

Abstract
Immortal epithelial cell lines were previously established after transduction of the HPV16-E6E7 genes into primary cultures of normal pancreatic duct epithelial cells. Single clones were isolated that demonstrated near normal genotype and phenotype. The proliferation of HPDE6-E6E7c7 and c11 cells is anchorage-dependent, and they were nontumorigenic in SCID mice. The cell lines demonstrated many phenotypes of normal pancreatic duct epithelium, including mRNA expression of carbonic anhydrase II, MUC-1, and cytokeratins 7, 8, 18, and 19. These cells have normal Ki-ras, p53, c-myc, and p16INK4A genotypes. Cytogenetic studies demonstrated losses of 3p, 10p12, and 13q14, the latter included the Rb1 gene. The wild-type p53 protein was detectable at very low levels consistent with the presence of E6 gene product, and the lack of functional p53 pathway was confirmed by the inability for γ-irradiation to up-regulate p53 and p21waf1/cip1 protein. The p110/Rb protein level was also not detectable consistent with the expression of E7 protein and haploid loss of Rb1 gene. Despite this, the proliferation of both c7 and c11 cells were markedly inhibited by transforming growth factor-β1. This was associated with up-regulation of p21cip1/waf1 but not p27kip1. Further studies showed that p130/Rb2 and cyclin D3 were expressed, suggesting that p130/Rb2 may have partially assumed the maintenance of G1 cell cycle checkpoint regulation. These results indicate that except for the loss of p53 functional pathway, the two clones of HPDE6-E6E7 cells demonstrated a near normal genotype and phenotype of pancreatic duct epithelial cells. These cell lines will be useful for future studies on the molecular basis of pancreatic duct cell carcinogenesis and islet cell differentiation.
Pancreatic cancer is the fourth commonest cause of cancer death in North America and has one of the worst prognoses. 1 Greater than 90% of these tumors arise from the pancreatic duct epithelium. These tumors are highly metastatic and only 20% of the patients are treated by surgical resection. 2 Even when the primary tumor is small and localized, the prognosis remains poor and chemotherapy or radiotherapy has demonstrated limited effectiveness. 3,4 Therefore, it seems that a significant improvement in pancreatic cancer mortality depends on the development of better treatment and preventive strategies, which require knowledge on the molecular biology and pathogenesis of this disease. Recent definitions of genetic changes that occur commonly in pancreatic cancer represent important first steps toward such a goal. 5 Nevertheless, the availability of dynamic models remains crucial to the study and understanding of the biological significance of these genetic changes, especially in the context of pancreatic duct epithelial cell carcinogenesis.
Our laboratory has previously reported the establishment of primary and immortal epithelial cell lines from normal human pancreatic ducts. 6 We also reported that in comparison with the pancreatic cancer cell lines, the human pancreatic duct epithelial (HPDE) cells demonstrated a gene expression pattern that more consistently resembled the phenotype of normal cells rather than cancerous duct cells in vivo. 7 These similarities included relatively low expression levels of various tyrosine kinase receptors, a wild-type Ki-ras genotype, and the retention and expression of p16INK4A gene. We have subsequently isolated several clones of these cell lines. We report here the phenotypic and genotypic characteristics of two of these cell lines that demonstrate anchorage-dependent growth requirement and that are nontumorigenic in immune-deficient mice.
Materials and Methods
Culture Media
The establishment of the HPDE6E76E7 (HPDE6) cell line was previously reported, 6 and it was routinely cultured in keratinocyte serum-free (KSF) medium supplemented by epidermal growth factor and bovine pituitary extract (Life Technologies, Inc., Grand Island, NY). Clones of HPDE6 cells were isolated using cloning rings. Primary cultures of normal human bronchial epithelial cells were also established in the supplemented KSF medium, as previously reported. 8
Cell Growth Assays
To determine the effect of transforming growth factor-β1 (TGF-β1; R & D Systems, Minneapolis, MN), ∼10 4 trypsin-dissociated cells were plated in replicate wells of Nunc’s 6-well tissue culture plates (Life Technologies, Inc.). Two days after plating, the culture medium was replaced with fresh supplemented KSF medium. Replicate plates were also replaced with the same medium containing 10 ng/ml of TGF-β1. Cells in triplicate wells were counted during the subsequent 6 to 8 days, using the Coulter’s ZM Cell/Particle Counter (Hialeah, FL). The effect of 10 ng/ml of TGF-β1 was also assessed at the colony-forming level by seeding 60-mm tissue culture plates with 500 cells. The plates were stained with 4% Giemsa solution (Sigma Chemical Co., St. Louis, MO), and colonies with >10 cells were counted using a dissecting microscope.
The effect of TGF-β1 concentration on the proliferation of HPDE cells was assessed by the [3H]-thymidine uptake assay. Briefly, 4,000 cells were plated into each well of Nunc’s 12-well tissue culture plates. Two days later, the medium of groups of four replicate wells was changed to fresh medium containing various concentrations of TGF-β1. The cells were then cultured for additional 4 days. One μCi/ml of [3H]-thymidine (NEN Life Science Products, Boston, MA) was then added to the medium of each well, and the cells were cultured for an additional 20 to 24 hours. To estimate the incorporation of [3H]-thymidine into the DNA, the cells were washed three times in cold phosphate-buffered saline, then were fixed in three changes of cold 5% aqueous solution of trichloroacetic acid (Sigma Chemical Co.) The cells were then serially dehydrated in ice-cold 70%, 95%, and 100% ethanol, and air-dried for at least 1 day. The incorporated radioactivity in each well was solubilized by an overnight incubation in 1 ml of 0.3 N NaOH at 37°C. After neutralization with 0.1 ml 3 N HCl, the solute was transferred into scintillation vials. After the addition of 5 ml Ecolite scintillation liquid (ICN, Costa Mesa, CA), they were counted using the Beckmann’s scintillation counter model LS6000SC.
