

Abstract
Programmed cell death plays critical roles in a wide variety of physiological processes during fetal development and in adult tissues. In most cases, physiological cell death occurs by apoptosis as opposed to necrosis. Defects in apoptotic cell death regulation contribute to many diseases, including disorders where cell accumulation occurs (cancer, restenosis) or where cell loss ensues (stroke, heart failure, neurodegeneration, AIDS). In recent years, the molecular machinery responsible for apoptosis has been elucidated, revealing a family of intracellular proteases, the caspases, which are responsible directly or indirectly for the morphological and biochemical changes that characterize the phenomenon of apoptosis. Diverse regulators of the caspases have also been discovered, including activators and inhibitors of these cell death proteases. Inputs from signal transduction pathways into the core of the cell death machinery have also been identified, demonstrating ways of linking environmental stimuli to cell death responses or cell survival maintenance. Knowledge of the molecular mechanisms of apoptosis is providing insights into the causes of multiple pathologies where aberrant cell death regulation occurs and is beginning to provide new approaches to the treatment of human diseases.
Apoptosis is a morphological phenomenon. As viewed with the assistance of the light (or, preferably, the electron) microscope, the characteristics of the apoptotic cell include chromatin condensation and nuclear fragmentation (pyknosis), plasma membrane blebbing, and cell shrinkage. Eventually, the cells breaks into small membrane-surrounded fragments (apoptotic bodies), which are cleared by phagocytosis without inciting an inflammatory response. The release of apoptotic bodies is what inspired the term “apoptosis” from the Greek, meaning “to fall away from” and conjuring notions of the falling of leaves in the autumn from deciduous trees. 1
Apoptosis Is Caused by Caspases
What causes these morphological changes that we recognize as apoptosis and the biochemical changes often associated with this phenomenon? The answer is proteases. Specifically, activation of a family of intracellular cysteine proteases which cleave their substrates at aspartic acid residues, known as caspases for Cysteine Aspartyl-specific Proteases. 2 These proteases are present as inactive zymogens in essentially all animal cells, but can be triggered to assume active states, generally involving their proteolytic processing at conserved aspartic acid (Asp) residues. During activation, the zymogen pro-proteins are cleaved to generate the large (∼20 kd) and small (∼10 kd) subunits of the active enzymes, typically liberating an N-terminal prodomain from the processed polypeptide chain. The active enzymes consist of heterotetramers composed of two large and two small subunits, with two active sites per molecule. 3,4 Analysis of the structures of the active sites of these enzymes, experiments with combinatorial peptide libraries, and other data suggest that caspases recognize the Asp residues they cleave within the context of tetrapeptide motifs, where the most proximal (N-terminal) residue recognized is designated P4 (position #4) and target Asp is P1 (position #1), and where cleavage occurs at the peptidyl bond distal (C-terminal) to the targeted Asp. This information about the structures and mechanisms of caspases has been exploited for developing small-molecule inhibitors, which are finding their way into clinical trials for stroke, liver failure, inflammatory diseases, and a wide variety of other ailments. 5,6
The observation that caspases cleave their substrates at Asp residues and are also activated by proteolytic processing at Asp residues makes evident that these proteases collaborate in proteolytic cascades, whereby caspases activate themselves and each other. In humans and mice, approximately 14 caspases have been identified. They can be subgrouped according to either their amino acid sequence similarities or their protease specificities. From a functional perceptive, it is useful to view the caspases as either upstream (initiator) caspases or downstream (effector) caspases. 7 The proforms of upstream initiator caspases possess large N-terminal pro-domains, which function as protein interaction modules, allowing them to interact with various proteins that trigger caspase activation. In contrast, the proforms of downstream effector caspases contain only short N-terminal prodomains, serving no apparent function. Downstream caspases are largely dependent on upstream caspases for their proteolytic processing and activation. Accordingly, the sequence of the cleavage sites separating the large and small subunits of the zymogen forms of the effector caspases generally match the preferred tetrapeptide specificities of the upstream initiator caspases. Similarly, examination of the cleavage sites of multiple cellular proteins, which have been identified as caspase substrates and which are known to undergo processing during apoptosis, reveals (in most instances) coincidence with the preferred tetrapeptide sequences cleaved by the effector caspases. 8 These substrates of effector caspases include protein kinases (often separating the autorepressing regulatory domains from catalytic domains) and other signal transduction proteins, cytoskeletal and nuclear matrix proteins, chromatin-modifying (eg, polyADP ribosyl polymerase) and DNA repair proteins, and inhibitory subunits of endonucleases (CIDE family proteins). 3,4,7
Though most caspases are directly involved in cell death, a few are not, at least in mammals and higher eukaryotes. A subgroup of caspases, including caspase-1,- 4, and -5 in humans, is involved in processing of pro-inflammatory cytokines such as pro-interleukin-1β (pro-IL-1β) and pro-IL-18. Unlike the effector caspases, which induce apoptosis, the tetrapeptide specificities of these cytokine-processing proteases do not match the cleavage sites of most of the proteins known to undergo cleavage during apoptosis, but they do coincide with the sequences of the cleavage sites within pro-cytokines. 6,7
Caspase Activation Mechanisms
A diversity of mechanisms exists for activating initiator caspases, thus setting the wheels of the apoptotic machinery in motion. However, fundamentally, the biochemical mechanisms appear to be remarkably similar and can be explained by a single model, known as the induced proximity model. 9 The induced proximity model is predicated on the empirical observation that the zymogen forms of unprocessed caspases are not entirely inactive but rather possess weak protease activity (measured in some cases at ∼1% of the fully active enzymes). When brought into close apposition through protein interactions, the zymogens can trans-process each other, producing the fully active proteases.
Though many pathways for activating caspases may exist, only two have been elucidated in detail. One of these centers on tumor necrosis factor (TNF) family receptors, which use caspase activation as a signaling mechanism, thus connecting ligand binding at the cell surface to apoptosis induction. 7,10,11 The other involves the participation of mitochondria, which release caspase-activating proteins into the cytosol, thereby triggering apoptosis. 12,13 The death receptor and mitochondrial pathways for caspase activation are sometimes referred to as the extrinsic and intrinsic apoptosis pathways (Figure 1) ▶ , respectively, though this is an oversimplification. Also, though commonly viewed as separate pathways and capable of functioning independently, cross-talk can occur between these pathways at multiple levels, depending on the repertoire of apoptosis-modulating proteins expressed.
Figure 1.

Pathways for caspase activation. Two of the major pathways for caspase activation in mammalian cells are presented, the extrinsic (left) and intrinsic (right). The extrinsic pathway can be induced by members of the TNF family of cytokine receptors, such as TNFR1 and Fas. These proteins recruit adapter proteins to their cytosolic DDs, including Fadd, which then binds DED-containing pro-caspases, particularly pro-caspase-8. The intrinsic pathway can be induced by release of cytochrome c from mitochondria, induced by various stimuli, including elevations in the levels of pore-forming pro-apoptotic Bcl-2 family proteins such as Bax. In the cytosol, cytochrome c binds and activates Apaf-1, allowing it to bind and activate pro-caspase-9. Active caspase-9 (intrinsic) and caspase-8 (extrinsic) have been shown to directly cleave and activate the effector protease, caspase-3. Other caspases can also become involved in these pathways (not shown); thus, the schematic represents an oversimplification of the events that occur in vivo.
