

Abstract
T cells seem to be responsible for liver damage in any type of acute hepatitis. Nevertheless, the importance of Kupffer cells (KCs) for T-cell-dependent liver failure is unclear. Here we focus on the role of KCs and tumor necrosis factor (TNF) production after T cell stimulation in mice. T-cell- and TNF-dependent liver injury were induced either by Pseudomonas exotoxin A (PEA), by concanavalin A (Con A), or by the combination of subtoxic doses of PEA and the superantigen Staphylococcus enterotoxin B (SEB). KCs were depleted by clodronate liposomes. Although livers of PEA-treated mice contained foci of confluent necrosis and numerous apoptotic cells, hardly any apoptotic cells were observed in the livers of Con A-treated mice. Instead, large bridging necroses were visible. Elimination of KCs protected mice from PEA-, Con A-, or PEA/SEB-induced liver injury. In the absence of KCs, liver damage was restricted to a few small necrotic areas. KCs were the main source of TNF. Hepatic TNF mRNA and protein production were strongly attenuated because of KC-depletion whereas plasma TNF levels were unaltered. Our results suggest that KCs play an important role in T cell activation-induced liver injury by contributing TNF. Plasma TNF levels are poor diagnostic markers for the severity of TNF-dependent liver inflammation.
Kupffer cells (KCs) are the most abundant macrophage (MΦ) population in the body. They are activated by invading bacteria, particularly by the cell wall constituent lipopolysaccharide (LPS) of gram-negative germs. As a consequence, KCs produce proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6. 1-3 TNF is an important immune mediator that has been implicated in the pathogenesis of septic shock in laboratory animals, 4 certain autoimmune diseases, 5,6 inflammatory organ damage, including heart 7 and acute experimental liver failure, 8 and other disorders. However, the importance of KCs for TNF-dependent diseases still remains unclear, and even the results of experiments on the role of KCs and splenic MΦ in LPS shock are controversial. 9,10 For functional studies, MΦ can be eliminated in experimental animals by administration of silica particles, by injection of gadolinium chloride (GdCl3), by carrageenan, or by the liposome-mediated macrophage suicide approach using liposome-encapsulated dichloromethylene-bisphosphonate (Cl2MBP). 11 The use of silica particles, GdCl3, or carrageenan has disadvantages because these drugs by themselves can activate KCs (all three), depress lymphocyte reactions (carrageenan), or induce a mitotic phenotype in hepatocytes (GdCl3). 11 Hence, any hepatoprotective potency may at least partially be explained by these activities. The liposome-mediated macrophage suicide approach is the most effective and best accepted method of MΦ depletion without stimulating the production of proinflammatory cytokines and/or nitric oxide by MΦ. 11 Cl2MBP liposomes eliminate MΦ depending on their dosage and their route of administration. 12
Liver damage occurring as a consequence of T cell activation is a serious health problem worldwide. The most common causes of life-threatening T-cell-mediated liver damage in humans are infections with hepatitis B or C viruses and autoimmune hepatitis. Therefore, different animal models of T-cell-mediated liver injury have been developed, including acute liver failure in mice induced by intravenous injection of the T-cell-stimulatory plant lectin concanavalin A (Con A). 13 Recently, we showed that T cells also contribute to liver injury induced by P. aeruginosa exotoxin A (PEA), an important virulence factor of the nosocomial gram-negative pathogen P. aeruginosa, in mice. 14 Furthermore, when given in small doses, PEA sensitizes mouse livers to superantigen-induced, T-cell-dependent liver injury, as shown after combined treatment of mice with a low dose of PEA together with Staphylococcus aureus enterotoxin B (SEB). 14,15 These results showed that the participation of T cells in liver cell destruction is a common mechanism. TNF plays a critical role in the aforementioned 14-22 and several other 23,26 mouse models of T cell activation-induced liver injury.
KCs are the primary source of intrahepatic TNF induced by either LPS 3 or PEA 14 in rodents. In the case of PEA, TNF production by KCs depends on the presence of T cells. 14 However, clear functional data on the role of KCs in T-cell-dependent liver injury and intrahepatic TNF production is still missing. Carrageenan was used to analyze KC function in a transgenic mouse model of hepatitis B, in which mice overexpressing the hepatitis B surface antigen (HBsAg) were injected with previously activated CD8-positive cytotoxic T lymphocytes directed against the viral antigen. Extensive pretreatment with carrageenan attenuated liver injury in this animal model. 27 However, activation of KCs by carrageenan 11 and induction of hepatocellular resistance before the hepatotoxic challenge might have been responsible for the protective effect. Moreover, controversial results exist for the hepatotoxic potency of Con A in mice pretreated with GdCl3, because both protective effects 28 and ineffectiveness 29 of GdCl3 pretreatment have been described. Hence, the aim of this study was to analyze the effect of KC depletion by Cl2MBP liposomes on T-cell-mediated hepatic damage and TNF production.
Materials and Methods
Mice
Male BALB/c mice were obtained from Charles River, Sulzfeld, Germany. Animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health. The legal requirements in Germany were met as well. Mice were maintained under controlled conditions (22°C, 55% humidity, 12-hour day/night rhythm) and were fed a standard laboratory chow (Altromin 1313, Altromin, Lage, Germany) ad libitum.
Animal Treatments
For depletion of KCs, BALB/c mice were injected with 100 μl of Cl2MBP liposomes intravenously 48 hours before challenge. 12 Cl2MBP liposomes were prepared as described previously, 30 and diluted in pyrogen-free saline. Cl2MBP was a gift of Roche Diagnostics, Mannheim, Germany. In control experiments, BALB/c mice were pretreated with saline instead of liposome-encapsulated Cl2MBP. Saline liposomes were not used because liposomes themselves block macrophage phagocytosis for certain periods of time. 30 The toxins were administered as follows: PEA (Sigma, St. Louis, MO), 85 μg/kg i.v.; Con A (Sigma), 20 mg/kg i.v.; SEB (Sigma), 2.5 mg/kg i.p.; recombinant murine (rmu)TNF (kindly provided by Dr. G. R. Adolf, Bender & Co., Vienna, Austria), various doses; recombinant murine interferon-γ (rmuIFN-γ) (kindly provided by Dr. G. R. Adolf), 50 μg/kg i.v. In case of co-administration, 10 μg/kg of PEA were given intravenously 15 minutes before SEB. rmuTNF was administered 60 minutes and rmuIFN-γ 75 minutes after toxin challenge.
Sampling of Material
Mice were lethally anesthetized with 150 mg/kg i.v. pentobarbital, containing 15 mg/kg heparin. From anesthetized mice blood was withdrawn for plasma cytokine determination or analysis of plasma transaminases. Livers were excised and divided into three parts. One small part was frozen in liquid nitrogen for preparation of RNA and subsequent real-time reverse transcriptase-polymerase chain reaction (RT-PCR), a second small part was embedded in Tissue Embedding Medium (Slee, Mainz, Germany) and frozen at −50°C for immunofluorescent staining and confocal laser imaging, and the rest of the livers was disintegrated in ice-cold Ripa buffer (150 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 50 mmol/L Tris, pH 7.4) containing protease inhibitors, DNase, and detergents (0.3% Triton X-100, 0.03% sodium dodecyl sulfate, 0.3% sodium deoxycholate), resulting in a 50% (w/w) liver homogenate. The homogenates were incubated on ice for 30 minutes and centrifuged at 15,000 × g for 30 minutes at 4°C. The supernatants were subjected to a second centrifugation at 15,000 × g for 20 minutes at 4°C. The resulting supernatants were then stored at −75°C for later quantification of intrahepatic TNF protein using an enzyme-linked immunosorbent assay. For histopathological determination of liver damage, mouse livers were perfused with 4% formalin/phosphate-buffered saline (PBS) via the portal vein before excision of the organ and storage at 4°C in 4% formalin/PBS.
Analysis of Plasma Transaminases and Plasma Cytokines
Liver injury was quantified by determination of plasma transaminase activities. The activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in plasma were determined using an automated procedure, according to Bergmeyer. 31 The plasma concentrations of TNF, IL-6, IL-2, and IFN-γ were determined with the help of specific enzyme-linked immunosorbent assays (ELISAs) purchased from PharMingen, Hamburg, Germany.
Hematoxylin and Eosin (H&E) Staining of Liver Sections
Formalin-fixed liver tissue was embedded in paraffin, sliced, and stained with H&E using a standard protocol.
