

Abstract
Testicular germ cell tumors are important neoplasms and seminoma accounts for 40 to 50% of these tumors. Little is known concerning the molecular events underlying the development of these malignancies. In the present study we used a modified differential display approach to compare gene expression between seminoma and normal testicular parenchyma, both of which are mixed tissues. We first analyzed mRNA (cDNA) expression by differential display and then directly used differentially expressed cDNAs for the synthesis of radiolabeled riboprobes to attribute differential expression to specific cell types in tissue sections by in situ hybridization. Using this approach along with real-time quantitative reverse transcriptase-polymerase chain reaction analysis we found an overexpression of eukaryotic initiation factor 3 p110 mRNA (EIF3S8) in seminoma cells compared to normal germ cells of testicular tubules. The elF3-p110 subunit may promote seminoma development by generally increasing translation leading to enhanced cellular growth and division rates.
Testicular germ cell tumors are common malignancies between the age of 15 and 35 years. They arise by transformation of early germ cell precursors and can differentiate into either embryonal or extra-embryonal directions. Seminoma is considered the most undifferentiated and is the most frequent of these tumors accounting for 40 to 50% of testicular germ cell tumors. Little is known about the molecular events underlying the development of these important neoplasms. In seminomas it is the KIT oncogene that is overexpressed and in nonseminomas it is the mast cell growth factor (MGF). Tumor suppressor genes supposed to be involved in testicular germ cell tumor development include DCC (deleted in colon cancer), RB1 (retinoblastoma), as well as NME1 and 2 (nucleoside diphosphate kinase). 1,2
Differential display is one of several methods, which allows the identification of new cancer genes by comparing gene expression of normal and neoplastic cells. 3,4 Application of this method to most human tumors is nevertheless hampered by the fact that tumor tissue not only contains neoplastic but also stromal cells (such as fibroblasts, endothelial, or inflammatory cells), which can mask differences in gene expression between neoplastic cells and their normal counterparts. Seminoma, in particular, contains a great number of inflammatory cells (mainly lymphocytes and macrophages) in the stroma. Moreover normal testicular parenchyma is also a mixture of germinal and other cell types such as Sertoli and Leydig cells. Comparison of mRNA expression between seminoma and normal testicular tissue by differential display and Northern blotting alone would therefore not be sufficient to attribute expression differences to specific cell types. In the present investigation we solved this problem by first comparing mRNA expression of normal testicular parenchyma and seminomas by conventional differential display and then localizing differentially expressed transcripts to specific cell types by in situ hybridization with riboprobes, which we synthesized directly from the differentially expressed cDNAs. In addition we verified differential expression by real-time quantitative reverse transcriptase-polymerase chain reaction (RT-PCR), with which we could demonstrate a significant overexpression of eukaryotic initiation factor 3 (elF3-p110) mRNA in seminoma cells compared to that of normal germ cell precursors in testicular tubules. Overexpression of this elF3 subunit may promote seminoma development by generally increasing translation leading to enhanced cellular growth and division rates.
Materials and Methods
Tissue Samples
We examined 25 human testicular seminomas and compared them with seven normal testicular biopsies taken during infertility diagnosis for other than testicular reasons. Patients were 19 to 49 years old. We immediately froze parts of each sample in −150°C isopentane (precooled in liquid nitrogen) for RNA extraction, to preserve histomorphology. In parallel, we fixed parts of the biopsies in 4% paraformaldehyde (for 24 hours at 4°C) for in situ hybridization and routinely embedded these samples into paraffin. Histological evaluation was done on frozen or paraffin sections stained with hematoxylin and eosin (H&E). In all cases biopsies were evaluated as normal and free of preneoplastic germ cell alterations.
Cryo-Microdissection and RNA Extraction
We microscopically selected areas from normal testicular parenchyma or seminomas on frozen sections and trimmed the tissue blocks to the size of these areas at −20°C using a sterile scalpel. We then cut 30 20-μm-thick sections from these blocks using a cryostat and immediately placed them into liquid nitrogen. Histology was again verified during this process at regular intervals on H&E-stained sections.
We isolated total cellular RNA from microdissected tissues with Trizol (Gibco-BRL, Karlsruhe, Germany) after evaporation of the liquid nitrogen using 750 μl of Trizol for 30 slides. We performed extraction according to the manufacturer’s instructions. We then treated RNA with DNase I (Roche, Mannheim, Germany) to remove genomic DNA. We quantified RNA by spectrophotometry and verified RNA integrity by electrophoresis of 4 μg of total RNA in a 2% agarose gel that had been stained with ethidium bromide.