The ability of cells to grow anchorage-independently was assayed by suspending them in KSF medium containing 0.3% Bacto-agar (Difco, Detroit, MI), as described previously. 9 Cells were seeded at 10 4 or 5 × 10 4 cells per 60-mm plates, and allowed to form colonies in 4 weeks.
G-Banding, Spectral Karyotyping (SKY), and Fluorescent in Situ Hybridization (FISH)
Cytogenetic preparations were made according to standard protocols using colcemid and KCl hypotonic treatment. 10 The slides were G-banded with trypsin (Difco) and Leishman’s stain (Sigma) and scanned for metaphase spreads. 10 For each cell line, 20 G-banded metaphases were karyotyped and their descriptions were provided according to ISCN 95 nomenclature. 11
The SKY kit probe cocktail from Applied Spectral Imaging (ASI, Carlsbad, CA) was hybridized to the cytogenetic preparation from the C7 clone according to standard protocols 12,13 and the manufacturer’s instructions (ASI). Briefly, the slide was treated with pepsin, fixed with 1% formaldehyde, dehydrated using an ascending ethyl alcohol series, and denatured for 2 minutes in 70% formamide/2× standard saline citrate at 70°C. The SKY probe was denatured for 7 minutes at 75°C, re-annealed at 37°C for 1 hour, and hybridized to denatured slide for 36 hours at 37°C. The posthybridization washes and detection steps were performed per manufacturer’s instructions (ASI). The slide was counterstained by 4,6-diamidino-2-phenylindole (Sigma).
The metaphase images were captured using a SD 200 spectral bio-imaging system (ASI Ltd., MigdalHaemek, Israel) attached to a Zeiss microscope (Axioplan 2) and stored on a SKY image-capture workstation. The images were analyzed using the SKYView software version 1.5 (ASI), which resolves individual fluorochrome spectra by Fourier spectroscopy and distinguishes the spectral signatures for each chromosome to provide a unique pseudocolor for each chromosome (classified image). The determination of the position of rearrangement breakpoints was performed according the G-banding as well as spectral patterns.
Single color metaphase and interphase FISH analysis using different centromeric probes was performed on routine cytogenetic preparations from three different passages (P4, P14, P35) of clone C7 to determine the proportion of aneusomies as an overall measure of segregation anomalies associated with chromosomal stability. Probes for centromeres of chromosome 1 and 17 from Roche-Boehringer Mannheim (Dorval, Québec, Canada) were used for interphase and metaphase FISH. For each probe and each passage, 200 metaphase/interphase nuclei were scanned and the number of signals per cell counted independently by two individuals. To determine copy number of the Rb1 gene, the retinoblastoma DNA probe from Ventana was hybridized to a cytogenetic preparation derived from P35 of clone C7.
Comparative Genomic Hybridization (CGH) and Imaging
Cell line DNA was extracted using the standard phenol-chloroform extraction method. Phytohemagglutinin-stimulated normal lymphocytes were prepared as targets for CGH experiments using standard protocol. 14 Slides were aged for 2 weeks before denaturation and proteinase K treatment. CGH was preformed according to the standard protocols. 14,15 Briefly, reference and test DNA were labeled with digoxigenin and biotin, respectively. Biotinylated DNA was detected using avidin-fluorescein isothiocyanate (Oncor Inc., Gaithersburg, MD) whereas digoxigenin-labeled DNA was detected with anti-dig-rhodamine (Boehringer Mannheim, Mannheim, Germany). At least five images were captured per case using a Nikon Microphot microscope connected to a Photometrics (Tucson, AZ) SenSys KAF 1400 charge-coupled device (CCD) camera for the analysis. The QuipXL Genetics Workstation (Vysis Inc., Downers Grove, IL) was used for the image analysis. The image analyzing software calculates an average ratio of fluorescein isothiocyanate:rhodamine and expressed it as a green:red ratio for each metaphase with a 95% confidence limit. Gains or losses of chromosomal regions were detected when the fluorescent intensity ratio deviated from 1. The lower and upper limit of gain and loss was established by performing the control CGH experiments with DNA derived from normal male and female tissues. Based on these findings, the cut-off values were set at 1.25 and 0.80, with a 95% confidence limit. Analysis was excluded from the following regions: centromere, acrocentric p-arms, teleomere, and heterochromatic-rich areas. 16-18
RNA Expression Assays
Total cellular RNA was isolated using the acid guanidinium thiocyanate-phenol-chloroform technique, as previously reported. The RNA was digested with RNase-free DNase I and reverse-transcribed using the murine Moloney leukemia virus reverse transcriptase, and aliquots of the product was used as template for polymerase chain reaction. The quality of reverse transcriptase reaction was checked first by amplification using the β-actin primers. 19 The primer set for carbonic anhydrase-II consists of forward primer (nucleotides 56 to 77): 5′-GCTCTAGACCATGTCCCATCAC-3′ and reverse primer (nucleotides 541 to 520): 5′-GGAATTCTGAAGGCCCGGTTTAG-3′. The primers for MUC-1 were forward primer (nucleotides 2958 to 2979) 5′-GAAGATCTGCATCAGGCTCAGC-3′ and reverse primer (nucleotides 3645 to 3624) 5′-GGAATTCTTTCGGCGGCACTGAG-3′.