A wide variety of experimental evidence, including gene ablation (knockout) experiments in mice, has demonstrated that caspase-8 represents the apical caspase in the TNF family death receptor pathway, whereas caspase-9 serves as the apical caspase of the mitochondrial pathway. 14-17 With regard to the extrinsic pathway, a network of protein interactions, involving adaptor proteins such as Fadd (Mort1), indirectly links the cytosolic domains of TNF family death receptors such as Fas (Apo1/CD95) to the zymogen forms of caspase-8, resulting in recruitment of pro-caspase-8 to liganded death receptor complexes and causing caspase-8 activation through the induced proximity mechanism. 18-22 In the case of the intrinsic pathway, release of cytochrome c from mitochondria triggers caspase activation by binding to the caspase-activating protein Apaf-1. 23 The Apaf-1 protein normally resides in an inactive conformation in the cytosol, but on binding cytochrome c, an ATP/dATP-binding oligomerization domain within this protein mediates Apaf-1 aggregation. 24,25 The oligomerized complex then binds pro-caspase-9, and facilitates trans-processing of caspase-9 zymogens via the induced proximity mechanism. 26 However, unlike caspase-8, where the N-terminal prodomain of the zymogen is cleaved off and the active protease is released into the cytosol, 20 the caspase-9 enzyme must remain bound to Apaf-1 for full activity, and its N-terminal prodomain is retained. 27,28
Protein Domains Associated with Apoptosis Regulation
The proteins that directly control the intrinsic, extrinsic, and other less understood caspase activation pathways often exist as families that can be recognized based on their amino acid sequence and/or structural similarity. Moreover, interactions among these proteins are commonly mediated by domains that are intimately associated with apoptosis regulation, including caspase-associated recruitment domains (CARDs), death domains (DDs), death effector domains (DEDs), Bcl-2 homology (BH) domains of Bcl-2 family proteins, baculovirus inhibitor of apoptosis proteins (IAP) repeat (BIR) domains of IAP family proteins, and NB-ARC domains representing the nucleotide-binding oligomerization domains of CED-4/Apaf-1 family proteins (Table 1) ▶ . A summary of the proteins that constitute the known members of these families of apoptosis regulators follows, along with information about their mechanisms and some examples of their relevance to diseases.
Table 1.
Domains Associated with Apoptosis
| 1. Caspase (catalytic) Domains |
| 2. Death Domains (DDs) |
| 3. Death Effector Domains (DEDs) |
| 4. Caspase-Associated Recruitment Domains (CARDs) |
| 5. BIR Domains (IAPs) |
| 6. Bcl-2 Homology (BH) Domains |
Death Domain Proteins
The DD is a protein interaction module consisting of a compact bundle of six α-helices. 29 DDs bind each other, probably forming oligomers of unknown stoichiometry. Specificity for partner selection among DDs is dictated by differences in surface residues. Several of the members of the TNF family of cytokine receptors contain DDs in their cytosolic regions, including TNFR1, Fas (Apo1), DR3 (Apo2), DR4 (TrailR1), DR5 (TrailR2), DR6 in humans, mice, and probably other mammals (Figure 2) ▶ . The p75 nerve growth factor receptor (p75-NGFR) also contains a modified (type II) DD 30 and has been reported to induce apoptosis under some circumstances. 31 The TNF family receptors, TNFR1, DR3, and DR6, are known to bind an adapter protein, Tradd, via its homologous DD. 32-34 The DD of Tradd is capable of binding certain other DD-containing proteins, including the adapter protein Fadd. The Fadd (Mort1) protein contains two protein interaction modules, a DD and a DED. Fadd links TNF family death receptors to caspases, using its DD to bind Tradd or to interact directly with the cytosolic DD of the TNFR family member Fas, and employing its DED to bind DED-containing caspases (see below). Experimental evidence has demonstrated the presence of Fadd within the receptor complexes of all known DD-containing members of the TNF family except p75-NGFR. Thus, this protein plays a central role in linking caspases to TNF family death receptors, a notion borne out by gene ablation studies in mice, which have demonstrated an inability of TNFR1, Fas, and other death receptors tested thus far to induce apoptosis in the absence of Fadd. 35-38
Figure 2.


Human death domain (DD) family proteins. The domain arrangements of the known members of the human DD family are presented. Numbers represent amino acid residue positions.
An analogous mechanism for recruiting pro-caspases to death receptor complexes has been revealed in the case of Raidd (Cradd). This adapter protein contains a DD in combination with a CARD, allowing it to bind the corresponding CARD found within the prodomain of pro-caspase-2. 39,40 Based on the aforementioned Fadd knockout studies, however, it remains questionable how important this alternative pathway for caspase activation is.
Additional DD-containing proteins have been implicated in apoptosis, such as the DAP kinase, which modulates apoptosis induction by TNF family death receptors through mechanisms that are not understood. 41 This kinase has been implicated in suppression of metastasis. Several cytoskeleton-associated ankryin family proteins contain DDs, but their relevance to apoptosis remains uncertain. Given evidence, however, that caspase-8 activation is triggered by suspension of adherent epithelial cells, 42,43 an event that disturbs the cytoskeleton, it is intriguing to speculate a possible role in the phenomenon of anoikis (ie, apoptosis induced by depriving cells of integrin-mediated attachments to extracellular matrix). Avoidance of anoikis represents an important aspect of tumor invasion, metastasis, and angiogenesis. 44 It is also fundamental to correct positioning of cells during development, possibly accounting for the embryonic lethality of Fadd and caspase-8 gene ablation in mice.
DDs, however, are not always involved in caspase activation or apoptosis induction. In fact, DDs have been found in proteins involved in two other receptor signaling systems, namely Toll family receptors and UNC family receptors. Nevertheless, some of the signal transduction pathways in which non-caspase-activating DD proteins are involved at least indirectly regulate apoptosis through effects on NF-κB, suppressing rather than inducing apoptosis. For example, the RIP protein binds Tradd and activates kinases that induce degradation of IkB, thus releasing NF-κB so that it can translocate to the nucleus and fulfill its function as a transcription factor. 45-47 Among NF-κB-inducible genes are several that block apoptosis, including anti-apoptotic Bcl-2 family members Bfl-1 (A1) and Bcl-X, and IAP family member cIAP2 and possibly cIAP1 and XIAP. 48,50-54 The dual function of Tradd, as a partner for both caspase-activator Fadd and NF-κB activator RIP, causes many of the TNF family receptors to nullify their own apoptosis-inducing activity. Thus, TNFR1, DR3, and DR6 are uncertain apoptosis inducers unless NF-κB induction is inhibited. 55,56 In contrast, Fas and the Trail receptors DR4 and DR5 only rarely activate NF-κB, probably because these receptor complexes contain Fadd but not Tradd. 20,37,38,57 In fact, it is because Trail receptors (DR4 and DR5) do not induce NF-κB-mediated pro-inflammatory responses in vivo that the Trail ligand is under consideration for clinical use in the treatment of cancer, whereas NF-κB-inducing TNF-α proved to be unacceptable.