Immunofluorescent Staining and Confocal Laser Imaging
Cryostat sections (12-μm thick) of livers were thawed onto poly-l-lysine-coated glass slides, air-dried, and fixed in acetone/methanol (1/1) for 10 minutes at 4°C before they were incubated in 3% bovine serum albumin/PBS for 30 minutes at room temperature. After the slides had been rinsed in PBS, incubation was continued with polyclonal rabbit anti-mouse TNF neat hyperimmune antiserum (Genzyme Virotech, Rüsselsheim, Germany; 1/750) together with a rat mAb directed against murine MΦ (clone BM 8, Dianova, Hamburg, Germany; 1/100) or a rat mAb directed against mouse CD4 (clone RM4–5, 1/50; PharMingen) in 3% bovine serum albumin/PBS overnight at 4°C. After rinsing with PBS, binding sites were detected by the use of appropriate secondary antibodies: fluorescein isothiocyanate-conjugated swine anti-rabbit IgG (1/30; DAKO, Hamburg, Germany) for staining of TNF, and Texas Red-conjugated goat anti-rat IgG (1/200; Dianova) for staining of MΦ or CD4+ cells. Secondary antibodies were diluted in 3% bovine serum albumin/PBS, and incubation was performed for 1 hour at room temperature. After rinsing with PBS, sections were coverslipped with 10% glycerol/PBS, pH 8.6. Sections processed for immunofluorescence were examined by confocal laser scanning microscopy (MRC 1000; Bio-Rad, Richmond, CA).
Real-Time RT-PCR for TNF mRNA in Liver Tissue
RNA was isolated from pieces of ∼25-mg liver tissue by the use of a RNA purification kit (Clontech, Heidelberg, Germany). For real-time RT-PCR, primers and probes were selected for murine β-actin and TNF (TIB Molbiol, Berlin, Germany). β-actin: 5′ TCACCCACACTGTGCCCATCTACGA; 3′ GGATGCCACAGGATTCCATACCCA. β-actin TaqMan probe: (FAM) TATGCTC (TAMRA) TCCCTCACGCCATCCTGCGT. TNF: 5′ TCTATGGCCCAGACCCTCAC; 3′ GACGGCAGAGAGGAGGTTGA. TNF TaqMan probe: (FAM) CTCAGATCATCTTCTCAAAATTCGAGTGACAAGC (TAMRA). Probes were 5′-labeled with 6-carboxyfluorescin (FAM) and internally with 6-carboxy-N,N,N′,N′-tetramethylrhodamine (TAMRA). Amplification and detection were done with an ABI 7700 system with the following profile: 2 minutes 50°C, 30 minutes 60°C, 5 minutes 95°C, and 45 cycles at 95°C for 15 seconds and 60°C for 1 minute. For more detailed information see User Bulletin 2 “ABI PRISM 7700 Sequence Detection System” by Perkin-Elmer’s Applied Biosystems (Perkin-Elmer, Emeryville, CA) describing the procedure of relative quantification of gene expression. β-actin was used as a housekeeping gene to normalize mRNA levels. The relative amounts of β-actin and TNF mRNA were determined and divided by each other. The resulting normalized values for TNF mRNA are arbitrary unitless numbers.
Quantification of Intrahepatic TNF
Liver lysates were prepared as described in Sampling of Material. They were directly used in a murine TNF ELISA kit purchased from R&D systems (Wiesbaden, Germany). Liver lysates were adjusted to equal protein concentrations after protein quantification by the Bradford method, as used in the Bio-Rad protein assay (Bio-Rad, Munich, Germany).
Statistical Analysis
The results were analyzed using the Student’s t-test or the Dunnett’s test. If variances were inhomogeneous, the data were transformed or analyzed using the Welsh test. Survival curves were compared using the log-rank test. All data in this study are expressed as the mean ± SEM. P < 0.05 was considered significant.
Results
Contribution of KCs to PEA-Induced Liver Injury and TNF Production
KCs turned out to be the major source of intrahepatic TNF after intravenous injection of PEA to mice and TNF is known to contribute to PEA-induced liver injury. 14 However, these correlating results did not prove the importance of KCs for PEA-induced liver injury. To this end, mice were depleted of KCs with the help of liposome-encapsulated Cl2MBP. 12 KC-deficient mice were treated with 85 μg/kg of PEA. Their susceptibility to PEA-induced liver injury was compared to that of control mice that had been pretreated with saline instead of liposome-encapsulated Cl2MBP. Liver damage was quantified by determination of plasma transaminase activities. Furthermore, livers were analyzed histopathologically. Pretreatment of mice with liposome-encapsulated Cl2MBP strongly inhibited PEA-induced release of transaminases. This protective effect of KC depletion was observed 12 hours and 17 hours after PEA challenge (Figure 1) ▶ . Histopathological examination of H&E-stained liver sections (Figure 2A) ▶ revealed that PEA induced single-cell necrosis of hepatocytes, morphologically resembling apoptosis, as well as focal confluent necrosis within 12 hours. The formation of apoptotic Councilman-like acidophil bodies was observed. Focal confluent necrosis with pale-stained hepatocytes predominantly appeared in the periportal areas. Immediately before death, ie, 17 hours after injection of PEA, livers were primarily injured. In these livers all stages of single-cell death could be observed, including formation of Councilman-like acidophil bodies, nuclear hyperchromasia, and karyorrhexis, in addition to pale-stained, confluent necrotic areas. In KC-depleted mice, PEA-induced hepatocellular death was strongly attenuated. Most strikingly, single-cell necrosis was not observed at all in livers of Cl2MBP liposome-pretreated, PEA-challenged mice. However, limited focal confluent necrosis was still detectable. Interestingly, limited focal confluent necrosis in the absence of KCs was not sufficient to induce significant transaminase release (Figure 1) ▶ .
Figure 1.

Importance of KCs for PEA-induced liver injury. Plasma transaminase activities were determined 12 and 17 hours after injection of PEA to BALB/c mice. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). *, P < 0.05.
Figure 2.
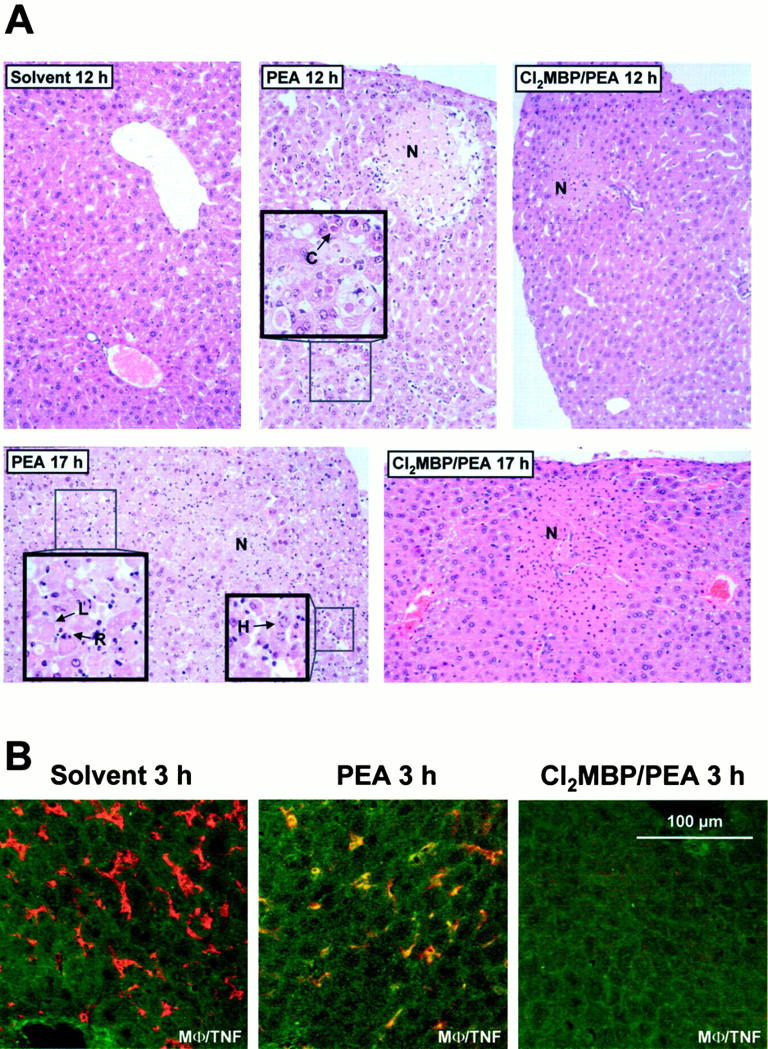
Importance of KCs for PEA-induced histopathological changes within mouse livers (A) and for intrahepatic production of TNF (B). Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. A: Liver sections were subjected to H&E staining and light microscopy 12 and 17 hours after injection of solvent (0.1% HSA) or PEA. C, Councilman-like acidophil body; N, confluent necrosis; R, karyorrhexis; L, karyolysis; H, nuclear hyperchromasia. Original magnification, ×90 (insets, ×180). B: Liver sections (12 μm) were subjected to immunofluorescent staining and confocal laser imaging 3 hours after injection of solvent (0.1% HSA) or PEA. Double staining was performed by use of antibodies specific for TNF (fluorescein isothiocyanate, green) together with antibodies against macrophages (Texas Red, red). Co-staining is represented by yellow fluorescence.