Differential Display
We used six base-anchored oligonucleotide primers (5′-T11CA-3′, 5′-T11CG-3′, 5′-T11AC-3′, 5′-T11GC-3′, 5′-T11CC-3′, and 5′-T11GG-3′) to reverse-transcribe 4 μg of total cellular RNA into first strand cDNA. We subsequently amplified the cDNA using the same primers together with 10 arbitrary primers (5′-TACAACGAGG-3′, 5′-TGGATTGGTC-3′, 5′-CT-TTCTACCC-3′, 5′-TTTTGGCTCC-3′, 5′-GGAACCAATC-3′, AAACTCCGTC-3′, 5′-GATCTGACTG-3′, 5′-GATCATGGTC-3′, 5′-GATCATAGCG-3′, and 5′-GATCTAAGGC-3′). PCR conditions are as described by Liang. 5 We analyzed PCR products on 8% DNA sequencing gels after silver-staining. 6 We excised bands from the cDNA ladders, which exhibited differences of intensities between normal and tumor tissue DNA and extracted them by 15 minutes cooking. This was followed by precipitation with sodium acetate (0.25 mol/L final concentration, pH 4.6) and reamplification with the same primers under the same PCR conditions described above. We electrophoresed reamplified cDNAs in a 2% agarose gel, cut them out, and extracted them with the QIAEX DNA extraction kit (Qiagen, Hilden, Germany) for further cloning.
Cloning and Sequencing
We ligated reamplified cDNA fragments into the pCRII vector using the TA Cloning kit (Invitrogen, Groningen, The Netherlands). We then transformed competent cells (Escherichia coli TOP10F′) with the plasmids by heat shock. Transformed cells were plated on β-X-Gal LB-agar and incubated for 18 hours at 37°C. We picked white colonies containing plasmids with inserts, transferred them into 3 ml of Luria-Bertani medium (supplemented with 70 μg/ml ampicillin), and incubated them for 12 hours at 37°C.
We then prepared plasmids by using the Qiagen Plasmid Purification Kit. We amplified individual plasmids for cycle sequencing with the M13-forward and the M-13 reverse sequencing primer using the ABI prism Terminator Cycle Sequencing Ready Reaction Kit (PE Biosystems, Weiterstadt, Germany). We sequenced at least 20 clones of one transformation using an ABI Prism 310 sequencer (PE Biosystems). Sequences were aligned to the human genome database with the BLAST program. 7
In Situ Hybridization
We cut out the elF3-p110 insert from pCRII and subcloned it in pBluescript to synthesize radiolabeled riboprobes by in vitro transcription, 8-10 Riboprobes were labeled with 35S-CTP (70 μCi/μl; Du Pont-New England Nuclear Research Products, Boston, MA) using an in vitro transcription kit (Promega, Mannheim, Germany). For in vitro transcription we linearized 10 μg of the recombinant plasmid containing the 200-bp elF-3 insert with either EcoRV or BamHI to generate sense or antisense riboprobes. Probes were synthesized for 2 hours (at 37°C in 1× transcription buffer) using SP6 or T7 RNA polymerase. We measured probe activity in a scintillation counter (Canberra-Packard, Zürich, Switzerland).
For in situ hybridization we routinely embedded the fresh tissue samples, which had been immediately fixed in 4% paraformaldehyde (for 24 hours at 4°C) into paraffin. We transferred 5-μm-thick paraffin sections on silanized slides (Superfrost; Menzel, Heidelberg, Germany) and dried them overnight at 42°C. After deparaffinization and rehydration, we incubated slides in 0.1 mol/L glycine and 0.2 mol/L Tris-HCl (pH 7.4) for 10 minutes at room temperature, treated them with proteinase K (1 μg/ml, 15 minutes at 37°C; Gibco-BRL) and refixed them for 15 minutes in 4% paraformaldehyde in phosphate-buffered saline (PBS). Slides were then washed in PBS, acetylated, and dehydrated. 11 We denaturated probes immediately before hybridization at 80°C for 2 minutes and diluted them in hybridization buffer 11 (riboprobe activity: 20 × 103cpm/μl). Hybridization was performed for 16 hours at 60°C in sealed boxes. We then washed slides in 4× standard saline citrate (SSC) at room temperature and treated them with RNase A (10 μg/ml, 30 minutes at 37°C; Roche). We performed stringent washes in 2× SSC and 0.1× SSC at 60°C for 10 minutes each. We finally dehydrated slides, dipped them in nuclear track emulsion (NTB2; Kodak, Rochester, NY), and exposed them at 4°C for 10 to 15 days for autoradiography. After development we counterstained nuclei with bisbenzimidine (Hoechst 33258) 12 and evaluated results in fluorescent light under dark-field illumination (with Axioscop 50, Zeiss, Göttingen, Germany) to visualize silver grains.
Real-Time Quantitative RT-PCR of EIF3S8 mRNA and the Housekeeping Gene TATA-Binding Protein (TBP)
To quantify EIF3S8 mRNA expression in addition to in situ hybridization we needed to establish a real-time quantitative RT-PCR approach for both the EIF3S8 gene and for the housekeeping gene TBP (TATA binding protein), because the precise amount of total RNA added to each reaction and its quality are difficult to estimate. No retropseudogenes are known for TBP and the expression level is in the range of the EIF3S8 target gene. 13 The amount of targets and endogenous reference was determined from standard curves for each experimental sample. To obtain a normalized target value the target amount was divided by the endogenous reference amount (EIF3S8/TBP). During real-time quantitative RT-PCR analysis of EIF3S8 expression, we compared seminoma samples with tumor-free testicular parenchyma from the same patients (n = 7).