Multiple mechanisms for modulating signaling by TNF family death receptors probably exist. One that acts directly on DDs involves the silencer of death domains protein (SODD), also know as BAG4. 58,59 SODD contains a C-terminal BAG domain that allows it to bind Hsc70/Hsp70 family molecular chaperones. 59 An N-terminal domain in SODD mediates its interactions with the DDs of TNFR1 and DR3. Overexpression of SODD prevents TNFR1 and DR3 from spontaneously aggregating and signaling in the absence of ligand. 58 Though unproven, it has been suggested that recruitment of Hsp70/Hsc70 to these death receptors induces conformational changes in the DD that prevent self-oligomerization until the receptors are appropriately triggered by cognate ligands. 60
Another resistance mechanism has been attributed to FAP, a protein tyrosine phosphatase that uses PDZ domains to interact with the C-termini of Fas and p75-NGFR, suppressing apoptosis signaling by these receptors through uncertain mechanisms. 61,62 Overexpression of FAP-1 in tumors has been associated with resistance to Fas-induced apoptosis. 63,64
Defects in the regulation, structure, or function of DD proteins are associated with multiple human diseases. For example, elevations in Fas expression are induced by hypoxia in cultured cardiomyocytes. 65 Inappropriate expression of Fas and Fas ligand (FasL) on lymphocytes and other immune cells has also been documented in patients with HIV infection and has been implicated in the loss of lymphocytes that characterizes this immunodeficiency syndrome. 66 Conversely, hereditary mutations in the DD of the FAS (APO1) gene cause an autoimmune lymphoproliferative syndrome in humans and mice. 67,68 The accumulation of autoreactive lymphocytes in these patients can presumably be explained by the important role the Fas plays in ensuring death of potentially autoreactive immune cells (through activation-induced lymphocyte apoptosis) and in down-regulation of lymphocyte numbers after immune responses. 69,70 Somatic mutations and deletions of the FAS gene have also been described in some malignancies, affording cancer cells resistance to immune-mediated attack. 71 Production of a soluble version of Fas has also been associated with the autoimmune disease lupus. This secreted isoform of Fas lacking the transmembrane domain is generated by alternative mRNA splicing, and increases in levels in association with exacerbation of symptoms in patients with lupus. 72 Soluble Fas competes with membrane-associated Fas for binding to ligand, thus interfering with FasL-mediated apoptosis. Similarly, soluble FasL has been shown to interfere with some Fas-mediated responses relevant to immune responses against tumors, 73 probably because FasL is normally expressed on the surface of cytolytic T cells (CTLs), where it can induce higher-order aggregation (cross-linking) of Fas on target cells. A novel member of the TNF family that is also overexpressed in some tumors, DcR3, encodes a decoy receptor that competes with Fas for binding of FasL. 74 Additionally, three decoy receptors have been identified for the death ligand, Trail, which protect DR4- and DR5-expressing cells from Trail-induced apoptosis. 75-77 Surprisingly, it seems that most normal cells express sufficient levels of Trail decoy receptors to be spared from apoptosis, whereas many tumor cells do not. 78,79 Finally, p53 has been reported to induce transcription of the death receptors Fas and DR5 in some types of tumor cells, thus linking the extrinsic pathway for apoptosis to genotoxic injury in specific cell contexts. 80
Death Effector Domain (DED) Proteins
The structure of the DED is similar to the DD, comprised of 6 α-helices. 81 DEDs are found in the initiator caspases, caspase-8 and -10 in humans (a caspase-10 orthologue has yet to be identified in mice). The prodomain regions of pro-caspase-8 and -10 contain two tandem DEDs, which are responsible their interactions with the DED of Fadd, and thus mediate their recruitment to death receptor complexes. Multiple DED-containing modulators of apoptosis have been identified (Figure 3) ▶ . Flash, for example, is a large protein containing two DED-like domains which reportedly enhances caspase-8 activation by Fas. DEDD (DEFT) is another DED-containing protein which reportedly enhances Fas-induced apoptosis. DEDD contains an N-terminal DED and a C-terminal histone-like domain. 82,83 DEDD is present in the cytosol, but during Fas-induced apoptosis it translocates to nuclei in a caspase-dependent manner, localizing to nucleoli and possibly shutting off ribosomal RNA gene transcription. 82 DEDD associates with Fadd, and to some extent pro-caspase-8, via DED-mediated interactions. 82,83
Figure 3.

Death effector domain (DED) family proteins. The domain arrangements of some of the known members of the DED family are presented. Numbers represent amino acid residue positions. Included in the diagram are the Drosophila caspase, Dronc, and the viral DED proteins from Kaposi’s sarcoma virus and MCV.
In contrast to Flash and DEDD (DEFT), which enhance Fas-induced apoptosis, the DED-containing protein Flip (also known as Flame, CASH, Clarp, MRIT, Casper, I-Flice, Usurpin) is capable of suppressing caspase-8 activation by Fas and other death receptors. Flip shares extensive amino acid sequence similarity with pro-caspase-8 and -10, containing two N-terminal DEDs followed by a pseudo-caspase domain that lacks critical residues required for protease activity, including the catalytic cysteine. 84,85 Flip associates with pro-caspase-8 and also competes with pro-caspases-8 and -10 for binding to Fadd, thus squelching death receptor signaling. Interestingly, some tumors have been reported to contain inappropriately elevated levels of Flip, rendering them resistant to apoptosis induction by Fas-expressing CTLs. 86 Though controversial, Flip-mediated resistance to Fas may even permit tumor cells to tolerate expressing FasL, using this death ligand as a weapon against neighboring normal cells and triggering apoptosis of immune cells. 87-89 Interestingly, some viruses also encode DED-containing proteins analogous to Flip that similarly suppress apoptosis induced by Fas. 90-93 By thwarting Fas-induced apoptosis, viruses presumably can avoid CTL-mediated eradication of the host cells they infect, allowing more time for viral replication.
Decreased expression of the DED-containing apoptosis suppressor, Flip (Usurpin), occurs in myocardial tissue damaged by ischemia-reperfusion injury, 96 suggesting increased sensitivity to apoptosis induction via the death receptor pathway.
In contrast to Flip, which is a soluble cytosolic protein, membrane-anchored DED-containing proteins have also been identified that are capable of modulating caspase-8 activation. Bap31 resides in the membranes of the endoplasmic reticulum (ER), possessing three putative membrane-spanning domains and a cytosolic DED-like domain. 94 BAR is found in the membranes of mitochondria and ER, with the bulk of the protein oriented toward the cytosol. The BAR protein contains a C-terminal membrane-anchoring domain and a DED which binds pro-caspase-8 and -10, suppressing Fas-induced apoptosis. 95 Both Bap31 and BAR contain additional domains that mediate their direct or indirect association with anti-apoptotic members of the Bcl-2 family such as Bcl-2 and Bcl-XL, thus defining points of cross-talk in the extrinsic (death receptor) and intrinsic (mitochondrial) pathway.
CARD Family Proteins
Several pro-caspases contain N-terminal CARDs in their prodomains, including caspases 1, 2, 4, 5, and 9 in humans and caspases 1, 2, 9, 11, and 12 in mice (Figure 4) ▶ . (To date, orthologues of human caspases 4 and 5 have not been observed in mice and conversely orthologues of murine caspases 11 and 12 have not been found in humans.) Roles for CARD-carrying caspases in diseases have been revealed through gene ablation studies in mice. For example, caspase-1 knockout mice exhibit marked resistance to endotoxin-induced sepsis. 97 Caspase-1, as well as caspase-2 and caspase-11, knockout mice also suffer less tissue loss in stroke models. 98-100 Inhibition of caspase-1 also slows progression in a mouse model of Huntington’s disease. 101 In addition, cells from caspase-12 knockout mice are resistant to apoptosis induced by amyloid β-peptide, 102 a finding of potential relevance to Alzheimer’s disease. In this regard, caspase-12 appears to be associated with the endoplasmic reticulum (ER) and becomes specifically activated by ER stress, thus linking ER damage to a caspase activation pathway independently of the mitochondrial (cytochrome c) and death receptor (TNF family) pathways. 102
Figure 4.