To determine a possible mechanism of how KCs might contribute to PEA-induced liver injury, it was tested whether depletion of these cells with the help of liposome-encapsulated Cl2MBP affects PEA-induced intrahepatic synthesis of TNF. Intrahepatic TNF was visualized in situ using immunofluorescent staining and confocal laser imaging 3 hours after challenge. As shown in Figure 2B ▶ , KCs were the predominant TNF-producing cells on injection of PEA and the absence of KCs prevented the induction of TNF within livers of PEA-treated mice. To get quantitative results, the amount of intrahepatic TNF in liver lysates was determined using an ELISA. As shown in Figure 3A ▶ , the induction of intrahepatic TNF by PEA could be ascertained quantitatively. We also attempted to confirm these results by quantifying intrahepatic TNF mRNA with the help of real-time RT-PCR. As shown in Figure 3B ▶ , the absence of KCs significantly inhibited the accumulation of TNF mRNA within mouse livers 2 hours after challenge with PEA.
Figure 3.
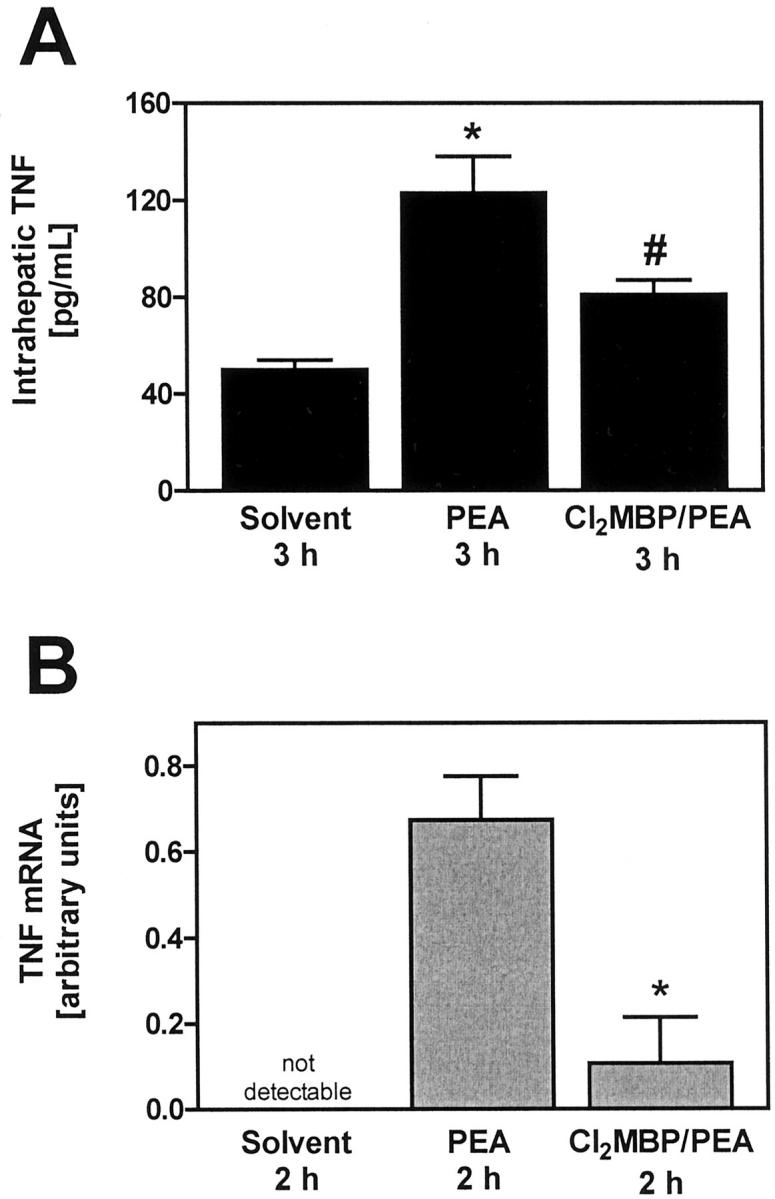
Importance of KCs for intrahepatic accumulation of TNF protein (A) and TNF mRNA (B) in PEA-treated mice. A: Intrahepatic TNF was quantified within liver lysates with the help of ELISA. Data are expressed as the mean ± SEM (n = 6). *, P < 0.05 versus solvent; #, P < 0.05 versus PEA 3 hours. B: Intrahepatic TNF mRNA was quantified by means of real-time RT-PCR specific for TNF. Real-time RT-PCR was also performed for the housekeeping gene product β-actin to normalize mRNA levels. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). #, P < 0.05 versus PEA.
To determine whether the missing TNF might be responsible for reduced sensitivity to PEA-induced liver injury, KC-depleted mice were treated with 85 μg/kg i.v. PEA followed by 0.5 μg/kg i.v. rmuTNF 1 hour later. This very low dose of rmuTNF, known to be nontoxic to mice when given alone, 32,33 was able to cause liver injury in otherwise resistant PEA-treated, KC-depleted mice, as determined by release of plasma transaminases 12 hours after injection of PEA (Table 1) ▶ . Hence, as little as 0.5 μg/kg of rmuTNF was able to overcome the protective effect of KC depletion. Even with 0.1 μg/kg of rmuTNF liver injury could be restored (data not shown).
Table 1.
Restoration of Sensitivity to PEA-Induced Liver Injury by Injection of rmuTNF to KC-Depleted Mice
| ALT [U/L] | AST [U/L] | |
|---|---|---|
| Solvent | 40 ± 5 | 80 ± 15 |
| PEA | 1,890 ± 570 | 2,540 ± 530 |
| Cl2MBP…PEA | 205 ± 15* | 520 ± 40* |
| Cl2MBP…PEA/rmuTNF | 3,310 ± 1,190 | 4,980 ± 1,780 |
Plasma transaminase activities were determined 12 hours after injection of solvent (0.1% HSA), 0.5 μg/kg of rmuTNF, 85 μg/kg of PEA, or combinations thereof to BALB/c mice. Depletion or KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). *, Protection from PEA-induced liver injury as a result of KC-depletion was significant (P < 0.05).
To prove whether KCs also contribute to lethality caused by PEA, survival of PEA-treated, KC-depleted mice was determined and compared to the survival of PEA-treated normal mice. Furthermore, survival was also determined with PEA-treated, KC-depleted mice that additionally received 0.5 μg/kg or 0.1 μg/kg of rmuTNF. As shown in Figure 4 ▶ , survival after injection of PEA was significantly prolonged by pretreatment with liposome-encapsulated Cl2MBP. The enhanced survival of the KC-depleted animals points to a prominent role of KCs for PEA-induced toxicity. Interestingly, 0.5 μg/kg and even as little as 0.1 μg/kg of rmuTNF significantly reduced the beneficial effect on survival conferred by KC depletion. Higher doses of rmuTNF caused death at earlier stages. For example, all mice died within 4 hours, if 10 μg/kg of rmuTNF were administered to KC-depleted, PEA-treated mice.
Figure 4.

Contribution of KC to PEA-induced lethality. Survival of normal and KC-depleted BALB/c mice was monitored after PEA injection. Groups of KC-depleted mice received rmuTNF in addition to PEA, as indicated (n = 3 to 5). Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. *, P < 0.05 versus PEA; #, P < 0.05 versus Cl2MBP… PEA.