Global gene expression was also studied using the Atlas Human Cancer cDNA Expression Array filter (Clonetech, Palo Alto, CA) as per instruction provided in user’s manual. 32P-labeled cDNA was generated by reverse-transcription using 5 μg of total cellular RNA as template. The labeled probe was purified with a ChromaSpin-200 (Clontech) column, and hybridized to the Atlas membrane at 68°C overnight. After repeated washing as recommended, the filter was exposed to X-ray film overnight.
Protein Immunoblot and Immunoprecipitation Assays
Antibodies to HPV16-E7 (ED17), p21waf1 (C-19), and Smad4 (B-8) were purchased from Santa Cruz Biotechnology (Santa Cruz Biotechology, Santa Cruz, CA). Antibodies to Rb, Rb2/p130, and p27Kip1/p2, cyclin D3, cyclin B, and cdk-4 were obtained from Transduction Laboratory (Mississauga, Ontario, Canada). Western blot was performed as previously described. 7
Antibodies to p53 protein were purified from supernatants of hybridoma clones. These included pAb 240 that was specific for mutant p53 protein, and pAb 421 and pAb 1620 that were reactive only to wild-type p53 protein. To perform immunoprecipitation, cellular proteins were extracted with cell lysis buffer, and the cleared supernatant was incubated with the appropriate p53 antibody at 4°C for 2 hours, then 30 μl of protein A Sepharose was added. After a further 1 hour of incubation at 4°C, the protein-A beads were centrifuged and washed, and then boiled in 30 μl of loading buffer containing 100 mmol/L dithiothreitol. After electrophoretic separation in polyacrylamide gel, the proteins were transferred onto polyvinylidene difluoride filter membrane (Boehringer Mannheim), then immunoblotted with p53 antibody, and revealed by the Chemiluminescence Western Biotin kit of Boehringer Mannheim.
γ-Irradiation
Five hundred thousand cells were grown to confluence in replicate 60-mm tissue culture dishes, then cultured for a further 2 days but with a daily change of fresh medium. The culture plates were then exposed to 5-Gy γ-irradiation from a 137Cs source Gammacell-40 Exactor irradiator (Nordion International Inc., Kanata, Ontario, Canada). Sham control plates were brought to room temperature for the same length of time. The plates were then returned to the tissue culture incubator, and paired irradiated and control plates were removed at varying times for protein extraction.
Biochemical Analysis of TGF-β1-Treated Cells
Replicate plates of cultured cells growing to ∼80% confluence were used in this series of experiments. Treatment was initiated by replacing the medium with fresh medium containing 20ng/ml TGF-β1 (R & D Systems). The control plates were concurrently replaced with fresh medium only. Total cellular protein from both TGF-β1-treated and control plates was serially collected at 3, 6, 9, and 24 hours later.
Results
Establishment of Clonal HPDE Cell Lines
As previously reported, the HPDE6-E6E7 cells underwent growth crisis at approximately passage 12, but immortal clones emerged and have subsequently been propagated for >40 passages. 6 At passage 18, the cells were plated at a colony forming density of 100 cells per 100-mm plate. The colonies that formed were isolated using steel cloning rings, and four clones were chosen randomly for further studies. Two clones (c7 and c11) that showed paradiploid DNA index by flow cytometry were also characterized for their karyotypic changes and growth properties. These two clones have been cultured for up to 35 passages and have shown no appreciable changes in both the morphology and growth characteristics. These cell lines also demonstrate telomerase activity as detected by the telomere repeat amplification (TRAP) assay (data not shown). These clones are herewith referred to as HPDE6c7 and HPDE6c11 cell lines.
Gene Expression of HPDE6-E6E7 Clones
Reverse transcriptase-polymerase chain reaction demonstrated that the pre-immortalized HPDE6-E6E7 cells at passage 6 (P6), its immortal line (P17), and clones isolated from the latter expressed mRNAs of carbonic anhydrase-II and MUC-1 (Figure 1A) ▶ . Northern blot analyses confirmed the mRNA expression of p16INK4A and c-myc (Figure 1C) ▶ . The levels of p16INK4A mRNA expression in cells transduced with the E6E7 genes were much higher than that in the primary cultured HPDE6 cells, but the transcript was normal in size. Southern blot analyses demonstrated that all clones retained a normal copy number of p16, c-myc, and p53 genes (Figure 1D) ▶ . Using the Atlas cDNA array analyses, we also demonstrated high-level expression of cytokeratin-7, -14, and -18 (Figure 1B) ▶ . Lower level expression of cytokeratin-2, -8, and -19 was also noted.
Figure 1.

Gene expression in HPDE6-E6E7 cells and their clones. A: Reverse transcriptase-polymerase chain reaction analyses showed the mRNA expression of carbonic anhydrase-II and MUC-1 genes in the pre-immortalized HPDE6-E6E7 cells and its immortal clones (c2–c11). B: cDNA microarray hybridization was used to demonstrate the expression of various cytokeratin mRNA in HPDE6-E6E7c7 cells. C: The immortal HPDE6-E6E7 (P17) cells and the clones expressed high levels of p16 mRNA expression, whereas the primary cultured HPDE6 cells that were not exposed to the E6E7 retrovirus showed low levels of p16 mRNA. D: Southern blot analysis confirmed the presence of same copy number of p16, p53, and c-myc genes in the various HPDE6-E6E7 clones.