Construction of cRNA Standards for EIF3S8 and TBP
After reverse transcription of 4 μg of total cellular RNA from each of the seminoma and or normal testicular parenchyma tissue into first strand cDNA we performed specific PCR-amplification with the designed primers shown in Table 1 ▶ . We then constructed oligonucleotides as described in Totzke et al 14 with a T7 promotor at the 5′ PCR primers and an oligo-dT at the 3′ primers to perform in vitro transcription of EIF3S8 and TBP cDNAs. We reamplified EIF3S8 and TBP cDNA with these primers and visualized fragments on a 1.5% agarose gel. We then used 1 μg of cDNA from each PCR reaction for in vitro transcription (MEGAscript T7 kit; Ambion, Austin, TX), after which we removed cDNA by digestion with RNase-free DNase I (for 15 minutes at 37°C). We stopped reactions and precipitated cRNAs by adding RNase-free dH2O and lithium chloride (incubation for at least 30 minutes at −20°C). We finally quantified cRNAs (based on absorbance) and made a dilution series from 10 11 to 10 5 transcripts μl−1 in 20-μl tRNA samples (1:100 dilution of brewer’s yeast tRNA in diethyl pyrocarbonate-dH2O; Roche).
Table 1.
Primers Used for the Amplification and Construction of a cRNA Standard from EIF3S8 and TBP and Fluorescence Hybridization Probes for Quantification of EIF3S8 and TBP mRNA Expression in Tissue
| Primer | EIF3 | TBP |
|---|---|---|
| 5′-PCR primer | 5′-GCTAAGAAGAAGCACGACAGGAAAT-3′ | 5′-GCTCTTCCACTCACAGACTC-3′ |
| 3′-PCR primer | 5′-CCAGGTTGGGGTTGTAGTCATAG-3′ | 5′-GCCAGTCTGGACTGTTCTTC-3′ |
| 5′-cRNA construction-primer | 5′-GGATCCTAATACGACTCACTATAGGGAGG GCTAAGAAGAAGCACGACAGGAAAT-3′ | 5′-GGATCCTAATACGACTCACTATAGGGAGG GCTCTTCCACTCACAGACTC-3′ |
| 3′-cRNA construction-primer | 5′-TTTTTTTTTTTTTTTCCAGGTTGGGGTTG TAGTCATAG-3′ | 5′-TTTTTTTTTTTTTTTGCCAGTCTGGACTG TTCTTC-3′ |
| FL-hybridization probe | 5′-GACCCTTTCCCACTCCCCGCCTT-FL-3′ | 5′-CCGTGGTTCGTGGCTCTCTTATCCTC-FL-3′ |
| LCR-hybridization probe | 5′-LCR-TGTCCTCCTCCTCCTCATCCAGGC-3′ | 5′-LCR-TGATTACCGCAGCAAACCGCTTG-3′ |
FL, fluorescein; LCR, light-cycler-red 640.
Real-Time PCR Quantification
We first reverse-transcribed 4 μg of total RNA from seminoma and normal testicular tissues and decreasing amounts of EIF3S8 and TBP cRNA in the range from 10 11 to 105. To achieve this we incubated samples with Moloney murine leukemia virus reverse transcriptase (200 U/μl) for 5 minutes at 25°C, 5 minutes at 30°C, 90 minutes at 37°C, and 5 minutes at 95°C in a total reaction volume of 40 μl containing 1× RT-buffer (50 mmol/L Tris-HCl, pH 8.3, 75 mmol/L KCl, 3 mmol/L MgCl2), 10 mmol/L dithiothreitol, 0.5 mmol/L of each dNTP, 50 U of RNA-guard (Pharmacia, Freiburg, Germany), and 100 pmol of random hexamer primers.
We next performed PCR on a Light-Cycler (Roche) additionally using hybridization probes in combination with the Light-Cycler DNA Master Hybridization Probes Kit (Roche). Primers used for amplification and hybridization (TIB Molbiol, Berlin, Germany) of EIF3S8 and TBP cDNAs are given in Table 1 ▶ . In principle, hybridization is performed in addition to primer annealing during Light-Cycler PCR with two different short oligonucleotides that hybridize to two adjacent internal sequences of the PCR fragment during the annealing phases. One probe is 5′-labeled with Light-Cycler-Red fluorophore and the other with fluorescein. Probes come into close proximity after specific hybridization resulting in fluorescence resonance energy transfer between the two fluorophores when the donor fluorophore, fluorescein, is stimulated by the light source of the cycler. Parts of the absorbed energy are transferred to the acceptor fluorophore, Light-Cycler-Red, the emitted fluorescence of which is finally measured. The PCR reaction mixture contained 2 mmol/L MgCl2, 20 pmol of both PCR primers, 1 pmol of each hybridization probe, 2 μl of Light-Cycler DNA Master Hybridization Mix (Roche), and 2 μl of reverse-transcribed samples in a final volume of 20 μl. After 3 minutes of denaturation (at 94°C) we performed 45 PCR cycles with 3 seconds of denaturation at 94°C, 20 seconds for EIF358 and 15 seconds for TBP (EIF3S8/TBP) of annealing at 63/60°C (EIF3S8/TBP), and 20 seconds of extension at 72°C. The larger the starting quantity of the target molecule, the earlier a significant increase in fluorescence caused by fluorescence resonance energy transfer is detected during on-line monitoring of PCR by the Light-Cycler. This point of first detection is the main parameter for quantitative measurements during real-time PCR. The online software system analyzes the spectral data collected during each cycle and plots fluorescence intensity versus cycle number. The cycle number at the first PCR signal is measured and RNA concentration calculated by the Light-Cycler Software (Roche).