Importance of KCs for PEA/SEB-Induced Liver Injury and TNF Production
The superantigen SEB exerts its toxicity through activation of a subset of T cells and subsequent production of TNF. 15,23,34-36 Prerequisite for the induction of apoptotic liver injury by SEB is the presence of inhibitors of transcription (GalN) 23 or translation (PEA). 14,15 Dendritic cells rather than macrophages are considered essential for SEB-induced clonal expansion of Vβ8-specific T cells 37 and local expression of cytokine mRNAs within the spleen. 38 Functional studies on the role of macrophages in SEB-induced toxicity are missing. We wondered whether KCs are required to mediate a SEB-triggered hepatotoxic response in mice and whether KC-depletion has an impact on the intrahepatic TNF response. We used a mixed intoxication model with a low dose of PEA as sensitizing agent plus SEB. This treatment causes T-cell- and TNF-dependent liver injury in mice within 12 hours, whereas the single toxins given alone are nontoxic. 14,15
KC-depleted mice were completely protected from PEA/SEB-induced liver injury as assessed by a significant inhibition of transaminase release 12 hours after challenge (Figure 5) ▶ .
Figure 5.

Importance of KCs for PEA/SEB-induced liver injury. Plasma transaminase activities were determined 12 hours after injection of PEA/SEB to BALB/c mice. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). *, P < 0.05.
To correlate this protection with the production of the most important toxic mediator of SEB, ie, TNF, we determined PEA/SEB-induced TNF plasma concentrations as well as locally produced TNF within the liver of normal and Cl2MBP liposome-pretreated mice 3 hours after challenge. At this stage, SEB-induced TNF plasma concentrations are maximal. 34,36 Depletion of KCs did not cause a significant reduction of the systemic TNF response to PEA/SEB (PEA/SEB: TNF, 60 ± 5 pg/ml; Cl2MBP/PEA/SEB: TNF, 85 ± 30 pg/ml) or to SEB alone (SEB: TNF, 25 ± 1 pg/ml; Cl2MBP/SEB: TNF, 115 ± 9 pg/ml). Hence, plasma TNF levels did not correlate with liver disease. Immunofluorescent staining of liver sections revealed that PEA/SEB induced hepatic TNF within 3 hours (Figure 6) ▶ . This TNF production was mainly co-localized with liver MΦ and also with some individual CD4+ cells. Depletion of KCs clearly prevented PEA/SEB-induced TNF production within the liver. Quantification of intrahepatic TNF in liver lysates corroborated these results. PEA/SEB-induced production of intrahepatic TNF was significantly attenuated in the absence of KCs (Figure 7A) ▶ . Similar results were obtained by real-time RT-PCR. TNF mRNA was clearly detectable within livers of PEA/SEB-treated mice (Figure 7B) ▶ . This induction of TNF mRNA was significantly impaired in livers of Cl2MBP liposome-pretreated mice (Figure 7B) ▶ .
Figure 6.

Importance of KCs for intrahepatic production of TNF in PEA/SEB-challenged mice. Liver sections (12 μm) were subjected to immunofluorescent staining and confocal laser imaging 3 hours after injection of solvent (0.1% HSA) or PEA/SEB. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Double staining was performed by use of antibodies specific for TNF (fluorescein isothiocyanate, green) together with antibodies against macrophages (Texas Red, red) or CD4+ cells (Texas Red, red). Co-staining is represented by yellow fluorescence.
Figure 7.
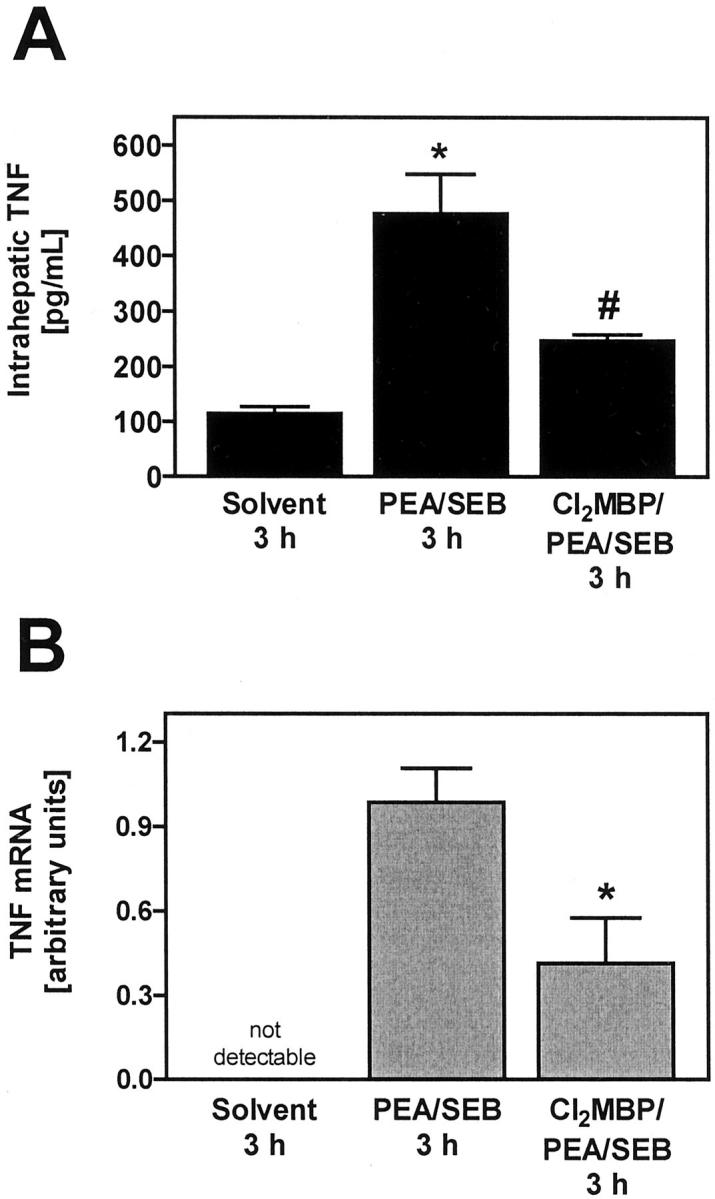
Importance of KCs for intrahepatic accumulation of TNF protein (A) and TNF mRNA (B) in PEA/SEB-treated mice. A: Intrahepatic TNF was quantified within liver lysates with the help of ELISA. Data are expressed as the mean ± SEM (n = 3). *, P < 0.05 versus solvent; #, P < 0.05 versus PEA/SEB 3 hours. B: Intrahepatic TNF mRNA was quantified by means of real-time RT-PCR specific for TNF. Real-time RT-PCR was also performed for the housekeeping gene product β-actin to normalize mRNA levels. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). #, P < 0.05 versus PEA/SEB.
To find out whether rmuTNF is able to re-establish liver injury in KC-depleted, PEA/SEB-treated mice, 0.5 μg/kg of rmuTNF was injected to Cl2MBP liposome-pretreated mice 1 hour after challenge with PEA/SEB. This treatment resulted in liver injury whereas in the absence of rmuTNF, animals were protected from liver damage (PEA/SEB: ALT, 1,535 ± 520 U/L; Cl2MBP… PEA/SEB: ALT, 340 ± 165 U/L*; Cl2MBP… PEA/SEB/rmuTNF: ALT, 2,320 ± 745 U/L; n = 3; * P < 0.05). Hence, TNF seems to be the KC-produced factor that mediates the hepatotoxic actions of KCs in the presence of PEA and SEB.
Importance of KCs for Con A-Induced Liver Injury and TNF Production
It is well established that intravenous injection of Con A induces CD4+ T-cell-, TNF-, and IFN-γ-dependent liver injury in mice 8 hours after challenge. 13,16-22 Early production of TNF 2 hours after Con A injection has been proven by determination of TNF in plasma, 16-18 and by Western blot analysis of liver tissue. 19 However, the identity of the TNF-producing cells in this model is unknown. Therefore, we wondered whether KCs are the primary sources of Con A-induced intrahepatic TNF and whether KCs play a role in Con A hepatitis.
Mice depleted of KCs were protected from Con A-induced liver injury, as assessed by significantly reduced plasma transaminase activities 8 hours after challenge (Figure 8) ▶ . Histopathological analysis of liver sections (Figure 9) ▶ revealed that Con A induced very large confluent areas of necrosis connecting several hepatic lobules (bridging necrosis). Single-cell necroses and the formation of Councilman-like acidophil bodies, ie, characteristic of livers in PEA-treated mice, was not observed at all. Pretreatment with Cl2MBP liposomes limited the spreading of focal confluent necroses induced by Con A, resulting in a more restricted pattern similar to the histopathology of livers of KC-depleted, PEA-treated mice (Figure 2A) ▶ . Again, the restricted formation of confluent necrotic areas was not sufficient to cause a significant increase in plasma transaminase activities.
