell carcinoma cell lines. Arch Dermatol Res 1994, 286:396-407 [DOI] [PubMed] [Google Scholar]
- 22.Aggarwal BB, Singh S, LaPushin R, Totpal K: Fas antigen signals proliferation of normal human diploid fibroblast and its mechanism is different from tumor necrosis factor receptor. FEBS Lett 1995, 364:5-8 [DOI] [PubMed] [Google Scholar]
- 23.Freiberg RA, Spencer DM, Chaote KA, Duh HJ, Schreiber SL, Crabtree GR, Khavari PA: Fas signal transduction triggers either proliferation or apoptosis in human fibroblasts. J Invest Dermatol 1997, 108:215-219 [DOI] [PubMed] [Google Scholar]
- 24.Jelaska A, Korn JH: Anti-Fas induces apoptosis and proliferation in human dermal fibroblasts: differences between foreskin and adult fibroblasts. J Cell Physiol 1998, 175:19-29 [DOI] [PubMed] [Google Scholar]
- 25.Hockenbery DM, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ: Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348:334-336 [DOI] [PubMed] [Google Scholar]
- 26.Nunez G, London L, Hockenbery D, Alexander M, McKearn JP, Korsmeyer SJ: Deregulated Bcl-2 gene expression selectively prolongs survival of growth factor deprived hematopoietic cell lines. J Immunol 1990, 144:3602-3610 [PubMed] [Google Scholar]
- 27.Itoh N, Tsujimoto Y, Nagata S: Effect of bcl-2 on Fas antigen-mediated cell death. J Immunol 1993, 151:621-627 [PubMed] [Google Scholar]
- 28.Shinoura N, Yoshida Y, Nishimura M, Muramatsu Y, Asai A, Kirino T, Hamada H: Expression level of Bcl-2 determines anti- or proapoptotic function. Cancer Res 1999, 59:4119-4128 [PubMed] [Google Scholar]
- 29.Cohen GM: Caspases: the executioners of apoptosis. Biochem J 1997, 326:1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicholson DW, Thornberry NA: Caspases: killer proteases. Trends Biochem Sci 1997, 22:299-306 [DOI] [PubMed] [Google Scholar]
- 31.Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P: Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science 1995, 269:1885-1888 [DOI] [PubMed] [Google Scholar]
- 32.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidle DR, Poirier GG, Salvesen GS, Dixit VM: Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase. Cell 1995, 81:801-809 [DOI] [PubMed] [Google Scholar]
- 33.Su X, Zhou T, Wang Z, Yang P, Jope RS, Mountz JD: Defective expression of hematopoietic protein tyrosine phosphatase (HCP) in lymphoid cells blocks Fas-mediated apoptosis. Immunity 1995, 2:353-362 [DOI] [PubMed] [Google Scholar]
- 34.Miyawaki T, Uehara T, Nibu R, Tsuji T, Yachie A, Yonehara S, Taniguchi N: Differential expression of apoptosis-related Fas antigen on lymphocyte subpopulations in human peripheral blood. J Immunol 1992, 149:3753-3758 [PubMed] [Google Scholar]
- 35.Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LT: Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 1997, 278:294-298 [DOI] [PubMed] [Google Scholar]
- 36.Chau D, Mancoll JS, Lee S, Zhao J, Phillips LG, Gittes GK, Longaker MT: Tamoxifen downregulates TGF-β production in keloid fibroblasts. Ann Plast Surg 1998, 40:490-493 [DOI] [PubMed] [Google Scholar]
- 37.Younai S, Venters G, Vu S, Nichter L, Nimni ME, Tuan TL: Role of growth factors in scar contraction: an in vitro analysis. Ann Plast Surg 1996, 36:495-501 [DOI] [PubMed] [Google Scholar]
- 38.Suzawa H, Kikuchi S, Arai N, Koda A: The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Jpn J Pharmacol 1992, 60:91-96 [DOI] [PubMed] [Google Scholar]
- 39.Kawakami A, Eguchi K, Matsuoka N, Tsuboi M, Kawabe Y, Aoyagi T, Nagataki S: Inhibition of Fas antigen-mediated apoptosis of rheumatoid synovial cells in vitro by transforming growth factor beta 1. Arthritis Rheum 1996, 39:1267-1276 [DOI] [PubMed] [Google Scholar]