Northern Hybridization
We separated total cellular RNAs of normal or seminoma tissue on denaturating 1.3% agarose gels (1.5% formaldehyde, v/v) and transferred them to nylon membranes (Hybond N; Amersham, Braunschweig, Germany). To obtain a probe for hybridization we cut out the EIF3S8 insert by EcoRI digestion and labeled it with 32P-dCTP using the Rediprime II DNA labeling kit (Amersham). We hybridized membranes with the probe overnight at 42°C. 28S and 18S bands in the gel before blotting were used as a control for equal loading. We performed washes in 2× SSC, 0.1% sodium dodecyl sulfate, and in 1× SSC, 0.1% sodium dodecyl sulfate at 60°C (15 minutes each), and exposed membranes during autoradiography for 1 to 7 days at −70°C.
Results
Overexpression of elF3-p110 mRNA (EIF3S8) in Seminomas Found by Differential Display
We first performed differential display RT-PCR to detect differences in gene expression between normal testicular parenchyma and seminomas. Using the 60 primer pairs we found a total of 78 bands which showed slight to strong differences of intensity between seminoma and normal testicular tissues. Using the primer pair 5′-T11CG-3′ and 5′-TACAACGAGG-3′ we detected large amounts of a cDNA fragment (of ∼200 bp), in all seminoma samples, whereas only weak or no signals were seen in normal testicular parenchyma (Figure 1) ▶ . Because this fragment showed the greatest difference between seminoma and normal testicular tissue, we subcloned and sequenced it and aligned the sequence to the human genome database using the BLAST program. 7 The sequence was 100% homologous (133 of 133 bases) to the mRNA of the p110 subunit of the eukaryotic initiation factor 3 (EIF3S8) (accession number: U46025).
Figure 1.

Comparison of mRNA (cDNA) expression between seminomas (n = 9) and normal testicular parenchyma (n = 6) by differential display. Differential display was performed after retrotranscription of total cellular mRNA into cDNA and PCR using a 5′ arbritary primer (5′-TACAACGAGG-3′) and a 3′ anchored primer (5′-TTTTTTTTTTTCG-3′). PCR conditions were as described in Liang. 5 Amplified cDNA fragments were separated in an 8% acrylamide gel. A fragment of ∼200 bp (arrow) is strongly displayed in all seminoma samples (T) but only weakly discernible in normal testicular parenchyma (N). Subcloning and sequencing of this fragment revealed a 100% homology (133 of 133 bases) to the mRNA of the elF3-p110 subunit.
Cellular Localization of elF3-p110 mRNA and Confirmation of Differential Expression Using in Situ Hybridization
We next wanted to identify the cell types that express elF3-p110 mRNA in normal testis and in seminomas and to confirm differential expression. We therefore synthesized 35S-CTP-labeled riboprobes by in vitro transcription using the sequenced elF3-p110 (EIF3S8) cDNA subclone as a template. We analyzed 10 seminomas and compared them to seven normal testicular biopsies. In situ hybridization revealed that elF3-p110 transcripts are expressed by the normal germinal epithelium of the testicular tubules and by the tumor cells of seminomas. On the other hand the interstitial testicular tissue as well as the seminoma stroma exhibited only weak signals or none at all (Figure 2) ▶ . In six of the 10 seminomas the intensity of signals turned out to be clearly stronger in seminoma cells than in normal germ cells (Figure 2) ▶ .
Figure 2.