Figure 8.

Importance of KCs for Con A-induced liver injury. Plasma transaminase activities were determined 8 hours after injection of Con A to BALB/c mice. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). *, P < 0.05.
Figure 9.

Importance of KCs for Con A-induced histopathological changes within mouse livers. Liver sections were subjected to H&E staining and light microscopy 8 hours after injection of solvent (saline) or Con A. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Original magnification, ×45.
To correlate the protective effect of Cl2MBP liposome pretreatment with alterations in the Con A-induced cytokine response, we determined the plasma levels of cytokines that are typically produced on Con A injection 2 hours after intervention. 13,16,17 As in the case of PEA/SEB, the amount of circulating TNF was not reduced in KC-depleted, Con A-challenged mice (Figure 10) ▶ . The levels of Con A-induced circulating IL-2 and IFN-γ were only partially reduced in KC-depleted mice (Figure 10) ▶ . However, the release of IL-6 was strongly impaired (Figure 10) ▶ . Hence, although KCs were the main cell population producing Con A-induced intrahepatic TNF (see below), depletion of KCs had no influence on Con A-induced plasma TNF levels. Immunofluorescent staining of liver sections revealed that Con A-induced intrahepatic TNF is mainly produced by KCs and to a lesser extent by some individual CD4+ T cells (Figure 11) ▶ . Markedly less TNF was observed in livers of Cl2MBP pretreated as compared to livers of normal mice, although, despite the absence of KCs, some individual CD4+ cells still produced TNF (Figure 11) ▶ . Hence, depletion of KCs inhibited Con A-induced intrahepatic TNF production. Quantification of intrahepatic TNF in liver lysates corroborated these results. Con A-induced production of intrahepatic TNF was significantly attenuated in the absence of KCs (Figure 12A) ▶ . Similar results were obtained by real-time RT-PCR. Intravenous injection of Con A clearly induced intrahepatic TNF mRNA within 2 hours (Figure 12B) ▶ . Pretreatment with Cl2MBP liposomes significantly diminished the intrahepatic induction of TNF mRNA by Con A (Figure 12B) ▶ .
Figure 10.

Contribution of KCs to the plasma concentrations of TNF, IL-2, IFN-γ, and IL-6, induced by Con A. Plasma cytokine levels were determined 2 hours after injection of Con A to BALB/c mice. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). *, P < 0.05.
Figure 11.
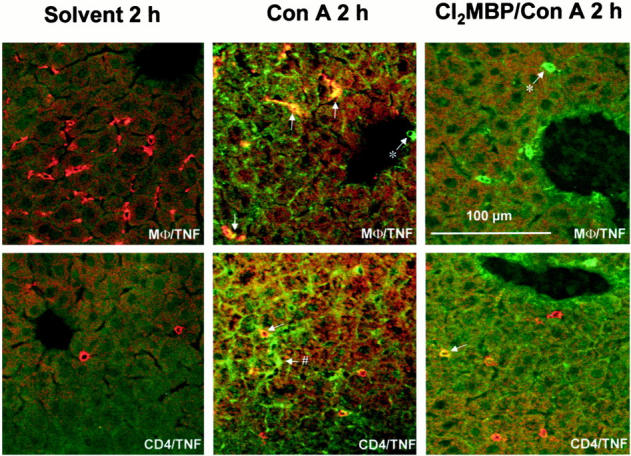
Importance of KCs for Con A-induced intrahepatic production of TNF. Liver sections (12 μm) were subjected to immunofluorescent staining and confocal laser imaging 2 hours after injection of solvent (saline) or Con A. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Double staining was performed by use of antibodies specific for TNF (fluorescein isothiocyanate, green) together with antibodies against macrophages (Texas Red, red) or CD4+ cells (Texas Red, red). Co-staining is represented by yellow fluorescence. *, TNF-positive cell that is not MΦ; #, TNF-positive cell that is not CD4+.
Figure 12.
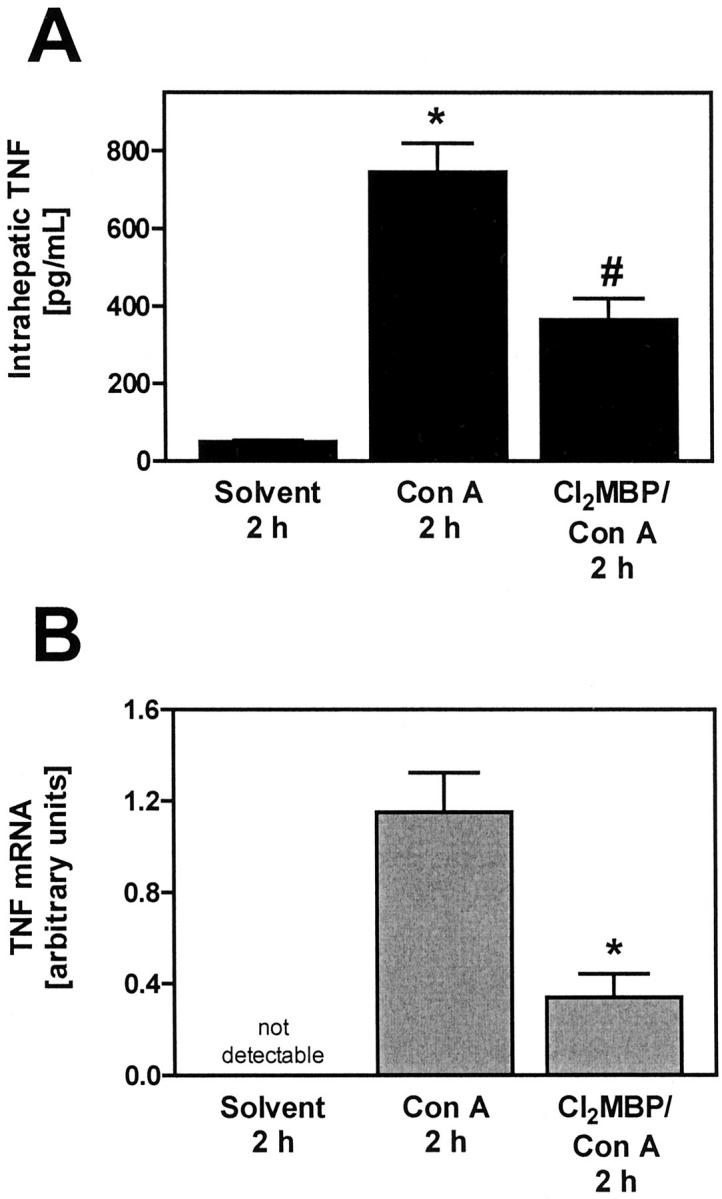
Importance of KCs for intrahepatic accumulation of TNF protein (A) and TNF mRNA (B) in Con A-treated mice. A: Intrahepatic TNF was quantified within liver lysates with the help of ELISA. Data are expressed as the mean ± SEM (n = 3). *, P < 0.05 versus solvent; #, P < 0.05 versus PEA 3 hours. B: Intrahepatic TNF mRNA was quantified by means of real-time RT-PCR specific for TNF. Real-time RT-PCR was also performed for the housekeeping gene product β-actin to normalize mRNA levels. Depletion of KCs was achieved by pretreatment with liposome-encapsulated Cl2MBP. Data are expressed as the mean ± SEM (n = 3). #, P < 0.05 versus PEA.
We attempted to restore the hepatotoxicity of Con A in Cl2MBP liposome-pretreated mice by administration of rmuTNF. However, bolus injections of as much as 10 μg/kg of rmuTNF did not abolish the protective effect of KC-depletion; even the combination of 10 μg/kg of rmuTNF together with 50 μg/kg of rmuIFN-γ was ineffective to render KC-depleted mice susceptible to Con A-induced liver injury (Con A: ALT, 1,370 ± 180 U/L; Cl2MBP/Con A: ALT, 110 ± 23 U/L; Cl2MBP/Con A/rmuTNF: ALT, 97 ± 25; Cl2MBP/Con A/rmuTNF/rmuIFN-γ: ALT, 156 ± 39 U/L). Because membrane-bound TNF is also involved in Con A hepatitis, 19 it is conceivable that this form of TNF might be required to mediate Con A-induced liver disease despite the presence of high amounts of soluble TNF and IFN-γ (see Discussion).