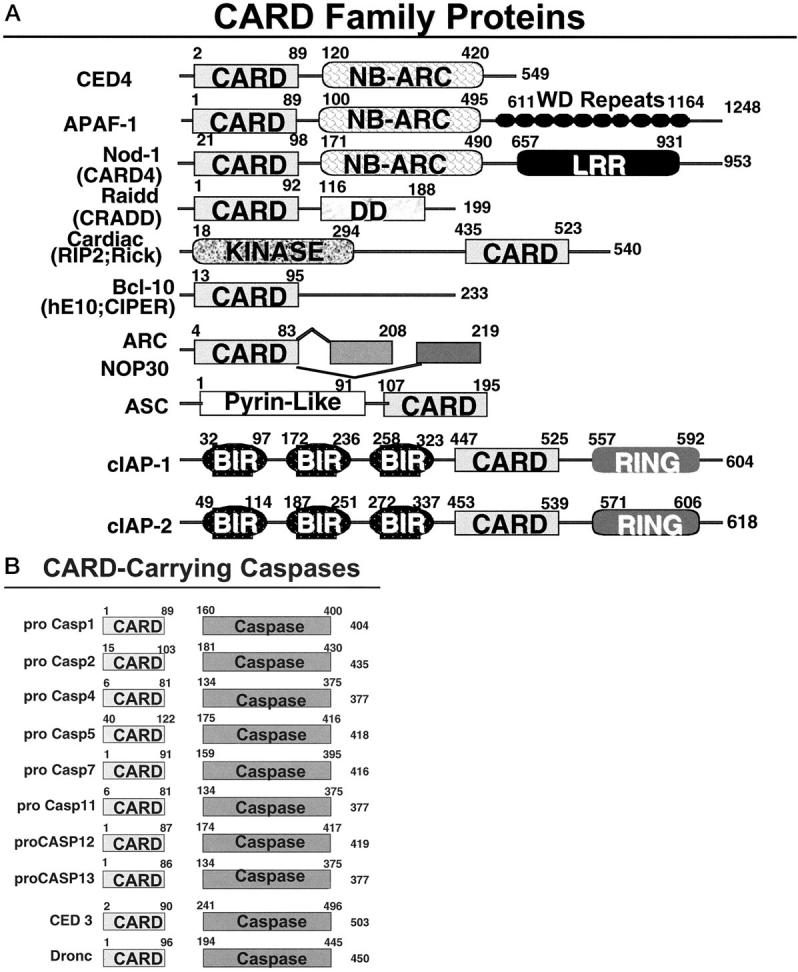
Caspase-associated recruitment domain (CARD) family proteins. Non-caspase CARD family proteins are shown. The domain arrangements of the known members of the CARD family are presented. Numbers represent amino acid residue positions. In addition to human proteins, the diagram includes the murine caspase-11 and caspase-12 proteins, C. elegans CED-3 and CED-4 proteins, and the Drosophila Dronc protein. Additional family members in lower organisms are not presented.
The overall structure of the CARD is similar to DDs and DEDs, comprised of 6 α-helices. 103-106 Homotypic interactions among CARD-carrying proteins play important roles in caspase activation throughout animal evolution. One of the paradigms used for caspase activation is embodied in CED-4/Apaf-1 family proteins. These proteins contain a CARD domain in combination with a nucleotide-binding oligomerization domain, known as a NB-ARC (NACHT) domain for Nucleotide-Binding domain homologous to Apaf-1, CED-4 and plant R gene products. 107 The N-terminal CARDs of Apaf-1 in humans and CED-4 in the nematode Caenorhabditis elegans mediate interactions with the CARDs of specific initiator caspases, pro-caspase-9 and pro-CED-3, respectively. 26,108,109 Oligomerized Apaf-1 and CED-4 activate caspases by the induced proximity method. 24,25,110
With the CED-4 protein of C. elegans, binding and activation of the caspase pro-CED-3 is spontaneous. In contrast, the human and Drosophila Apaf-1 proteins contain an additional regulatory domain, comprised of several WD repeat domains, which renders them dependent on cytochrome c. 12,111 Though less is known about the fly protein, biochemical analysis of human Apaf-1 using in vitro reconstituted systems with purified components indicates that cytochrome c, in combination with dATP, induces oligomerization of Apaf-1 molecules, followed by binding to and activation of pro-caspase-9 24,25 (Figure 4) ▶ . Several mechanisms for suppressing caspase activation by Apaf-1 have been identified, including expression of a shorter noncatalytic isoform of pro-caspase-9 that competes with full-length caspase-9 for binding Apaf-1, phosphorylation of human caspase-9 by the kinase Akt, overexpression of heat shock proteins, and alkaline pH. 112-116
Additional CARD-carrying proteins are also involved caspase activation. Nod-1 (CARD4) reportedly activates caspases when overexpressed in cells. 117,118 This protein contains a CARD, followed by a nucleotide-binding domain and several leucine-rich repeats, which presumably represent protein interaction domains somehow involved in the regulation of this protein. The NB domain of Nod-1 can self-associate, suggesting parallels with the mechanisms used by Apaf-1 and CED-4 for activating caspases. Because Nod-1 readily co-immunoprecipitates with pro-caspase-9, it may represent an alternative activator of this initiator caspase. However, because the phenotypes of apaf-1 and pro-caspase-9 knockout mice are highly similar, 16,17,119 Nod-1 presumably substitutes for Apaf-1 in only rare circumstances in vivo. Also, unlike Apaf-1, Nod-1 is a potent inducer of NF-κB, suggesting additional actions besides caspase activation. 117,118
Another CARD family protein that associates with pro-caspase-9 is Bcl-10 (huE10; CIPER). 120-122 In addition to its CARD, Bcl-10 contains a proline-rich domain that mediates associations directly or indirectly with pro-caspase-9. When overexpressed, Bcl-10 induces apoptosis and activates NF-κB. Mutant versions of Bcl-10 lacking the C-terminal domain implicated in pro-caspase-9 binding have been reported in cancers, though the frequency of such gene mutations is debated. 120,123,124 These truncated forms of Bcl-10 retain NF-κB-inducing activity but fail to induce apoptosis when overexpressed in cells, instead manifesting transforming activity in classical rodent cell-based assays in vitro. 120 The herpes equine virus encodes a very similar protein, called E10. 122
Another CARD-carrying caspase activator is Cardiak (RIP2; Rick) (Figure 3) ▶ . This protein contains a CARD and a protein kinase domain similar to that found in the NF-κB-inducing DD-containing protein RIP1 (Figure 2) ▶ and in the NF-κB-modulator RIP-3. 125-128 The CARD of Cardiak binds the CARD-containing prodomain of pro-caspase-1 and induces caspase-1 activation, presumably through the induced-proximity method. Cardiak has been reported to bind certain TRAF family proteins, which may link it to signaling by various TNF and Toll/IL-1/LPS family receptors, 127 thus explaining the ability of certain cytokine to induce pro-IL-1β processing, yielding bioactive IL-1β. 6 Though Cardiak can induce apoptosis when overexpressed, it is questionable whether this represents a physiological role for this protein. Activation of pro-caspase-2 by the CARD-carrying protein, Raidd (Cradd), was mentioned above (Figures 2 and 4) ▶ ▶ in the context of TNFR1 signaling where the DD of these adapter proteins links them to certain DD-containing TNF family receptors and the CARD domain interacts with the CARD-containing pro-domain of pro-caspase-2. 39,40
IAP Family Proteins
The IAPs represent a family of evolutionarily conserved apoptosis suppressors. 129-131 IAPs are found in the genomes of mammals, insects, and certain animal viruses. All members of this family, by definition, contain at least one copy of a so-called BIR (baculovirus iap repeat) domain, a zinc-binding fold 132-134 important for their anti-apoptotic activity. In addition to 1 to 3 copies of a BIR domain, many IAP family proteins also contain other domains, including RING zinc-fingers, CARDs, Ubiquitin-conjugating enzyme (E2s) domains, or putative nucleotide-binding domains (Figure 5) ▶ . Interestingly, the RINGs of IAPs have recently been implicated in interactions with the cellular components of the ubiquitination machinery, 135 thus controlling turnover of these proteins and possibly other proteins with which they associate. Also, the BIR-containing protein, Apollon (Bruce) contains a domain with ubiquitin-conjugating enzyme (E2) activity, further suggesting links of BIR family proteins to the cellular ubiquitination machinery. 136 Though the overall relevance to apoptosis of this protein remains uncertain, antisense-mediated down-regulation of Apollon reportedly can sensitize tumor cell lines to apoptosis induced by anti-cancer drugs. 137 In this regard, it bears noting that the mere presence of a BIR domain does not necessarily indicate anti-apoptotic activity. For example, BIR-containing proteins, which regulate mitosis and meiosis but have no apparent effect on cell death regulation, have been studied in yeast and C. elegans. 138,139
Figure 5.