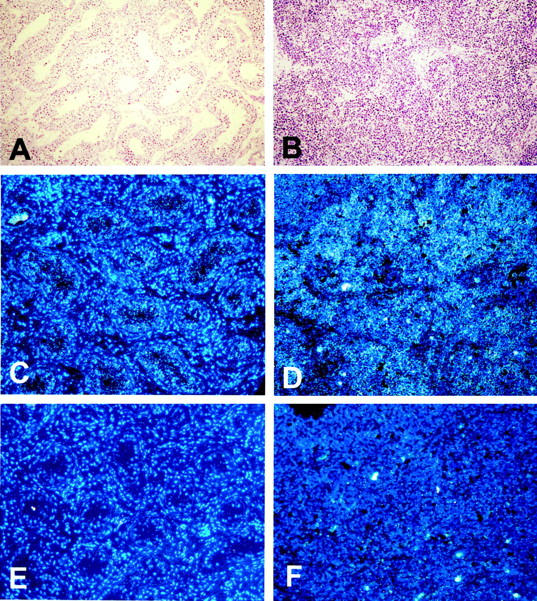
Localization of elF3 (p110 subunit) mRNA in normal testicular parenchyma and in a seminoma by in situ hybridization. Positive signals (light grains, dark-field illumination) are weakly seen in normal intratubular germ cells (C) whereas seminoma tumor cells display a strong expression (D). Normal interstitial tissues (C) and seminoma stroma (D) are, in contrast, negative. No positive signals are observed after hybridization of sections with a negative control sense probe (E and F). Nuclei have been counterstained with fluorescent bisbenzimidine (Hoechst 33258) and signals evaluated in dark-field illumination. A and B: Morphology of normal testicular tissue (A) and of seminoma (B) in H&E-stained sections. Original magnification, ×100.
Verification of elF3-p110 (EIF3S8) mRNA Overexpression in Seminomas by Quantitative Real-Time RT-PCR
For real-time RT-PCR analysis of EIF3S8 expression we established standard curves for EIF3S8 and TBP using the serially diluted cRNA (10 5 to 10 11 transcripts) obtained by in vitro transcription. Figure 3A ▶ shows the standard curves for the EIF3S8-gene. The dynamic range was wide (at least five orders of magnitude). A strong linear relationship between the fractional cycle number and the log of the starting copy number was demonstrated (Figure 3B) ▶ . For determination of the total amount of EIF3S8 transcripts in 2 μg of total tissue RNA, we reverse-transcribed and amplified tissue samples during real-time RT-PCR together with the standard samples (Figure 3A) ▶ . We applied the same approach for the analysis of TBP housekeeping gene expression and determined the amounts of both EIF3S8 and TBP mRNAs in the samples from the standard curves. To obtain a normalized value for EIF3S8 mRNA for each tissue sample we divided EIF3S8 amounts by TBP amounts.
Figure 3.

Analysis of elF3-p110 mRNA (ElF3S8) expression in seminomas and normal testicular parenchyma by real-time quantitative RT-PCR analysis. A: A serial dilution of ElF3S8 standard cRNA over a range from 10 5 to 10 11 was performed. For determination of the total amount of ElF3S8 transcripts in 2 μg of total tissue RNA, tissue samples were reverse-transcribed and amplified during real-time PCR together with the standard samples. B: A strong linear relationship between the fractional cycle number and the log of the starting copy number was always demonstrated in standard samples (A).
We found a mean relative amount for EIF3S8 transcripts of 6.225 ± 1.075 (EIF3S8/TBP, SD±SEM) in seven seminoma samples, compared to 3.161 ± 0.513 in seven normal testicular tissues. Results are given in Table 2 ▶ . The difference between the values for seminoma and normal samples was significant with a z-value of −2.364 (P < 0.05). 15
Table 2.
EIF3S8 mRNA Levels in Seminomas and Normal Testicular Tissues
| Patient no. | Tissue type* | EIF/TBP | Copy no. EIF3S8 | Copy no. TBP |
|---|---|---|---|---|
| 1 | S | 10.582 | 16920000 | 1599000 |
| N | 3.538 | 7949000 | 2247000 | |
| 2 | S | 9.573 | 19070000 | 1992000 |
| N | 3.367 | 2938000 | 872600 | |
| 3 | S | 5.999 | 14620000 | 2437000 |
| N | 1.594 | 3783000 | 2374000 | |
| 4 | S | 5.651 | 2010000 | 355700 |
| N | 2.265 | 1685000 | 744000 | |
| 5 | S | 5.014 | 13860000 | 2764000 |
| N | 4.997 | 14870000 | 2976000 | |
| 6 | S | 2.942 | 3831000 | 1302000 |
| N | 1.669 | 2003000 | 1200000 | |
| 7 | S | 3.817 | 304000 | 79700 |
| N | 4.702 | 3498000 | 744000 | |
| Sum | S (n = 7) | 6.225 ± 1.075 (SD ± SEM) | ||
| N (n = 7) | 3.161 ± 0.513 (SD ± SEM) | |||
| Z = −2.364; P < 0.05 |
The EIF3S8 mRNA copy number was divided by the TBP mRNA copy number for each sample in order to obtain a normalized EIF3S8/TBP value.
*S, seminoma; N, normal.
Verification of elF3-p110 (EIF3S8) Overexpression in Seminomas by Northern Blotting
In four cases of seminoma and normal testicular biopsies sufficient amounts of mRNA were left after quantitative RT-PCR for Northern blot analyses. Using radiolabeled elF3-p110 probes, which had been synthesized by random priming, we found that elF-3-p110 transcripts were present in larger amounts in tumor tissue than in normal parenchyma. Results are shown in Figure 4 ▶ .
Figure 4.