Chromosomal Constitution of c7 and c11 Clones
The chromosomal compositions of HPDE6c7 and HPDE6c11 cells were studied in more detail using both G-banding and SKY. Both clones demonstrated an almost identical hypodiploid karyotype (45/44, XX) with numerical and some structural changes (Figure 2, A and C) ▶ . The common aberrations were der (3;14) (q10;q10), del(10p)(p11), der (17)t(13;17)(q21;p13), and +20. The c7 clone additionally demonstrated a der (8)t(8;9)(q24;?), whereas c11 clone showed a −22. The CGH studies were compatible with the karyotypic findings and demonstrated chromosomal losses on 3p, 10p12, and 13q14 in both cell lines, but a loss of chromosome 22 in c11 only (Figure 2, B and D) ▶ . A gain on 20 was also evident in both clones.
Figure 2.

Cytogenetic characterization of the HPDE6-E6E7 c7 and c11 cells. Analyses were performed using cells between passage 4 to 6. A: SKY of c7 cells showing the various chromosomal translocations. B and D: The results of CGH analyses on the c7 and c11 cells. C: Inverse-DAPI karyotyping of c11 clones showing results that are consistent with both SKY and CGH findings.
Chromosomal Stability
FISH analysis using centromeric DNA probes of two chromosomes (1 and 17) on P4, P14, and P35 cells of HPDE6c7 line showed relatively stable karyotype with passaging (Figure 3, A–C) ▶ . P35 cells of HPDE6c7 line showed relatively stable karyotype with passaging (Figure 3, A–C) ▶ . Some cells with one to four chromosome 1 signals were noted but the frequency did not change with increasing passage number. Such variation was not seen with chromosome 17. FISH analysis also confirmed the loss of one Rb1 allele as a consequence of an unbalanced translocation of 13q to chromosome 17 (Figure 3D) ▶ .
Figure 3.

FISH analysis to demonstrate chromosomal stability of HPDE6c7 cells. A and B: The distribution of nuclei with various numbers of chromosome 1 and 17 in cells at three different passages (4, 14, and 35). C: A representative image of interphase and metaphase FISH using the probe for chromosome 1 centromere in HPDE6C7 cells at passage 14. D: FISH demonstration of haploid loss of Rb gene with interphase nuclei showing doublet staining for cells in G2 phase.
Growth Properties of c7 and c11 Cells
In KSF medium, the HPDE6c7 and c11 clones proliferated with population doubling times of 24 to 32 hours. Similar to the parental HPDE6-E6E7 cells, the growth of these immortal HPDE clones were inhibited by TGF-β1 (Figure 4, A and B) ▶ . Inhibition was evident starting at 0.1 ng/ml, and became maximal at ∼1 ng/ml concentration (Figure 4C) ▶ . At 4 × 10 4 cells/60-mm plate seeding density, both clones failed to form colonies in soft agar. The implantation of two million cells into the neck fat pad of SCID mice did not result in tumor formation for up to 6 months.
Figure 4.
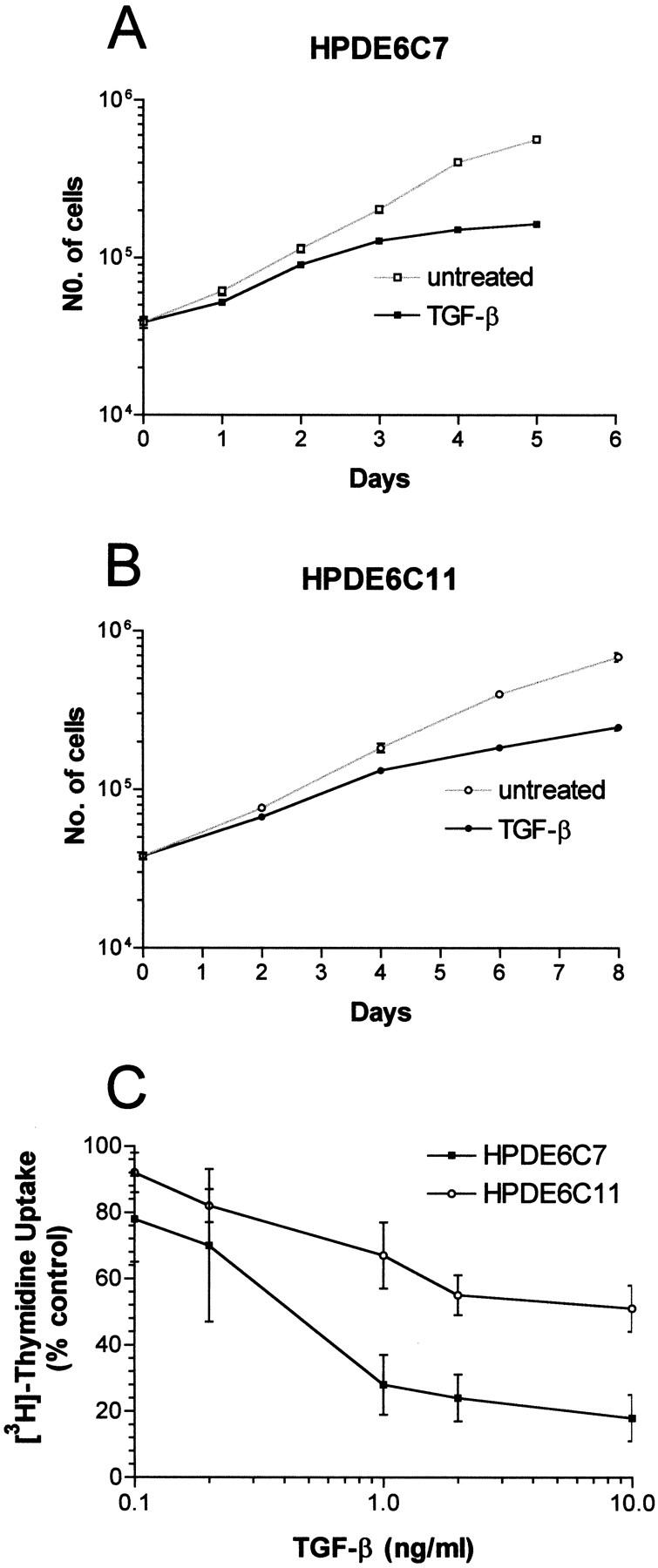
The inhibition of proliferation of HPDE6c7 and c11 cells by TGF-β1. Shown are the effects of 10 ng/ml of TGF-β1 on the growth of these cells (A and B), and the inhibitory response of DNA synthesis to various concentrations of TGF-β1 (C).