Human IAP family proteins. The domain arrangements of the known members of the human IAP family are presented. Numbers represent amino acid residue positions. Shown are BIR, nucleotide-binding (NB), ubiquitin-conjugating (Ubc), and RING zinc-finger domains.
Though IAP family proteins may possess other functions, 129 several of them have been shown to bind and potently inhibit activated caspases. Among the caspases inhibited by human IAP family members XIAP, cIAP1, and cIAP2 are the effector caspases-3 and -7, as well as the initiator caspase-9. 140-142 Suppression of the effector caspases maps to the N-terminal half of the protein in the region encompassing the first two BIR domains, BIR1 and BIR2, 132,143 whereas the third BIR domain is required for suppression of caspase-9. 142 Importantly, IAPs are selective caspase inhibitors and lack activity against many members of the caspase family of cell death proteases. This stands in marked contrast to the baculovirus p35 protein, an apoptosis suppressor that displays broad activity against caspases but for which no cellular homologue has been identified. 144
Human IAPs can arrest apoptosis induced via either the intrinsic (mitochondrial) or extrinsic (death receptor) pathways, probably because they target effector caspases common to both pathways. 145 In contrast, insect IAPs may inhibit primarily initiator caspases within the mitochondria-dependent pathway, possibly reflecting the later evolution of the death receptor (TNF family) pathway. 146
Alterations in the expression of IAPs in association with diseases have been observed. For example, the first of the human IAP family genes to be discovered, NAIP, suffers inactivating mutations in a subgroup of patients with severe forms of spinal muscular atrophy (SMA), a motor-neuron degenerative disease. 147 In addition, overexpression of the IAP member Survivin has been documented in many common types of cancer. 148 Survivin is highly expressed in the developing fetus but largely absent from normal adult tissues. 149 Through unclear mechanisms, Survivin expression appears to be deregulated in cancers. Indeed, genome-wide transcription profiling suggests that Survivin is among the most tumor-specific genes thus far identifiable. 150 The promoter of the Survivin gene is normally cell cycle-regulated, becoming activated specifically in late G2/M phase. 151 Moreover, the Survivin protein is associated with the mitotic chromosomes during mitosis and the mitotic spindle apparatus, where it appears to play a critical role in ensuring proper control chromosome segregation during anaphase and in the final execution of cytokinesis. Suppression of survivin expression using antisense methods or interference with Survivin function using dominant-inhibitory mutants results in polyploidy, aneuploidy, and apoptosis. 152 Apparent Survivin orthologues in yeast and C. elegans also participate in cell cycle regulation but lack effects on cell life/death, 139,153 suggesting that Survivin evolved this function later as a possible way of creating a cell cycle checkpoint to ensure apoptotic elimination of cells that fail to properly sort their genetic material during cell division. Elevated expression of other IAP family members in cancer also occurs. 154
Regulators of the IAPs have only recently been identified in mammalian cells, but several examples were previously derived from analysis of programmed cell death in Drosophila. 111,155 In the fly, the death gene-products Reaper, Grim, and Hid (and possibly Doom) directly bind insect IAPs, apparently precluding them from inhibiting caspases and enhancing apoptosis. 156-159 Reaper, Grim and Hid share a conserved ∼14 amino acid domain that is necessary and sufficient for binding IAPs and inducing apoptosis. In vitro, synthetic peptides corresponding to this region can directly suppress the caspase-inhibiting activity of insect IAPs. 146 Regulation of the expression of Reaper, Grim, and Hid can be controlled in interesting ways. For example, Reaper (rpr) is a p53-responsive gene in Drosophila, and is induced in response to x-irradiation. 160
Ectopic expression of Hid in mammalian cells induces apoptosis, suggesting a conserved mechanism. 161 However, humans and other mammals contain no recognizable Reaper, Grim, or Hid homologues, suggesting that IAP suppression can be accomplished using diverse protein structures that may have in common only small structural elements such as that embodied in the conserved ∼14 amino acid regions of the fly proteins mentioned above. In this regard, a mammalian IAP inhibitor Smac (Diablo) was recently described, which binds multiple IAP family members including XIAP, cIAP1, cIAP2, and Survivin, and which allows caspases to induce apoptosis. 162,163 In addition, Xenopus lavis has been demonstrated to possess a nuclear protein, Scythe, which binds the fly Reaper protein in vitro and which modulates apoptosis in some situations, 164 suggesting that it may also somehow be involved indirectly in the regulation of IAPs. Little is known about the human homologue of Scythe. Some inhibitors of IAP have begun to emerge through analysis of apoptosis in humans, and it seems likely that many more are yet to be discovered, given that flies have devoted at least 3 (possibly 4) genes to this task and that the human genome may contain as many as 9 times more genes than Drosophila.
Bcl-2 Family Proteins
The mitochondria-dependent pathway for apoptosis is governed by Bcl-2 family proteins. Bcl-2 family proteins are conserved throughout metazoan evolution, with homologues found in mammalian, avian, fish, and amphibian species, as well as in invertebrates such as C. elegans, Drosophila, and marine sponges. Several types of animal viruses also harbor Bcl-2 family genes within their genomes. Both pro- and anti-apoptotic Bcl-2 family proteins exist, and many of these proteins physically bind each other, forming a complex network of homo- and heterodimers. 165-169 The relative ratios of anti- and pro-apoptotic Bcl-2 family proteins dictate the ultimate sensitivity or resistance of cells to various apoptotic stimuli, including growth factor/neurotrophin deprivation, hypoxia, radiation, anti-cancer drugs, oxidants, and Ca2+ overload. Not surprisingly, then, alterations in the amounts of these proteins have been associated with a variety of pathological conditions, characterized by either too much (cell loss) or too little (cell accumulation) cell death. These diseases include cancer, autoimmune disorders such as lupus (where a failure to eradicate autoreactive lymphocytes occurs), immunodeficiency associated with HIV infection, and ischemia-reperfusion injury during stroke and myocardial infarction, among others. 170 For example, the Bcl-2 gene (anti-apoptotic) is activated by chromosomal translocations in the majority of non-Hodgkin’s lymphomas 171,172 and is also inappropriately overexpressed in many solid tumors, contributing to resistance to chemotherapy- and radiation-induced apoptosis. 173 Conversely, loss-of-function mutations have been identified in the BAX genes (pro-apoptotic) of human tumors 174 and analysis of bax gene knockout mice indicates that Bax is a tumor suppressor in vivo. 175 Transcription of the BAX genes is also indirectly regulated by p53, thus providing another connection of this important tumor suppressor to apoptosis pathways. 176 In contrast to cancer, where insufficient Bax expression occurs, it has been shown that Bax is induced in neurons after ischemia-reperfusion injury. 177 Furthermore, bax gene knockout mice display marked resistance to neuronal cell death induced by ischemia, axotomy, and other insults. 178,179 Bax is also increased in apoptosing neurons in patients with Alzheimer’s disease. 180 Many, many more examples of associations of diseases with changes in the relative amounts of Bcl-2 family proteins exist, besides the few provided here.