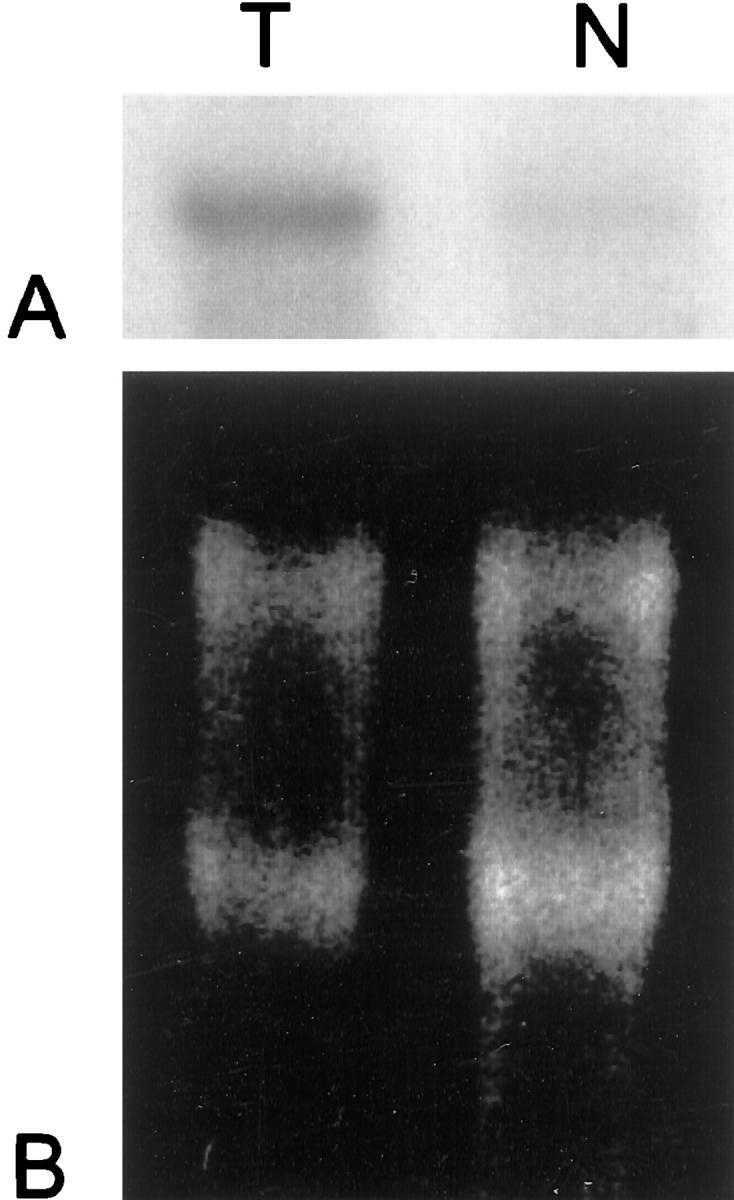
Demonstration of elF3 (p110 subunit) mRNA overexpression in seminomas by Northern blotting. A: Twenty μg of total cellular RNA from a seminoma sample (T) and from normal testicular parenchyma (N) from one patient have been electrophoretically separated, blotted onto a nylon membrane, and hybridized with a 32P-dCTP-labeled elF3-p110 cDNA probe. Hybridization signals are clearly stronger in the seminoma sample (T) compared to normal testicular control (N). B: Intensity of 28S and 18S bands in the agarose gel before blotting shows equal loading.
Discussion
Differential display is one of several suitable methods, which allows the comparison of gene expression at the mRNA level of different cells or tissues. Application of this technique is nevertheless generally restricted to a comparison of uniform cell lines. Most normal and tumor tissues, including testicular and seminomatous, are on the other hand composed of several cell types. Seminoma in particular, shows an intricate mixture of tumor cells and stromal inflammatory cells (mainly lymphocytes and macrophages). Differential display of seminoma and testicular cDNA followed by Northern blotting can, therefore, yield no information about the cell types which are responsible for expression differences. We solved this problem by first comparing cDNA expression of normal and seminoma tissues and then directly synthesizing RNA probes from differentially expressed cDNAs for in situ hybridization. Using this strategy we could attribute expression of elF3 (p110) mRNA (EIF3S8) to normal testicular germ cells and to the tumor cells of seminomas. At the same time we could confirm elF3-p110 mRNA overexpression in six of 10 tumors investigated. We additionally verified EIF3S8 overexpression in seminomas by quantitative real-time RT-PCR and Northern blot analyses.