Deregulation of p53 Pathway in HPDE6c7 Cells
HPDE6c7 cells demonstrated a very low level of p53 protein that was detectable by immunoprecipitation (Figure 5A) ▶ , and both p53 and 21wafl/cipl proteins (Figure 5B) ▶ and mRNA (data not shown) were not inducible by γ-ray irradiation. As a control, primary cultured normal human bronchial epithelial cells showed normal up-regulation of p53 and p21 after radiation (Figure 5B) ▶ . The low-level p53 protein that was detectable by immunoprecipitation was reactive with antibodies pAb1620 and pAb 421 that detect wild-type p53, but was not recognized by antibody pAb 240 that reacts exclusively with p53 protein with mutations in amino acids 213 to 217 (Figure 5A) ▶ . These results indicated that HPDE6c7 cells and most likely its sister clones expressed wild-type protein but at a markedly low level resulting from the expression of E6 gene that was transduced during the establishment of the HPDE6-E6E7 cell line.
Figure 5.

Inactivation of the p53 pathway in HPDE6-E6E7c7 cells. A: These cells showed low-level expression of wild-type p53 protein as recognized by both pAb 421 and pAb 1802 but not by pAb 240 antibodies. B: Irradiation with γ-ray failed to induce p53 and p21 proteins in HPDE6c7 cells. As control, normal bronchial epithelial cells showed the normal response to γ-irradiation.
Rb and TGF-β Pathways
Western blot analysis demonstrated a marked reduction of Rb protein level in HPDE6c7 cells (Figure 6A) ▶ , but high levels of Rb2/p130 protein. The low level of Rb protein is consistent with the presence of HPV16E7 protein (Figure 6B) ▶ . Interestingly, the p16INK4A mRNA levels in all HPDE6-E6E7 clones were markedly higher than that in the parental primary cultured HPDE6 cells (Figure 1C) ▶ .
Figure 6.

The Rb pathway in HPDE cells. A: The HPDE6-E6E7c7 cells demonstrated no detectable pRb/p110 level, but p130 was present at high levels. These cells also demonstrated Smad-2/3 and Smad-4 protein expression. As comparison and positive control for Rb/p110, the human bronchial epithelial cells were similarly analyzed. B: The HPV16-E7 protein was expressed in HPDE6-E6E7c7 cells and Kaski cervical squamous cell carcinoma cell line, but not in NIH3T3 cells. C: Treatment of the HPDE6c7 cells by 20 ng/ml of TGF-β1 induced up-regulation of p21 protein as early as 3 hours after treatment, but induction of p27 was delayed up to later than 6 hours. As expected, TGF-β1 failed to modulate the levels of cyclin D3, cyclin B, and cdk4.
The ability of TGF-β1 to inhibit the proliferation of the c7 and c11 clones of HPDE6-E6E7 cells indicated the preservation and expression of normal molecules involved in TGF-β signal transduction pathway. This was studied in greater detail with the c7 clone. Western blot analysis confirmed the expression of Smad-2/3 and Smad-4 proteins in these cells (Figure 6A) ▶ . Growth inhibition by TGF-β1 (10 ng/ml) was associated with an up-regulation of the p21wafl/cipl protein 3 hours after exposure to TGF-β1, and of the p27kipl 24 hours after treatment (Figure 6C) ▶ . No change in the levels of cyclin D3, cyclin B, and Cdk4 was noted.
Discussion
We have demonstrated that it is possible to obtain immortal HPDE cell lines with near-normal genotype and phenotype. These clonal lines were established from a primary culture of normal human pancreatic duct epithelium, and the E6 and E7 genes of human papilloma virus (HPV)-16 was used to immortalize these duct cells. Introduction of immortalizing genes such as the E6E7 or SV40 large T antigen have been necessary to establish immortal human epithelial cell lines, hence the establishment of a completely normal HPDE cell line is currently not yet possible. Recent reports indicated that the expression of the catalytic component of the telomerase (hTERT) gene is capable of immortalizing human fibroblasts, 20,21 but disruption of additional pathways, such as the one regulated by Rb/p16 seems essential to achieve this effect in keratinocytes or mammary epithelial cells. 22 However, the critical pathways and genes responsible for cellular immortalization are also commonly aberrant in human cancer cells, especially pancreatic ductal carcinoma. Inactivation of the G1 cell cycle checkpoint regulated by the Rb-p16 pathway seems to occur in almost all, and p53 mutations in 50 to 70% of this cancer. 5 Thus, despite the putative inactivation of the p53 and Rb pathways, HPDE6E6E7 clones will be very useful as in vitro models to study the molecular basis of human ductal carcinogenesis, and possibly also the differentiation of duct cells into islet cells.