Discussion
In the present study we show that KCs clearly contribute to T-cell-dependent liver injury induced by either PEA, PEA/SEB, or Con A. This was demonstrated by the prevention of transaminase release into the plasma of Cl2MBP liposome-pretreated mice and by histopathological analysis. The amount and route of administration of Cl2MBP liposomes we used, ie, 100 μl/mouse i.v., was suitable to deplete KCs in mice. Splenic MΦ are partially eliminated by this treatment as well. 12 However, because Con A is able to induce liver injury in mice after removal of the spleen (M. Leist, personal communication), the spleen does obviously not play an important role in T-cell-dependent liver injury. Hence, it is very likely that the hepatoprotective effect of 100 μl of intravenously administered Cl2MBP liposomes is because of the depletion of the KCs.
Histopathological studies revealed that pretreatment with Cl2MBP liposomes strongly attenuated liver injury induced by PEA or Con A. In both cases, this pretreatment restricted liver damage to some areas of focal confluent necrosis, which were not sufficient to cause significant release of transaminases. PEA- and Con A-induced liver injury were morphologically different. Although the livers of PEA-treated mice contained foci of confluent necrosis and cells with apoptotic morphology (single-cell necrosis), which have been morphologically identified by others as well, 39 apoptotic cell death could not be observed morphologically in livers of Con A-treated mice. Instead, very large bridging necroses were visible. With respect to the lack of apoptosis induction after injection of Con A, our results seem to be in contrast to an earlier study, in which apoptotic bodies were found in livers of Con A-treated mice. 17 However, our results are in line with a previous study, in which only a sparse occurrence of apoptotic cells has been described. 40 As demonstrated by Künstle et al, 40 Con A hepatitis is characterized by the presence of intrahepatic internucleosomal DNA fragmentation, being characteristic of apoptotic cell death, and a concomitant lack of morphological features of apoptosis as well as of activation of caspase-3-like proteases. Also, Küsters et al 18 demonstrated intrahepatic DNA laddering on Con A injection and Trautwein et al, 41 showed terminal dUTP nick-end labeling-positive staining of hepatocytes. Hence, Con A seems to induce TNF-dependent hepatocellular death characterized by internucleosomal DNA fragmentation by nonapoptotic morphology and without caspase-3 activation. The rarely observed apoptotic bodies within livers of Con A-challenged mice 17,40 might be derived from nonparenchymal cells such as T cells that undergo activation-induced cell death. Accordingly, caspase-inhibitors failed to prevent Con A-induced liver injury, 40 whereas PEA-induced liver damage was prevented by these agents. 14 Hence, it seems that in the PEA model, TNF induces caspase-dependent hepatic apoptosis, while in the Con A model TNF-dependent, caspase-3-independent necrosis was observed. It should be pointed out that PEA-induced, rather than Con A-induced, liver injury morphologically resembles viral hepatitis, which is characterized by the presence of numerous Councilman bodies. The prevalence of apoptotic cell death in viral hepatitis or PEA-induced liver injury could be explained by mechanisms of sensitization toward TNF. These include synthesis of certain viral gene products in the case of viral hepatitis 42 and significant inhibition of protein synthesis in the case of PEA-induced liver damage. 43 Furthermore, cytotoxic lymphocytes may contribute to the apoptotic morphology in viral hepatitis 27,44 or PEA-induced liver injury, 14 eg, by producing perforin. Accordingly, the requirement of sensitizing events, such as strong inhibition of protein synthesis, for induction of apoptosis provides an explanation why Con A-induced liver injury proceeds with hardly any morphological signs of apoptosis, whereas in the presence of the transcriptional inhibitor GalN the livers of Con A-treated mice contain significant numbers of apoptotic cells and are characterized by strongly induced caspase-3-like activity 40 (Figure 13) ▶ .
Figure 13.
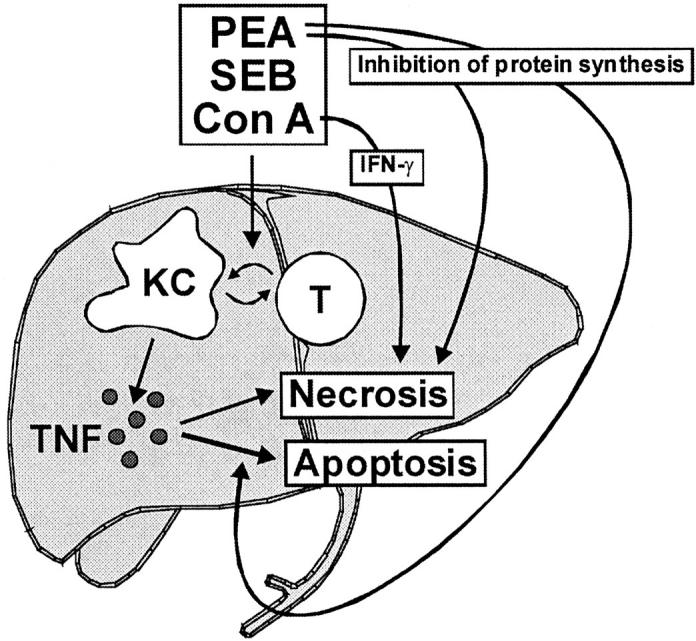
Proposed cascade of events deciding the severity and morphology of T-cell-dependent liver injury. Bacterial immunostimulators or Con A activates T cells and KCs. KCs are the main producers of TNF, which contributes to the enlargement of necrotic foci and to the induction of apoptotic cell death. Additional sensitizing events such as PEA-mediated inhibition of protein synthesis seem to be required for the induction of apoptotic morphology by TNF. In the absence of these events, eg, during Con A-induced TNF- and IFN-γ-mediated hepatitis, liver injury is characterized by large necroses without morphological signs of apoptosis.
We identified KCs as the main producers of intrahepatic TNF mRNA and TNF protein, thereby contributing to T-cell-mediated liver injury. Residual TNF production in case of Con A- or PEA/SEB-induced liver injury could be attributed to individual CD4+ cells. T cells have been described as sources of TNF in response to Con A and SEB by others as well. 38,45 Our results show that KCs cannot be replaced by T cells or professional antigen-presenting cells such as dendritic cells to induce relevant hepatotoxic amounts of TNF. In KC-depleted, toxin-treated mice, there was no residual TNF staining around the periportal areas, where most of the intrahepatic dendritic cells reside. 46,47 The functional importance of TNF production by KCs was indicated by the high susceptibility of KC-depleted, PEA- or PEA/SEB-treated mice to very low doses of exogenously administered rmuTNF. The relevance of KC-produced TNF for PEA toxicity was further supported by the prolonged survival of KC-depleted mice, which was negatively influenced by exogenously administered rmuTNF (Figure 4) ▶ . KC depletion was not life saving for PEA-treated mice, similar to T cell depletion that also prolonged survival of PEA-treated mice, but did not protect them from death at later stages. 14 This points to additional mechanisms leading to death in KC-depleted, PEA-treated animals at later stages, such as ongoing inhibition of protein synthesis in PEA-sensitive cells 43 and systemic TNF release, eg, by blood monocytes and by macrophages within other tissues, which are not affected by intravenously injected liposome-encapsulated Cl2MBP. 1,12,48 Indeed, systemic TNF levels, as determined in plasma 12 hours after injection of PEA to mice, 14 were even slightly enhanced in the absence of KCs (data not shown). It is conceivable that the clearance of PEA is disturbed in the absence of KC. Hence, continuously high levels of PEA might cause stronger systemic TNF release and more potent inhibition of protein synthesis and therefore lead to late-stage death in KC-depleted mice. TNF release into the circulation of KC-depleted mice might be further enhanced by missing KC-produced controllers such as IL-10 (see below). In contrast to the PEA model, rmuTNF and rmuIFN-γ were ineffective to restore the animals’ susceptibility to Con A. An explanation is the absence of contemporaneous strong inhibition of protein synthesis by Con A. 49 Furthermore, Con A-induced liver injury significantly depends on the expression of transmembrane TNF, 19 which cannot be simulated by a bolus injection of rmuTNF. Transmembrane TNF might have to be present to sustain Con A hepatitis in KC-depleted mice. Additionally, the macrophage cytokine IL-18 might have to be present to allow Con A toxicity in KC-depleted mice. An active role of IL-18 in Con A hepatitis has been described recently. 22
Despite depletion of KCs and strongly inhibited production of intrahepatic TNF, the plasma TNF levels were neither reduced in Con A- nor in PEA/SEB-treated mice. Leukocytes (lymphocytes, neutrophils, monocytes, and/or MΦ) in other tissues or in the blood stream may account for this discrepancy. The fact that the plasma TNF levels in Con A- or PEA/SEB-treated mice even tended to be slightly higher in the absence of KCs suggests a controlling function of KCs. A similar phenomenon has also been observed in LPS-challenged mice, 1 and in PEA-challenged mice (see above). IL-10 may be such a KC-produced controller of TNF synthesis. The liver is the main producer of LPS-induced IL-10, 50 and pretreatment of mice with anti-IL-10 mAb aggravates LPS lethality 51,52 as well as Con A-induced liver injury, 53 associated with enhanced production of TNF. 51-53 Additionally, it cannot be excluded that KCs participate in the removal of either the toxins PEA, SEB, or Con A or of TNF. In contrast to TNF, the plasma concentration of Con A-induced IL-6 strongly correlated with the presence or absence of KCs. However, the protection caused by KC depletion is probably not related to impaired IL-6 production, because IL-6-deficient mice were highly susceptible to Con A-induced liver injury. 54
In conclusion, T-cell- and TNF-dependent liver injury clearly depends on the activation of KCs. We propose that KCs contribute to the rapid spreading of liver necrosis and to the induction of apoptosis by producing TNF. Importantly, T-cell-, KC-, and TNF-mediated liver injury results in different morphological patterns indicating different mechanisms of hepatocellular death, ie, necrosis and “conventional” apoptosis or necrosis and “cryptic” caspase-3-independent apoptosis, as described by Künstle et al. 40 The development of morphologically visible apoptosis likely depends on the sensitization of the hepatocyte toward TNF. This observation will enforce further studies on the molecular and cellular prerequisites deciding the morphological character of liver disease.