Eukaryotic initiation factors (elFs) are a complex system of many cellular proteins, which are necessary for the first steps of translation. The elF3 complex (650 kd), in particular, is composed of 10 subunits, one of the largest of which is p110 (913 amino acids). 16,17 The whole elF3 complex recognizes the first AUG initiation codon closest to the 5′ end of the mRNA. 18 ElF2 carries the Met-tRNA to the 40S subunit of the ribosome. The elF4 complex consisting of four subunits, stabilizes the mRNA by binding to the cap (7-methyl guanosine, m7GpppN). 18,19 Its elF4G subunit is thought to establish a bridge between elF3 and elF4E. 20,21
Several lines of evidence suggest a role of eukaryotic initiation factors in tumor development. 22 NIH3T3 cells transfected with elF4E expression vectors undergo malignant phenotypic changes. 23 Moreover elF2α, elF4E, or elF4γ have been found to be overexpressed in transformed cell lines and in human tumors such as breast, lung, head, and neck cancers. 19,24-27 A different elF3 subunit (elF3-p40), from the one investigated in this study (EIF3S8), has recently been reported to be overexpressed in human breast and prostate cancer, underlining the concept that overexpression of eukaryotic initiation factors may be a common feature of different tumors. 28 A general increase in translation by overexpressed eukaryotic initiation factors is thought to accelerate cell growth leading to an early entrance of the cell into the cell cycle and consequently to an increased cell division rate. 22,29 We suggest that an augmented translation of oncogenic proteins may be another mechanism by which eukaryotic initiation factors promote tumor growth including that of seminomas. Functional studies are required to prove such a role or other functions for tumor development and progression.
EIF3S8 is located at 16p11.2. The possible causes of EIF3S8 overexpression in seminomas include gene amplification, translocation, or differential transcriptional regulation (for example by alterations of methylation). Instead of gains or amplifications losses of chromosomal material were found at 16p in seminomas in several comparative genomic hybridization and loss of heterozygosity studies. 30-32 Nothing is presently known concerning other possible mechanisms of EIF3S8 overexpression in seminomas. They will likewise have to be addressed in further studies.
Acknowledgments
We thank T. Pietsch (Institute of Neuropathology, Bonn University) for stimulating discussion; D. Schmidt and B. Koch for excellent technical assistance; and O. Landt (TIB Molbiol, Berlin, Germany) for the construction of the Light-Cycler hybridization probes.
Footnotes
Address reprint requests to Prof. Nicolas Wernert, M.D., Institute of Pathology, Laboratory of Molecular Pathology, University of Bonn, P.O. Box 2120, D-53011 Bonn, Germany. E-mail: wernert@meb.uni-bonn.de.
Supported by the BONFOR program (199 A/01) of the Faculty of Medicine, University of Bonn, Germany.
References
- 1.Murty VVVS, Chaganti RSK: A genetic perspective of male germ cell tumors. Semin Oncol 1998, 25:133-144 [PubMed] [Google Scholar]
- 2.Rothe M, Albers P, Wernert N: Loss of heterozygosity, differentiation and clonality in microdissected male germ cell tumors. J Pathol 1999, 188:389-394 [DOI] [PubMed] [Google Scholar]
- 3.Liang P, Pardee AB: Differential display of eucaryotic mRNA by means of the polymerase chain reaction. Science 1992, 257:967-971 [DOI] [PubMed] [Google Scholar]
- 4.Bauer D, Müller H, Reich J, Riedel H, Ahrenkiel V, Warthoe P, Strauss M: Identification of differentially expressed mRNA species by an improved display technique (DDRT-PCR). Nucleic Acids Res 1993, 21:4272-4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang P: Analysis of messenger RNA by differential display. A Laboratory Guide to RNA. Edited by PA Krieg. New York, Chichester, Brisbane, Toronto, Singapore, Wiley-Liss, Inc., 1996
- 6.von Deimling A, Bender B, Louis DN, Wiestler OD: A rapid and non-radioactive PCR based assay for the detection of allelic loss in human gliomas. Neuropathol Appl Neurobiol 1993, 19:524-529 [DOI] [PubMed] [Google Scholar]
- 7.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215:403-410 [DOI] [PubMed] [Google Scholar]
- 8.Vandenbunder B, Pardanaud L, Jaffredo T, Mirabel MA, Stéhelin D: Complementary patterns of expression of c-ets 1, c-myb and c-myc in the blood-forming system of the chick embryo. Development 1989, 107:265-274 [DOI] [PubMed] [Google Scholar]
- 9.Wernert N, Raes MB, Lasalle P, Gosselin B, Vandenbunder B, Stéhelin D: The c-ets 1 proto-oncogene is a transcription factor expressed in endothelial cells during tumor vascularization and other forms of angiogenesis in man. Am J Pathol 1992, 140:119-127 [PMC free article] [PubMed] [Google Scholar]