In humans, 20 members of the Bcl-2 family gene family have been described to date. These genes encode the anti-apoptotic proteins, Bcl-2, Bcl-XL, Mcl-1, Bfl-1 (A1), Bcl-W, and Boo (Diva), as well as the pro-apoptotic proteins Bax, Bak, Bok (Mtd), Bad, Bid, Bim, Bik, Hrk, Bcl-XS, APR (Noxa), p193, Bcl-G, Nip3, and Nix (BNIP). Some of the Bcl-2 family genes produce two or more proteins through alternative mRNA splicing, sometimes exerting opposing effects on cell death regulation (eg, Bcl-XL versus Bcl-XS). Also, some of these proteins may display anti-apoptotic activity in some cellular backgrounds and have pro-apoptotic functions in other cellular contexts (eg, Boo/Diva, Bcl-2, Bax). 181-184 Gene ablation studies in mice suggest that each of the Bcl-2 family members plays unique roles in controlling cell survival in vivo, reflecting their tissue-specific patterns of expression or cell-context-dependent requirements for these proteins.
Based on their predicted (or experimentally determined) three-dimensional structures, Bcl-2 family proteins can be broadly divided into two groups. One subset of these proteins is probably similar in structure to the pore-forming domains of bacterial toxins, such as the colicins and diphtheria toxin. 185-188 These α-helical pore/channel-like proteins include both pro-apoptotic proteins (Bcl-2, Bcl-XL, Mcl-1, Bfl-1, Bcl-W, and possibly Boo), as well as pro-apoptotic proteins (Bax, Bak, Bok, and Bid). Most of these protein in this subcategory can be recognized by conserved stretches of amino acid sequence homology, including the presence of Bcl-2 homology (BH) domains BH1, BH2, BH3, and sometimes BH4 (Figure 6) ▶ . However, this is not uniformly the case, as the Bid protein contains only a BH3 domain but has been determined to share the same overall protein fold with Bcl-XL. 186,187 Where tested, these proteins, including Bcl-2, Bcl-XL, Bax, and Bid, have all been shown to form ion-conducting channels in synthetic membranes in vitro. 189-193
Figure 6.

Bcl-2 family proteins. The domain arrangements of the known members of the Bcl-2 family are presented. The BH1, BH2, BH3, BH4, and transmembrane domains are indicated. In addition to human or murine proteins, included in the diagram are the chicken Nr13 protein, C. elegans proteins CED-9, EGL1, and CeBNIP, and the Drosophila proteins DBok (Drob1, dBorg 1, Debcl). Homologues from viruses, xenopas, and marine sponges are not depicted. Human noxa (APR) has only one BH3 domain.
The other subset of Bcl-2 family proteins appears to have in common only the presence of the BH3 domain, including Bad, Bik, Bim, Hrk, Bcl-GS, p193, and APR (Noxa). These proteins are all pro-apoptotic in their function, and their cell death-inducing activity depends on their ability to dimerize with anti-apoptotic Bcl-2 family members, typically functioning as trans-dominant inhibitors of proteins such as Bcl-2 and Bcl-XL. 169 In this regard, the BH3 domain has been shown to mediate dimerization among Bcl-2 family proteins. This domain consists of an amphipathic α-helix ∼16 amino acids in length that inserts into a hydrophobic crevice on the surface of anti-apoptotic proteins such as Bcl-XL. 194 Thus, mutations in the BH3 domain of proteins such as Bad, Bik, Bim, Bcl-GS, and Hrk that abolish their ability to bind other Bcl-2 family member also abrogate their capacity to induce apoptosis.
Nip3 and Nix (and their counterpart in C. elegans, CeBNIP) may define a third subgroup of Bcl-2 family proteins (Figure 6) ▶ . Though these proteins contain BH3-like domains, mutagenesis studies suggest that these domains do not entirely account for their pro-apoptotic function, and instead a carboxyl-terminal membrane-anchoring domain may play an important role. 195 It should also be noted that BH3-mediated interactions among Bcl-2 family proteins do not always result in antagonism. For example, binding of the BH3 domain of Bid to Bax appears to activate Bax, perhaps promoting its insertion into membranes and allowing it to assume a cytotoxic conformation. 196,197
Many Bcl-2 family proteins are constitutively localized to the membranes of mitochondria, whereas others are induced to target these organelles in response to specific stimuli. For example, Bcl-2, Bcl-XL, and many other members of the Bcl-2 family have a hydrophobic stretch of amino acids near their C-termini (transmembrane domains) that anchors them in the outer mitochondrial membrane. Some of these proteins also insert into endoplasmic reticulum and nuclear envelope, though their effects on cell death regulation in these compartments are poorly understood compared to those of mitochondria. 13 In contrast to Bcl-2, Bcl-XL, and many of the Bcl-2 family proteins, the pro-apoptotic proteins Bid, Bim, and BAD lack C-terminal transmembrane domains. These proteins are normally found in the cytosol but can be induced to target mitochondria. In the case of Bid, cleavage by caspase-8 is required, removing the N-terminal 52 amino acids and exposing both the BH3 dimerization domain and the hydrophobic core of the protein, which is believed to be responsible for its insertion into mitochondrial membranes. 193,198,199 This caspase-8-mediated activation of Bid represents an important mechanism accounting for cross-talk between the death receptor (extrinsic) and mitochondrial (intrinsic) pathways. 200 With Bim, isoforms of this protein have been found that associate with microtubules via direct binding to microtubule-associated dynein light-chain. 201 Disruption of these protein interactions frees Bim, allowing it to dimerize through its BH3 domain with anti-apoptotic Bcl-2 family proteins on the surface of mitochondria. In addition to cleavage by caspases (Bid) and sequestration by interacting proteins (Bim), targeting to mitochondria of some pro-apoptotic Bcl-2 family members is controlled by phosphorylation. The BH3-only protein BAD, for example, translocates between the cytosolic and mitochondria compartments depending on whether it is phosphorylated. Several protein kinases, including Akt (PKB), PKA, Raf1, Rsk1, and Pak1 have been reported to phosphorylate BAD, thus inactivating the protein so that it cannot dimerize with and antagonize Bcl-2 or Bcl-XL. 202,203 Phosphorylated BAD is therefore found in the cytosol, sometimes found in a complex with 14-3-3. Evidence has been presented that BAD is inactivated by this phosphorylation mechanism in many cancers. 203 Conversely, dephosphorylation of BAD has been implicated in apoptosis induction in response to agents that induce sustained elevation in intracellular cytosolic free Ca2+, causing activation of the Ca2+/calmodulin-dependent phosphatase, calcineurin. In hippocampal neurons, for example, the NMDA receptor agonist L-glutamate induces calcineurin-dependent BAD dephosphorylation and dimerization with Bcl-XL, correlating with translocation of BAD from cytosol to mitochondria. 204 Finally, at least one Bcl-2 family member (Bax) contains a C-terminal transmembrane domain but nevertheless displays regulated movement between cytosolic and mitochondrial compartments. Bax appears to assume a latent confirmation in the cytosol of many healthy cells, where its transmembrane domain is evidently masked. 205 On delivery of a variety of apoptotic stimuli, Bax undergoes a conformational change associated with translocation to and insertion into mitochondria membranes. The nature of the signal that controls Bax activation remains unclear, though changes in cellular pH have been suggested to play a role. 206
What do Bcl-2 family proteins do when they reach mitochondria? This question is still debated among workers in the field. 13,168,207 What can be stated with confidence is that Bcl-2 family proteins regulate the release of cytochrome c from mitochondria, with pro-apoptotic Bcl-2 family proteins inducing or making it easier to induce release of this caspase-activating protein and anti-apoptotic members of the family suppressing cytochrome c release. Still unclear are the issues of whether the control of cytochrome c release is directly attributable to the pore/channel-like properties of some Bcl-2 family proteins and whether cytochrome c release reflects a primary effect on the outer membrane in which proteins are allowed to escape versus a direct or indirect influence on the inner membrane, where changes in osmotic homeostasis dictate organelle volume regulation and consequently release of cytochrome c due to swelling and rupture of mitochondria. 13,168,207 It has also become clear that whatever the mechanism, it is caspase-independent and controls cell death commitment, though exceptions may exist in some lower organisms, particularly C. elegans. 166 In mammalian cells, even when caspases are completely inhibited and apoptosis is thus prevented, the Bcl-2-regulated changes in mitochondrial membrane barrier function responsible for deciding sequestration versus release of cytochrome c dictate cell life-death decisions (clonigenic survival). In such instances where caspases are inhibited, cell death occurs via necrosis but is nevertheless regulated by members of the Bcl-2 family.