Considering on the one hand the deleterious role of KCs and TNF in T cell-mediated liver injury, which can be induced by bacterial toxins or viruses, and on the other hand the protective role of KCs and TNF in host defense against bacteria 35,55-57 and viruses, 44,58 any therapeutic manipulations of KCs or TNF should be taken with great care. Therapeutic approaches in infectious liver diseases should rather be directed against TNF-dependent later events that solely play a role in the disease progression. However autoimmune hepatitis, which is not associated with infections, may be well controlled by neutralization of TNF or by attenuation of KC function.
Acknowledgments
We thank Dr. G. R. Adolf (Bender & Co., Vienna, Austria) for kindly providing recombinant murine TNF and IFN-γ, Dr. W. Neuhuber (Institute of Anatomy, University or Erlangen-Nürnberg, Erlangen, Germany) for experimental support regarding confocal laser scanning microscopy, and A. Agli and S. Heinlein for perfect technical assistance.
Footnotes
Address reprint requests to Prof. Dr. Gisa Tiegs, Institute of Experimental and Clinical Pharmacology and Toxicology, University of Erlangen-Nürnberg, Fahrstrasse 17, D-91054 Erlangen, Germany. E-mail: gisa.tiegs@pharmakologie.uni-erlangen.de.
Supported by Deutsche Forschungsgemeinschaft Grants Ti 169/3-4 and Ti 169/4-2 and by Interdisziplinäres Zentrum für Klinische Forschung, Universität Erlangen-Nürnberg.
References
- 1.Salkowski CA, Neta R, Wynn TA, Strassmann G, van Rooijen N, Vogel SN: Effect of liposome-mediated macrophage depletion on LPS-induced cytokine gene expression and radioprotection. J Immunol 1995, 155:3168-3179 [PubMed] [Google Scholar]
- 2.Luster MI, Germolec DR, Yoshida T, Kayama F, Thompson M: Endotoxin-induced cytokine gene expression and excretion in the liver. Hepatology 1994, 19:480-488 [PubMed] [Google Scholar]
- 3.Chensue SW, Terebuh PD, Remick DG, Scales WE, Kunkel SL: In vivo biologic and immunohistochemical analysis of interleukin-1 α, β and tumor necrosis factor during experimental endotoxemia. Kinetics, Kupffer cell expression, and glucocorticoid effects. Am J Pathol 1991, 138:395-402 [PMC free article] [PubMed] [Google Scholar]
- 4.Abraham E: Why immunomodulatory therapies have not worked in sepsis. Intensive Care Med 1999, 25:556-566 [DOI] [PubMed] [Google Scholar]
- 5.Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H, Woody JN: Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet 1994, 344:1105-1110 [DOI] [PubMed] [Google Scholar]
- 6.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ: A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor α for Crohn’s disease. Crohn’s disease cA2 study group. N Engl J Med 1997, 337:1029-1035 [DOI] [PubMed] [Google Scholar]
- 7.Deswal A, Bozkurt B, Seta Y, Parilti-Eiswirth S, Hayes FA, Blosch C, Mann DL: Safety and efficacy of a soluble P75 tumor necrosis factor receptor (Enbrel, etanercept) in patients with advanced heart failure. Circulation 1999, 99:3224-3226 [DOI] [PubMed] [Google Scholar]
- 8.Schümann J, Tiegs G: Pathophysiological mechanisms of TNF during intoxication with natural or man-made toxins. Toxicology 1999, 138:103-126 [DOI] [PubMed] [Google Scholar]
- 9.Tschaikowsky K, Brain JD: Effects of liposome-encapsulated dichloromethylene diphosphonate on macrophage function and endotoxin-induced mortality. Biochim Biophys Acta 1994, 1222:323-330 [DOI] [PubMed] [Google Scholar]
- 10.Groeneveld PHP, Claassen E, Kupfer CF, van Rooijen N: The role of macrophages in LPS-induced lethality and tissue injury. Immunology 1988, 63:521-527 [PMC free article] [PubMed] [Google Scholar]
- 11.Van Rooijen N, Sanders A: Elimination, blocking, and activation of macrophages: three of a kind? J Leukoc Biol 1997, 62:702-709 [DOI] [PubMed] [Google Scholar]
- 12.Van Rooijen N, Bakker J, Sanders A: Transient suppression of macrophage functions by liposome-encapsulated drugs. TIBTECH 1997, 15:178-185 [DOI] [PubMed] [Google Scholar]
- 13.Tiegs G, Hentschel J, Wendel A: A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 1992, 90:196-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schümann J, Angermüller S, Bang R, Lohoff M, Tiegs G: Acute hepatotoxicity of Pseudomonas aeruginosa exotoxin A in mice depends on T cells and TNF. J Immunol 1998, 161:5745-5754 [PubMed] [Google Scholar]
- 15.Schümann J, Bluethmann H, Tiegs G: Synergism of Pseudomonas aeruginosa exotoxin A with endotoxin, superantigen, or TNF results in TNFR1- and TNFR2-dependent liver toxicity in mice. Immunol Lett 2000, 74:165-172 [DOI] [PubMed] [Google Scholar]
- 16.Mizuhara H, O’Neill E, Seki N, Ogawa T, Kusunoki C, Otsuka K, Satoh S, Niwa M, Senoh H, Fujiwara H: T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med 1994, 179:1529-1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G: Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology 1995, 21:190-198 [DOI] [PubMed] [Google Scholar]
- 18.Küsters S, Gantner F, Künstle G, Tiegs G: Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996, 111:462-471 [DOI] [PubMed] [Google Scholar]
- 19.Küsters S, Tiegs G, Alexopoulou L, Pasparakis M, Douni E, Künstle G, Bluethmann H, Wendel A, Pfizenmaier K, Kollias G, Grell M: In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol 1997, 27:2870-2875 [DOI] [PubMed] [Google Scholar]
- 20.Ksontini R, Colagiovanni DB, Josephs MD, Edwards CK, III, Tannahill CL, Solorzano CC, Norman J, Denham W, Clare-Salzler M, MacKay SL, Moldawer LL: Disparate roles for TNF-α and Fas ligand in concanavalin A-induced hepatitis. J Immunol 1998, 160:4082-4089 [PubMed] [Google Scholar]
- 21.Künstle G, Hentze H, Germann PG, Tiegs G, Meergans T, Wendel A: Concanavalin A hepatotoxicity in mice: tumor necrosis factor-mediated organ failure independent of caspase-3-like protease activation. Hepatology 1999, 30:1241-1251 [DOI] [PubMed] [Google Scholar]
- 22.Faggioni R, Jones-Carson J, Reed DA, Dinarello CA, Feingold KR, Grunfeld C, Fantuzzi G: Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicity: role of tumor necrosis factor α and IL-18. Proc Natl Acad Sci USA 2000, 97:2367-2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gantner F, Leist M, Jilg S, Germann PG, Freudenberg MA, Tiegs G: Tumor necrosis factor-induced hepatic DNA fragmentation as an early marker of T cell-dependent liver injury in mice. Gastroenterology 1995, 109:166-176 [DOI] [PubMed] [Google Scholar]
- 24.Nagakawa J, Hishinuma I, Hirota K, Miyamoto K, Yamanaka T, Tsukidate K, Katayama K, Yamatsu I: Involvement of tumor necrosis factor-α in the pathogenesis of activated macrophage-mediated hepatitis in mice. Gastroenterology 1990, 99:758-765 [DOI] [PubMed] [Google Scholar]