We have previously reported that as compared to a series of pancreatic duct carcinoma cell lines, HPDE cell lines expressed significantly lower levels of several growth factors and tyrosine kinase receptors commonly overexpressed in pancreatic ductal carcinoma. 7 The findings indicated that the phenotypes of HPDE cells more closely resembled the normal duct epithelium than carcinoma cells in vivo. We demonstrate here that the HPDE cells also expressed several characteristic structural and functional genes expressed in normal pancreatic duct epithelium. These included the CK-7, -8, -18, and -19; carbonic anhydrase-II; and MUC-1 apomucin genes. 23-25 The expression of these CK genes are characteristic of simple epithelial cells, and was also demonstrated in primary cultured normal human pancreatic duct cells. 26-28 Our HPDE6c7 cells additionally also expressed high levels of CK14, which apparently was expressed only in <5% cells of normal duct epithelium. Several possibilities can be postulated to explain the expression of CK14 gene in HPDE-E6E7 cells. It is possible that this was an adaptive change in gene expression as a consequence of long-term culture, or as the result of HPV16-E6E7 gene expression. It is also possible that clone 7 cells originated from a subpopulation of human pancreatic ductal cells that constitutively expressed CK14. Note that epithelial cell lines established from adult rat pancreas expressed CK14 additional to CK7 and CK8, but adult rat pancreas is not known to express CK14. Further studies are necessary to clarify the importance of CK14 expression in immortal HPDE cell lines.
We have used both conventional and modern cytogenetic techniques to characterize the karyotypic and genomic changes in HPDE6-E6E7 c7 and c11 cells. Except for one copy gain of chromosome 20 and an additional copy loss of chromosome 22 in HPDE6c11 cells, other karyotypic changes involved small chromosomal losses that are associated with structural changes on 3p, 10p12, and 13q14. The latter included a haploid loss of the Rb gene. It is of interest to note that gain of whole chromosome 20 and deletions on 3p and 13q have been reported to occur commonly in primary pancreatic ductal carcinoma. 29-33 Homozygous intragenic deletions of the FHIT gene located in 3p14.2 have been reported in approximately one third of primary tumor and cell lines of pancreatic cancer. 34-36 Our results suggest that at least these chromosomal losses and gain in combination with HPV16-E6E7 genes are not sufficient for malignant transformation of human pancreatic duct cells.
The expression of E6E7 genes putatively should inactivate the p53 and Rb pathways, and these abnormalities clearly were not sufficient to induce malignant transformation of HPDE cells. Both clones of HPDE6E6E7 cells were neither tumorigenic in immune-deficient mice, nor were anchorage-independent in their growth requirement. This is consistent with previous results on the effect of E6E7 genes in various other human epithelial cells, and the need of other genetic abnormalities such as ras oncogene as a co-factor for the malignant transformation of human epithelial cells. 22,37 Previous reports, however, indicated that an intact Rb pathway is essential for mediating the mito-inhibitory effect of TGF-β1. 38 The proliferation of both HPDE6 clone 7 and 11 cells were partially but significantly inhibited by TGF-β1, and this was associated with the up-regulation of p21cip1 and p27kip1. The expression of Smads-2/3/4 was consistent with this preservation of the TGF-β1-signaling pathway. In most normal epithelial cells including Mv1 Lu mink lung epithelial cells and primary cultured human keratinocytes, growth inhibition by TGF-β1 resulted from increased expression of p15INK4B, p27kip1, and p21cip1 that caused the inhibition of the cyclinD:Cdk4/6 and cyclinE:Cdk2 activities. 38-40 The former requires the presence of Rb, hence HPDE6-E6E7 cells are expected to be resistant to TGF-β1. An Rb family member p130/Rb2 gene may also exert similar G1/S cell cycle regulatory function, and it is also phosphorylated by G1 cyclin-associated Cdks. 41 TGF-β1-induced p21cip1/waf1 up-regulation can also increase the levels and stability of hyperphosphorylated p130/Rb, hence induce G1 growth arrest. 42 In contrast to Rb, however, binding of E7 to p130/Rb does not lead to increase proteolytic degradation. 43 The results suggest that the G1 cell-cycle checkpoint regulation may still be intact in HPDE6-E6E7 cells, and the p130/Rb2 may have assumed this important function in place of the p110/Rb. This hypothesis remains to be tested.
We have also shown that despite the loss of wild-type p53 protein function, the HPDE6c7 cells demonstrated chromosomal stability. Chromosomal instability is defined as alterations of chromosome number involving losses or gains of whole chromosomes. 44-46 In contrast to microsatellite instability which is a recessive trait and is caused by defects in mismatch repair genes resulting in genomic losses at the nucleotide sequence level, chromosomal instability seems to be a dominant phenotype that is consistently associated with the loss of spindle checkpoint control. 44,45 The p53 gene has been considered as a “guardian” of the genome and its loss may result in genomic and/or chromosomal instability. 47 Gualberto et al 48 have reported that certain missense p53 mutants may cause the failure of cells to arrest at 4n after treatment with colcemid. The generation of polyploid cells may result in chromosomal instability through random losses of chromosomes and generation of aneuploidy. On the other hand, the loss of p53 protein caused by expression of the HPV16-E6 gene seemed to result in the maintenance of normal spindle checkpoint control. Our results also suggests that loss of the wild-type p53 protein and its function is not sufficient to induce chromosomal instability.
In conclusion, aside from the apparent loss of wild-type p53 protein function and the presence of a limited number of genetic changes, the chromosomally stable clones of HPDE6-E6E7 cells show phenotypic resemblance to normal pancreatic duct epithelial cells. These immortal cell lines will be useful as in vitro models for future studies in pancreatic duct cell carcinogenesis and differentiation.
Acknowledgments
We thank Dr. Lea Harrington for assistance in the TRAP assay.