In addition to cytochrome c, Bcl-2 family proteins have been reported to control the release of other proteins from mitochondria. These proteins include (i) certain caspases (caspase-2, -3, and -9) which reportedly are sequestered inside mitochondria in some types of cells, 208-210 (ii) apoptosis inducing factor (AIF), a flavoprotein implicated in nuclear manifestations of apoptosis via caspase-independent mechanisms, 211 and (iii) Smac/Diablo, the inhibitor of IAP family proteins. 162,163 All of these proteins are encoded within the nuclear genome, transported into mitochondria, and stored in the space between the inner and outer membranes, thus awaiting release into the cytosol upon breakdown of the outer membrane. Also, the proforms of cytochrome c, AIF, and Smac/Diablo are inactive in terms of cell death induction, requiring modifications such as attachment of prosthetic groups (heme for cytochrome c; flavin adenine dinucleotide (FAD) for AIF) and/or proteolytic processing of their N-terminal mitochondria-targeting leader peptides (AIF and Smac/Diablo), which occurs only within mitochondria. In this way, apoptosis is avoided during biosynthesis of the apoproteins and is functionally linked to disruption of mitochondrial membrane barrier function, providing cells with a suicide mechanism that can be triggered in response to mitochondrial damage.
Conclusions
Advances in elucidating the molecular mechanisms of apoptosis and its regulation have laid the foundation for a deeper understanding of the pathophysiology of many diseases. More importantly, this work has revealed strategies for therapeutic intervention in a wide range of ailments, including cancer, autoimmune disorders, immunodeficiency, inflammation, ischemic heart disease, stroke, and neurodegenerative diseases. 212 Small-molecule inhibitors of caspases, for example, are now in early clinical trials, with more to come in the near future. Anticancer therapies based on antisense oligonucleotide-mediated suppression of BCL-2 expression have advanced to Phase III trials. Biological agents that control apoptosis, such as Trail-ligand (Apo2L), will also soon enter clinical trials for patients with refractory metastatic cancers. This progress in translating knowledge about apoptosis mechanisms into the clinical arena suggests rich opportunities for new and more effective treatments for many of the medical illnesses for which adequate therapies currently do not exist.
Acknowledgments
I thank K. Doctor and K. Pawlowski for assistance with bioinformatics and R. Cornell for manuscript preparation. I dedicate this article and associated award to my mentor, Peter C. Nowell.
Footnotes
Address reprint requests to John C. Reed, M.D., Ph.D., The Burnham Institute, La Jolla, CA 92037. E-mail: jreed@burnham-inst.org.
References
- 1.Kerr JF, Wyllie AH, Currie AR: Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972, 26:239-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J: Human ICE/CED-3 protease nomenclature. Cell 1996, 87:171. [DOI] [PubMed] [Google Scholar]
- 3.Thornberry N, Lazebnik Y: Caspases: enemies within. Science 1998, 281:1312-1316 [DOI] [PubMed] [Google Scholar]
- 4.Cryns V, Yuan Y: Proteases to die for. Genes Dev 1999, 13:371. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson DW: ICE/CED3-like proteases as therapeutic targets for the control of inappropriate apoptosis. Nat Biotechnol 1996, 14:297-301 [DOI] [PubMed] [Google Scholar]
- 6.Reed J: Caspases and cytokines: roles in inflammation and autoimmunity. Adv Immunol 1998, 73:265-287 [DOI] [PubMed] [Google Scholar]
- 7.Salvesen GS, Dixit VM: Caspases: intracellular signaling by proteolysis. Cell 1997, 91:443-446 [DOI] [PubMed] [Google Scholar]
- 8.Thornberry N, Rano T, Peterson E, Rasper D, Timkey T, Garcia-Calvo M, Houtzager V, Nordstrom P, Roy S, Vaillancourt J, Chapman K, Nicholson D: A combinatorial approach defines specificities of members of the caspase family and granzyme B. J Biol Chem 1997, 272:17907-17911 [DOI] [PubMed] [Google Scholar]
- 9.Salvesen GS, Dixit VM: Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA 1999, 96:10964-10967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP: Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol 1999, 17:331-367 [DOI] [PubMed] [Google Scholar]
- 11.Yuan J: Transducing signals of life and death. Curr Opin Cell Biol 1997, 9:247-251 [DOI] [PubMed] [Google Scholar]
- 12.Reed JC: Cytochrome c: can’t live with it; can’t live without it. Cell 1997, 91:559-562 [DOI] [PubMed] [Google Scholar]
- 13.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 14.Varfolomeev E, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann J, Mett I, Rebrikov D, Brodianski V, Kemper O, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham K, Goncharov T, Holtmann H, Lonai P, Wallach D: Targeted disruption of the mouse caspase 8 gene ablates cell death inducation by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9:267-276 [DOI] [PubMed] [Google Scholar]
- 15.Juo P, Kuo CJ, Yuan J, Blenis J: Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol 1998, 8:1001-1008 [DOI] [PubMed] [Google Scholar]
- 16.Kuida K, Haydar TF, Kuan C-Y, Gu Y, Taya C, Karasuyama H, Su MS-S, Rakic P, Flavell RA: Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94:325-337 [DOI] [PubMed] [Google Scholar]
- 17.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW: Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 1998, 94:339-352 [DOI] [PubMed] [Google Scholar]
- 18.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM: FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81:505-512 [DOI] [PubMed] [Google Scholar]
- 19.Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D: A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem 1995, 270:7795-7798 [DOI] [PubMed] [Google Scholar]
- 20.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM: FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996, 85:817-827 [DOI] [PubMed] [Google Scholar]
- 21.Boldin MP, Goncharov TM, Goltsev YV, Wallach D: Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85:803-815 [DOI] [PubMed] [Google Scholar]
- 22.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM: An induced proximity model for caspase-8 activation. J Biol Chem 1998, 273:2926-2930 [DOI] [PubMed] [Google Scholar]
- 23.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X: Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90:405-413 [DOI] [PubMed] [Google Scholar]
- 24.Zou H, Li Y, Liu X, Wang X: An APAF-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 1999, 274:11549-11556 [DOI] [PubMed] [Google Scholar]
- 25.Saleh A, Srinivasula S, Acharya S, Fishel R, Alnemri E: Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem 1999, 274:17941-17945 [DOI] [PubMed] [Google Scholar]
- 26.Li P, Nijhawan D, Budihardjo I, Srinivasula S, Ahmad M, Alnemri E, Wang X: Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91:479-489 [DOI] [PubMed] [Google Scholar]
- 27.Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS: Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem 1998, 273:27084-27090 [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez A, Oliver H, Zou H, Chen P, Wang X, Abrams J: Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat Cell Biol 1999, 1:272-279 [DOI] [PubMed] [Google Scholar]