- 40.Kim ES, Kim RS, Ren RF, Hawver DB, Flanders KC: Transforming growth factor-β inhibits apoptosis induced by β-amyloid peptide fragment 25–35 in cultured neuronal cells. Mol Brain Res 1998, 62:122-130 [DOI] [PubMed] [Google Scholar]
- 41.Lee J, Park BJ, Park JH, Yang MH, Chi SG: TGF-beta 1 inhibition of apoptosis through the transcriptional up-regulation of BcL-X(L) in human monocytic leukemia U937 cells. Exp Mol Med 1999, 31:126-133 [DOI] [PubMed] [Google Scholar]
- 42.Genestier L, Kasibhatla S, Brunner T, Green DR: Transforming growth factor-β inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J Exp Med 1999, 189:231-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chin BY, Petrache I, Choi AM, Choi ME: Transforming growth factor beta 1 rescues serum deprivation-induced apoptosis via the mitogen-activated protein kinase (MAPK) pathway in macrophages. J Biol Chem 1999, 274:11362-11368 [DOI] [PubMed] [Google Scholar]
- 44.Dybedal I, Guan F, Borge OJ, Veiby OP, Ramsfjell V, Nagata S, Jacobsen SE: Transforming growth factor-β abrogates Fas-induced growth suppression and apoptosis of murine bone marrow progenitor cells. Blood 1997, 90:3395-3403 [PubMed] [Google Scholar]
- 45.Riedl E, Strobl H, Majdic O, Knapp W: TGF-β1 promotes in vitro generation of dendritic cells by protecting progenitor cells from apoptosis. J Immunol 1997, 158:1591-1597 [PubMed] [Google Scholar]
- 46.Cerwenka A, Kovar H, Majdic O, Holter W: Fas and activation-induced apoptosis are reduced in human T cells preactivated in the presence of TGF-beta 1. J Immunol 1996, 156:459-464 [PubMed] [Google Scholar]
- 47.Weller M, Fontana A: The failure of current immunotherapy for malignant glioma. Tumor-derived TGF-beta, T-cell apoptosis, and the immune privilege of the brain. Brain Res Brain Res Rev 1995, 21:128-151 [DOI] [PubMed] [Google Scholar]
- 48.Smith P: TGF-β2 activates proliferative scar fibroblasts. J Surg Res 1999, 82:319-323 [DOI] [PubMed] [Google Scholar]
- 49.Owen-Schaub LB, Radinsky R, Kruzel E, Berry K, Yonehara S: Anti-Fas on nonhematopoietic tumors: levels of Fas/APO-1 and bcl-2 are not predictive of biological responsiveness. Cancer Res 1994, 54:1580-1586 [PubMed] [Google Scholar]
- 50.Martin SJ, Green DR: Protease activation during apoptosis: death by a thousand cuts? Cell 1995, 82:349-352 [DOI] [PubMed] [Google Scholar]
- 51.Wolf BB, Green DR: Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem 1999, 274:20049-20052 [DOI] [PubMed] [Google Scholar]
- 52.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 53.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES: Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for pro-caspase-9 activation. J Biol Chem 1999, 274:17941-17945 [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Smith P, Pu LL, Kim YJ, Ko F, Robson MC: Exogenous transforming growth factor beta (2) modulates collagen I and collagen III synthesis in proliferative scar xenografts in nude rats. J Surg Res 1999, 87:194-200 [DOI] [PubMed] [Google Scholar]
- 55.Tamaoki T: Use and specificity of staurosporine, UCN-01 and calphostin C as protein kinase inhibitors. Methods Enzymol 1991, 201:340-347 [DOI] [PubMed] [Google Scholar]
- 56.Sanford LP, Ormsby I, Adriana C, Groot G, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T: TGF-β2 knockout mice have multiple developmental defects that are non-overlapping with other TGF-β knockout phenotypes. Development 1997, 124:2659-2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franklin HE: Role of transforming growth factor β in human disease. N Engl J Med 2000, 342:1350-1358 [DOI] [PubMed] [Google Scholar]
- 58.Tredget EE, Nedelec B, Scott PG, Ghahary A: Hypertrophic scars, keloids and contractures. The cellular and molecular basis for therapy. Wound Healing 1997, 77:701-730 [DOI] [PubMed] [Google Scholar]