- 10.Wernert N, Gilles F, Fafeur V, Bouali F, Raes MB, Pyke C, Dupressoir T, Seitz G, Vandenbunder B, Stéhelin D: Stromal expression of c-ets 1 transcription factor correlates with tumor invasion. Cancer Res 1994, 54:5683-5688 [PubMed] [Google Scholar]
- 11.Cox KH, De Leon DV, Angerer LM, Angerer RC: Detection of mRNAs in sea urchin embryos by in situ hybridisation using asymmetric RNA probes. Dev Biol 1984, 101:485-502 [DOI] [PubMed] [Google Scholar]
- 12.Hilwig J, Gropp A: Staining of constitutive heterochromatin in mammalian chromosomes with a new fluorochrome. Exp Cell Res 1972, 75:122-126 [DOI] [PubMed] [Google Scholar]
- 13.Bieche I, Laurendeau I, Tozlu S, Olivi M, Vidaud D, Lidereau R, Vidaud M: Quantitation of MYC gene expression in sporadic breast tumors with a real-time reverse transcription-PCR assay. Cancer Res 1999, 59:2759-2765 [PubMed] [Google Scholar]
- 14.Totzke G, Sachinidis A, Vetter H, Ko Y: Competitive reverse transcription/polymerase chain reaction for the quantification of p53 and mdm2 mRNA expression. Mol Cell Probes 1996, 10:427-433 [DOI] [PubMed] [Google Scholar]
- 15.Milton RC: An extended table of critical values for the Mann-Whitney (Wilcoxon) two-sample statistic. J Am Stat Assoc 1964, 59:925-934 [Google Scholar]
- 16.Flynn A, Proud C: The role of elF4 in cell proliferation. Cancer Surv 1996, 27:293-310 [PubMed] [Google Scholar]
- 17.Asano K, Kinzy TG, Merrick WC, Hershey JWB: Conservation and diversity of eukaryotic translation initiation factor elF3. J Biol Chem 1997, 10:1101-1109 [DOI] [PubMed] [Google Scholar]
- 18.Moldave K: Eukaryotic protein synthesis. Annu Rev Biochem 1985, 54:1109-1149 [DOI] [PubMed] [Google Scholar]
- 19.Li BD, Liu L, Dawson M, De Benedetti A: Overexpression of eukaryotic initiation factor 4E (elF4E) in breast carcinoma. Cancer 1997, 79(Suppl 12):S2385-S2390 [PubMed] [Google Scholar]
- 20.Lamphear BJ, Kirchweger R, Skern T, Rhoads RE: Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (elF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J Biol Chem 1995, 270:21975-21983 [DOI] [PubMed] [Google Scholar]
- 21.Morley SJ, McKendrick L, Bushell M: Cleavage of translation initiation factor 4G (elF4G) during anti-Fas IgM-induced apoptosis does not require signalling through the p38 mitogen-activated protein (MAP) kinase. FEBS Lett 1998, 438:41-48 [DOI] [PubMed] [Google Scholar]
- 22.De Benedetti A, Harris AL: elF4E expression in tumors: its possible role in progression of malignancies. Int J Biochem Cell Biol 1999, 31:59-72 [DOI] [PubMed] [Google Scholar]
- 23.Lazaris-Karatzas A, Montine K, Sonenberg N: Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 1990, 345:544-547 [DOI] [PubMed] [Google Scholar]
- 24.Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, De Benedetti A: The proto-oncogene/translation factor elF4E: a survey of its expression in breast carcinomas. Int J Cancer 1995, 64(Suppl 1):S27-S31 [DOI] [PubMed] [Google Scholar]
- 25.Rosenwald IB: Upregulated expression of the genes encoding translation initiation factors elF-4E and elF-2alpha in transformed cells. Cancer Lett 1996, 102:113-123 [DOI] [PubMed] [Google Scholar]
- 26.Nathan CA, Liu L, Li BD, Abreo FW, Nandy I, De Benedetti A: Detection of the proto-oncogene elF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene 1997, 15(Suppl 5):S579-S584 [DOI] [PubMed] [Google Scholar]
- 27.Brass N, Heckel D, Sahin U, Pfreundschuh M, Sybrecht GW, Meese E: Translation initiation factor elF-4gamma is encoded by an amplified gene and induces an immune response in squamous cell lung carcinoma. Hum Mol Genet 1997, 6:33-39 [DOI] [PubMed] [Google Scholar]
- 28.Nupponen NN, Porkka K, Kakkola L, Tanner M, Persson K, Borg A, Isola J, Visakorpi T: Amplification and overexpression of p40 subunit of eukaryotic translation initiation. Am J Pathol 1999, 154:1777-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonenberg N: Translation factors as effectors of cell growth and tumorigenesis. Curr Opin Cell Biol 1993, 5:955-960 [DOI] [PubMed] [Google Scholar]
- 30.Al-Jehani RMA, Povey S, Delhanty JDA, Parrington JM: Loss of heterozygosity on chromosome arms 5q, 11p, 11q, 13q, and 16p in human testicular germ cell tumors. Genes Chromosom Cancer 1995, 13:249-256 [DOI] [PubMed] [Google Scholar]
- 31.Summersgill B, Goker H, Weber-Hall S, Huddart R, Horwich A, Shipley J: Molecular cytogenetic analysis of adult testicular germ cell tumours and identification of regions of consensus copy number change. Br J Cancer 1998, 305–313 [DOI] [PMC free article] [PubMed]
- 32.Ottesen AM, Kirchhoff M, De-Meyts ER, Maahr J, Gerdes T, Rose H, Lundsteen C, Petersen PM, Philip J, Skakkebaek NE: Detection of chromosomal aberrations in seminomatous germ cell tumours using comparative genomic hybridization. Genes Chromosom Cancer 1997, 4:412-418 [PubMed] [Google Scholar]