Footnotes
Address reprint requests to Dr. Ming-Sound Tsao, 610 University Ave., Toronto, Ontario, Canada, M5G 2M9. E-mail: ming.tsao@uhn.on.ca.
Supported by the Medical Research Council of Canada grant MT-14359 (to M. S. T.) and the Academic Enrinchment Fund of UHN Pathology Department.
References
- 1.Parker SL, Tong T, Bolden S, Wingo PA: Cancer statistics. CA Cancer J Clin 1997, 47:5-27 [DOI] [PubMed] [Google Scholar]
- 2.Baumel H, Huguier M, Manderscheid JC, Fabre JM, Houry S, Fagot H: Results of resection for cancer of the exocrine pancreas: a study from the French Association of Surgery. Br J Surg 1994, 81:102-107 [DOI] [PubMed] [Google Scholar]
- 3.Warshaw AL, Castillo CF: Pancreatic carcinoma. N Engl J Med 1992, 326:455-465 [DOI] [PubMed] [Google Scholar]
- 4.Murr MM, Sarr MG, Oishi AJ, van Heerden JA: Pancreatic cancer. CA Cancer J Clin 1994, 44:304-318 [DOI] [PubMed] [Google Scholar]
- 5.Goggins M, Kern SE, et al: Progress in cancer genetics: lessons from pancreatic cancer. Ann Oncol 1999, 10(Suppl 4):S4-S8 [PubMed] [Google Scholar]
- 6.Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao M-S: Long term culture and immortalization of epithelial cells from normal adult human pancreatic ducts by transfection with the E6E7 gene of human papilloma virus 16. Am J Pathol 1996, 148:1763-1770 [PMC free article] [PubMed] [Google Scholar]
- 7.Liu N, Furukawa T, Kobari M, Tsao M-S: Comparative phenotypic studies of duct epithelial cell lines derived from normal human pancreas and pancreatic carcinoma. Am J Pathol 1998, 153:263-269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsao M-S, Viallet J: Pre-clinical models of lung cancer: cultured cells and organ culture. Kane MA Bunn P eds. Biology of Lung Cancer. 1998, :pp 215-246 Marcel Dekker, New York [Google Scholar]
- 9.Liu C, Tsao M-S, Grisham JW: Transforming growth factors produced by normal and neoplastically transformed rat liver epithelial cells in culture. Cancer Res 1988, 48:850-855 [PubMed] [Google Scholar]
- 10.Barch MJ, Knutsen T, Spurbeck JL: The AGT Cytogenetics Laboratory Manual. 1997. Lippincott-Raven Publishers, Philadelphia
- 11.Mitelman F: International System for Human Cytogenetic Nomenclature. 1995. S. Karger, New York
- 12.Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D, Garini Y, Ried T: Multicolor spectral karyotyping of human chromosomes. Science 1996, 273:494-497 [DOI] [PubMed] [Google Scholar]
- 13.Veldman T, Vignon C, Schrock E, Rowley JD, Ried T: Hidden chromosome abnormalities in histological malignancies detected by multicolor spectral karyotyping. Nat Genet 1997, 15:406-410 [DOI] [PubMed] [Google Scholar]
- 14.Dracopoli NC (Ed): Current Protocols in Human Genetics. New York, John Wiley & Sons, 1999
- 15.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258:818-821 [DOI] [PubMed] [Google Scholar]
- 16.Du Manoir S, Schrock E, Bentz M, Speicher MR, Joos S, Ried T, Lichter P, Cremer T: Quantitative analysis of comparative genomic hybridization. Cytometry 1995, 19:27-41 [DOI] [PubMed] [Google Scholar]
- 17.Lundsteen C, Maahr J, Christensen B, Bryndorf T, Bentz M, Lichter P, Gerdes T: Image analysis in comparative genomic hybridization. Cytometry 1995, 19:42-50 [DOI] [PubMed] [Google Scholar]
- 18.Piper J, Rutovitz D, Sudar D, Kallioniemi A, Kallioniemi OP, Waldman FM, Gray JW, Pinkel D: Computer image analysis of comparative genomic hybridization. Cytometry 1995, 19:10-26 [DOI] [PubMed] [Google Scholar]
- 19.Tsao M-S, Zhu H, Giaid A, Viallet J, Nakamura T, Park M: Hepatocyte growth factor/scatter factor is an autocrine factor for human normal bronchial epithelial and lung carcinoma cells. Cell Growth Differ 1993, 4:571-579 [PubMed] [Google Scholar]
- 20.Viziri H, Benchimol S: Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 1998, 8:279-282 [DOI] [PubMed] [Google Scholar]
- 21.Bodnar AG, Ouellete M, Frolkis M, Holt SE, Chiu C, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE: Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279:349-352 [DOI] [PubMed] [Google Scholar]
- 22.Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ: Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396:84-88 [DOI] [PubMed] [Google Scholar]
- 23.Schussler MH, Skoudy A, Ramaekers F, Real FX: Intermediate filaments as differentiation markers of exocrine pancreas. Am J Pathol 1992, 140:559-568 [PMC free article] [PubMed] [Google Scholar]
- 24.Real FX, Vila MR, Skoudy A, Ramaekers FC, Corominas JM: Intermediate filaments as differentiation markers of exocrine pancreas. II. Expression of cytokeratins of complex and stratified epithelia in normal pancreas and in pancreatic cancer. Int J Cancer 1993, 54:720-727 [DOI] [PubMed] [Google Scholar]