- 25.Tsuji H, Harada A, Mukaida N, Nakanuma Y, Bluethmann H, Kaneko S, Yamakawa K, Nakamura S-I, Kobayashi K-I, Matsushima K: Tumor necrosis factor receptor p55 is essential for intrahepatic granuloma formation and hepatocellular apoptosis in a murine model of bacterium-induced fulminant hepatitis. Infect Immun 1997, 65:1892-1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishimura T, Ohta A: A critical role for antigen-specific Th1 cells in acute liver injury in mice. J Immunol 1999, 162:6503-6509 [PubMed] [Google Scholar]
- 27.Ando K, Moriyama T, Guidotti LG, Wirth S, Schreiber RD, Schlicht HJ, Huang S-N, Chisari FV: Mechanism of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J Exp Med 1993, 178:1541-1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamoto T, Maeda O, Tsuzuike N, Hara K: Effect of gadolinium chloride treatment on concanavalin A-induced cytokine mRNA expression in mouse liver. Jpn J Pharmacol 1998, 78:101-103 [DOI] [PubMed] [Google Scholar]
- 29.Knolle PA, Gerken G, Löser E, Dienes HP, Gantner F, Tiegs G, Meyer zum Büschenfelde KH, Lohse AW: Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 1996, 24:824-829 [DOI] [PubMed] [Google Scholar]
- 30.Van Rooijen N, Sanders A: Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Meth 1994, 174:83-93 [DOI] [PubMed] [Google Scholar]
- 31.Bergmeyer HU: Methods of Enzymatic Analysis, ed. 3. 1984, Verlag Chemie, Weinheim
- 32.Tiegs G, Wolter M, Wendel A: Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol 1989, 38:627-631 [DOI] [PubMed] [Google Scholar]
- 33.Tiegs G, Niehörster M, Wendel A: Leukocyte alterations do not account for hepatitis induced by endotoxin or TNFα in galactosamine-sensitized mice. Biochem Pharmacol 1990, 40:1317-1322 [DOI] [PubMed] [Google Scholar]
- 34.Miethke T, Wahl C, Heeg K, Echtenacher B, Krammer PH, Wagner H: T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: critical role of tumor necrosis factor. J Exp Med 1992, 175:91-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M, Mak TW: Mice deficient for the 55 kD tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993, 73:457-467 [DOI] [PubMed] [Google Scholar]
- 36.Nagaki M, Muto Y, Ohnishi H, Yasuda S, Sano K, Naito T, Maeda T, Yamada T, Moriwaki H: Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology 1994, 106:450-458 [DOI] [PubMed] [Google Scholar]
- 37.Koesling M, Rott O, Fleischer B: Macrophages are dispensable for superantigen-mediated stimulation and anergy induction of peripheral T cells in vivo. Cell Immunol 1994, 157:29-37 [DOI] [PubMed] [Google Scholar]
- 38.Bette M, Schäfer MKH, van Rooijen N, Weihe E, Fleischer B: Distribution and kinetics of superantigen-induced cytokine gene expression in mouse spleen. J Exp Med 1993, 175:47-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavlovskis OR, Voelker FA, Shackelford AH: Pseudomonas aeruginosa exotoxin in mice: histopathology and serum enzyme changes. J Infect Dis 1976, 133:253-259 [DOI] [PubMed] [Google Scholar]
- 40.Künstle G, Hentze H, Germann P-G, Tiegs G, Meergans T, Wendel A: Concanavalin A hepatotoxicity in mice: tumor necrosis factor-mediated organ failure independent of caspase-3-like protease activation. Hepatology 1999, 30:1241-1251 [DOI] [PubMed] [Google Scholar]
- 41.Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G: Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 1998, 114:1035-1045 [DOI] [PubMed] [Google Scholar]
- 42.Su F, Schneider RJ: Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor α. Proc Natl Acad Sci USA 1997, 94:8744-8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavlovskis OR, Shackelford AH: Pseudomonas aeruginosa exotoxin in mice: localization and effects on protein synthesis. Infect Immun 1974, 9:540-546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari F: Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 1996, 4:25-36 [DOI] [PubMed] [Google Scholar]
- 45.Cao Q, Batey R, Pang G, Russell A, Clancy R: IL-6, IFN-γ and TNF-α production by liver-associated T cells and acute liver injury in rats administered concanavalin A. Immunol Cell Biol 1998, 76:542-549 [DOI] [PubMed] [Google Scholar]
- 46.Markus AMA, Köckerling F, Neuhuber WA: Close anatomical relationships between nerve fibers and MHC class II-expressing dendritic cells in the rat liver and extrahepatic bile duct. Histochem Cell Biol 1998, 109:409-415 [DOI] [PubMed] [Google Scholar]
- 47.Thomson AW, Drakes ML, Zahorchak AF, O’Connell PJ, Steptoe RJ, Qian S, Lu L: Hepatic dendritic cells: immunobiology and role in liver transplantation. J Leukoc Biol 1999, 66:322-330 [DOI] [PubMed] [Google Scholar]
- 48.Qian Q, Jutila MA, van Rooijen N, Cutler JE: Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J Immunol 1994, 152:5000-5008 [PubMed] [Google Scholar]
- 49.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A: Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol 1995, 146:1220-1234 [PMC free article] [PubMed] [Google Scholar]
- 50.Barsig J, Küsters S, Vogt K, Volk H-D, Tiegs G, Wendel A: Lipopolysaccharide-induced interleukin-10 in mice: role of endogenous tumor necrosis factor-α. Eur J Immunol 1995, 25:2888-2893 [DOI] [PubMed] [Google Scholar]
- 51.Ishida H, Hastings R, Thompson-Snipes L, Howard M: Modified immunological status of anti-IL-10 treated mice. Cell Immunol 1993, 148:371-384 [DOI] [PubMed] [Google Scholar]
- 52.Marchant A, Bruyns C, Vandenabeele P, Ducarme M, Gerard C, Delvaux A, De Groote D, Abramovic D, Velu T, Goldman M: Interleukin-10 controls interferon-gamma and tumor necrosis factor production during experimental endotoxemia. Eur J Immunol 1994, 24:1167-1171 [DOI] [PubMed] [Google Scholar]
- 53.Louis H, Moine O, Peny MO, Quertinmont E, Fokan D, Goldman M, Deviere J: Production and role of interleukin-10 in concanavalin A-induced hepatitis in mice. Hepatology 1997, 25:1382-1389 [DOI] [PubMed] [Google Scholar]
- 54.Tagawa Y-I, Matthys P, Heremans H, Dillen C, Zaman Z, Iwakura Y, Billiau A: Bimodal role of endogenous interleukin-6 in concanavalin A-induced hepatitis in mice. J Leukoc Biol 2000, 67:90-96 [DOI] [PubMed] [Google Scholar]
- 55.Bodey GP, Bolivar R, Fainstein V, Jadeja L: Infections caused by Pseudomonas aeruginosa. Rev Infect Dis 1983, 5:279-313 [DOI] [PubMed] [Google Scholar]
- 56.Hultgren O, Eugster H-P, Sedgwick JD, Körner H, Tarkowski A: TNF/lymphotoxin-α double-mutant mice resist septic arthritis but display increased mortality in response to Staphylococcus aureus. J Immunol 1998, 161:5937-5942 [PubMed] [Google Scholar]
- 57.Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H: Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364:798-802 [DOI] [PubMed] [Google Scholar]
- 58.Ruby J, Bluethmann H, Peschon JJ: Antiviral activity of tumor necrosis factor (TNF) is mediated via p55 and p75 receptors. J Exp Med 1997, 186:1591-1596 [DOI] [PMC free article] [PubMed] [Google Scholar]