- 25.Balagué C, Gambus G, Carrato C, Porchet N, Aaubert JP, Kim YS, Real FX: Altered expression of MUC2, MUC4 and MUC5 gene in pancreas tissue and cancer cell lines. Gastroenterology 1994, 106:1054-1061 [DOI] [PubMed] [Google Scholar]
- 26.Vila MR, Lloreta J, Real FX: Normal human pancreas cultures display functional ductal characteristics. Lab Invest 1994, 71:423-431 [PubMed] [Google Scholar]
- 27.Oda D, Savard CE, Nguyen TD, Swenson ER, Lee SP: Culture of human main pancreatic duct epithelial cells. In Vitro Cell Dev Biol Anim 1998, 34:211-216 [DOI] [PubMed] [Google Scholar]
- 28.Kolar C, Caffrey T, Hollingsworth M, Scheetz M, Sutherlin M, Weide L, Lawson T: Duct epithelial cells cultured from human pancreas processed for transplantation retain differentiated ductal characteristics. Pancreas 1997, 15:265-271 [DOI] [PubMed] [Google Scholar]
- 29.Solinas-Toldo S, Wallrapp C, Muller-Pillasch F, Bentz M, Gress T, Lichter P: Mapping of chromosomal imbalances in pancreatic carcinoma by comparative genomic hybridization. Cancer Res 1996, 56:3803-3807 [PubMed] [Google Scholar]
- 30.Hahn SA, Seymour AB, Hoque AT, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ: Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995, 55:2670-2675 [PubMed] [Google Scholar]
- 31.Griffin CA, Hruban RH, Morsberger LA, Ellingham T, Long PP, Jaffee EM, Hauda KM, Bohlander SK, Yeo CJ: Consistent chromosome abnormalities in adenocarcinoma of the pancreas. Cancer Res 1995, 55:2394-2399 [PubMed] [Google Scholar]
- 32.Johansson B, Bardi G, Heim S, Mandahl N, Mertens F, Bak-Jensen E, Andren-Sandberg A, Mitelman F: Nonrandom chromosomal rearrangements in pancreatic carcinomas. Cancer 1992, 69:1674-1681 [DOI] [PubMed] [Google Scholar]
- 33.Bardi G, Johansson B, Pardis N, Mandahl N, Bak-Jensen E, Andren-Sandberg A, Mitelman F, Heim S: Karyotypic abnormalities in tumours of the pancreas. Br J Cancer 1993, 67:1106-1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorio C, Baron A, Orlandini S, Zamboni G, Pederzoli P, Huebner K, Scarpa A: The FHIT gene is expressed in pancreatic ductular cells and is altered in pancreatic cancers. Cancer Res 1999, 59:1308-1314 [PubMed] [Google Scholar]
- 35.Simon B, Bartsch D, Barth P, Prasnikar N, Munch K, Blum A, Arnold R, Goke B: Frequent abnormalities of the putative tumour suppressor gene FHIT at 3p14.2 in pancreatic carcinoma cell lines. Cancer Res 1998, 58:1583-1587 [PubMed] [Google Scholar]
- 36.Shridhar R, Shridhar V, Wang X, Paradee W, Dugan M, Sarkar F, Wilke C, Glover TW, Vaitkevicius VK, Smith DI: Frequent breakpoints in the 3p14.2 fragile site, FRA3B, in pancreatic tumors. Cancer Res 1996, 56:4347-4350 [PubMed] [Google Scholar]
- 37.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA: Creation of human tumor cells with defined genetic elements. Nature 1999, 400:464-468 [DOI] [PubMed] [Google Scholar]
- 38.Hannon G, Beach D: p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature 1994, 371:257-261 [DOI] [PubMed] [Google Scholar]
- 39.Polyak K, Kato J, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A: P27kip1, a cyclin-Cdk inhibitor, links transforming growth factor-b and contact inhibition to cell cycle arrest. Genes Dev 1994, 8:9-22 [DOI] [PubMed] [Google Scholar]
- 40.Robson CN, Gnanapragasam V, Byrne RL, Collins AT, Neal DE: Transforming growth factor-β1 up-regulates p15, p21 and p27 and blocks cell cycling in G1 in human prostate epithelium. J Endocrinol 1999, 160:257-266 [DOI] [PubMed] [Google Scholar]
- 41.Grana X, Garriga J, Mayol X: Role of retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 1998, 17:3365-3383 [DOI] [PubMed] [Google Scholar]
- 42.Yoo YD, Choi J-Y, Lee S-J, Kjm JS, Min B-R, Lee YI, Kang Y-K: TGF-□-induced cell cycle arrest through the p21waf1/cip1-G1 cyclin/cdks-p130 pathway in gastric carcinoma cells. Int J Cancer 1999, 83:512-517 [DOI] [PubMed] [Google Scholar]
- 43.Berezutskaya E, Yu B, Morozov A, Raychaudhuri P, Bagchi S: Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16E7 oncoprotein. Cell Growth Differ 1997, 8:1277-1286 [PubMed] [Google Scholar]
- 44.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 45.Cahill DP, Lengauer C, Yu J, Riggins GJ, Wilson JK, Markowitz SD, Kinzler KW, Vogelstein B: Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392:300-303 [DOI] [PubMed] [Google Scholar]
- 46.Lengauer C, Kinzler KW, Vogelstein B: Genetic instability in colorectal cancers. Nature 1997, 386:623-627 [DOI] [PubMed] [Google Scholar]
- 47.Lane DP: p53, guardian of the genome. Nature 1992, 358:15-16 [DOI] [PubMed] [Google Scholar]
- 48.Gualberto A, Aldepe K, Kozakiewicz K, Tlsty TD: An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc Natl Acad Sci USA 1998, 95:5166-5171 [DOI] [PMC free article] [PubMed] [Google Scholar]