- 29.Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW: NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 1996, 384:638-641 [DOI] [PubMed] [Google Scholar]
- 30.Liepinsh E, Ilag LL, Otting G, Ibanez CF: NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 1997, 16:4999-5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bredesen D, Ye X, Tasinato A, Sperandio S, Assa-Munt N, Rabizadeh S: P75NTR and the concept of cellular dependence: seeing how the other half die. Cell Death Differ 1998, 5:365-371 [DOI] [PubMed] [Google Scholar]
- 32.Hsu H, Xiong J, Goeddel DV: The TNF receptor 1-associated protein TRADD signals cell death and NF-kappaB activation. Cell 1995, 81:495-504 [DOI] [PubMed] [Google Scholar]
- 33.Chinnaiyan AM, O’Rourke K, Yu G-L, Lyons RH, Garg M, Duan R, Xing L, Gentz R, Ni J, Dixit VM: Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science 1996, 274:990-992 [DOI] [PubMed] [Google Scholar]
- 34.Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM: Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett 1998, 431:351-356 [DOI] [PubMed] [Google Scholar]
- 35.Yeh W, De La Pompa J, El-Deiry W, Lowe S, Goeddel D, Mak T: FADD essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998, 279:1954-1958 [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Cado C, Chen A, Kabra N, Winoto A: Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort 1. Nature 1998, 392:296-299 [DOI] [PubMed] [Google Scholar]
- 37.Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, Krammer PH, Walczak H: FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12:599-609 [DOI] [PubMed] [Google Scholar]
- 38.Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A: Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12:611-620 [DOI] [PubMed] [Google Scholar]
- 39.Duan H, Dixit VM: RAIDD is a new ‘death’ adaptor molecule. Nature 1997, 385:86-89 [DOI] [PubMed] [Google Scholar]
- 40.Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T, Alnemri ES: CRADD, a novel human apoptotic adaptor molecule for caspase-2 and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res 1997, 57:615-619 [PubMed] [Google Scholar]
- 41.Levy-Strumpf N, Kimchi A: Death associated proteins (DAPs): from gene identification to the analysis of their apoptotic and tumor suppressive functions. Oncogene 1998, 17:3331-3340 [DOI] [PubMed] [Google Scholar]
- 42.Frisch S: Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr Biol 1999, 9:1047-1049 [DOI] [PubMed] [Google Scholar]
- 43.Rytomaa M, Martins L, Downward J: Involvement of FADD and caspase-8 signaling in detachment-induced apoptosis. Curr Biol 1999, 9:1043-1046 [DOI] [PubMed] [Google Scholar]
- 44.Frisch SM, Ruoslahti E: Integrins and anoikis. Curr Opin Cell Biol 1997, 9:701-706 [DOI] [PubMed] [Google Scholar]
- 45.Stanger BZ, Leder P, Lee T-H, Kim E, Seed B: RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 1995, 81:513-523 [DOI] [PubMed] [Google Scholar]
- 46.Hsu H, Huang J, Shu H, Baichwal V, Goeddel DV: TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4:387-396 [DOI] [PubMed] [Google Scholar]
- 47.Ting AT, Pimentel-Muinos FX, Seed B: RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J 1996, 15:6189-6196 [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G: NF-κB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci USA 1999, 96:9136-9141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.(Deleted in proof)
- 50.Stroka D, Badrichani A, Bach F, Ferran C: Overexpression of A1: an NF-κB-inducible, anti-apoptotic Bcl gene that inhibits endothelial cell activation. Blood 1999, 93:3803-3810 [PubMed] [Google Scholar]
- 51.Grumont R, Rourke I, Gerondakis S: Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev 1998, 13:400-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stehlik C, de Martin R, Kumabashiri I, Schmid A, Binder B, Lipp J: Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor α-induced apoptosis. J Exp Med 1998, 188:211-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stehlik C, de Martin R, Binder B, Lipp J: cytokine induced expression of porcine inhibitor of apoptosis protein (iap) family member is regulated by NF-κB. Biochem Biophys Res Commun 1998, 243:827-832 [DOI] [PubMed] [Google Scholar]
- 54.Wang C, Mayo M, Korneluk R, Goeddel D, Baldwin A: NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress capase-8 activation. Science 1998, 281:1680-1683 [DOI] [PubMed] [Google Scholar]
- 55.Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM: Suppression of TNF-α-induced apoptosis by NF-κB. Science 1996, 274:787-789 [DOI] [PubMed] [Google Scholar]
- 56.Liu Z-G, Hsu H, Goeddel DV, Karin M: Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 1996, 87:565-576 [DOI] [PubMed] [Google Scholar]
- 57.Sabbatini P, Han J, Chiou S-K, Nicholson DW, White E: Interleukin 1β converting enzyme-like proteases are essential for p53-mediated transcriptionally dependent apoptosis. Cell Growth Differ 1997, 8:643-653 [PubMed] [Google Scholar]
- 58.Jiang Y, Woronicz J, Liu W, Goeddel D: Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science 1999, 283:543-546 [DOI] [PubMed] [Google Scholar]
- 59.Takayama S, Xie Z, Reed J: An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J Biol Chem 1999, 274:781-786 [DOI] [PubMed] [Google Scholar]
- 60.Tschopp J, Martinon F, Hofmann K: Apoptosis: silencing the death receptors. Curr Biol 1999, 9:R381-R384 [DOI] [PubMed] [Google Scholar]
- 61.Sato T, Irie S, Kitada S, Reed JC: FAP-1: A protein tyrosine phosphatase that associates with Fas. Science 1995, 268:411-415 [DOI] [PubMed] [Google Scholar]
- 62.Irie S, Hachiya T, Rabizadeh S, Maruyama W, Mukai J, Li Y, Reed J, Bredesen D, Sato T: Functional interaction of Fas-associated phosphatase-1 (FAP-1) with p75 and their effect on NF-kB activation. FEBS Lett 1999, 460:191-198 [DOI] [PubMed] [Google Scholar]
- 63.Yanagisawa J, Takahashi M, Kanki H, Yano-Yanagisawa H, Tazunoki T, Sawa E, Nishitoba T, Kamishohara M, Kobayashi E, Kataoka S, Sato T: The molecular interaction of Fas and FAP-1: A tripeptide blocker of human Fas interaction with FAP-1 promotes Fas-induced apoptosis. J Biol Chem 1997, 272:8539-8545 [DOI] [PubMed] [Google Scholar]
- 64.Myc A, Arscott PL, Bretz JD, Thompson NW, Baker JRJ: Characterization of FAP-1 expression and function in throid follicular cells. Endocrinology 1999, 140:5431-5434 [DOI] [PubMed] [Google Scholar]
- 65.Tanaka M, Ito H, Adachi S, Akimoto H, Nishikawa T, Kasajima T, Marumo F, Hiroe M: Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ Res 1994, 75:426-433 [DOI] [PubMed] [Google Scholar]
- 66.Gougeon M, Montagnier L: Apoptosis in AIDS. Science 1993, 260:1269-1270 [DOI] [PubMed] [Google Scholar]
- 67.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middelton LA, Lin AY, Strober W, Lenardo MJ, Puck JM: Dominant interfering fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81:935-946 [DOI] [PubMed] [Google Scholar]
- 68.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S: Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992, 356:314-317 [DOI] [PubMed] [Google Scholar]
- 69.Green DR, Bissonnette RP, Glynn JM, Shi Y: Activation-induced apoptosis in lymphoid systems. Semin Hematol 1992, 4:379-388 [PubMed] [Google Scholar]
- 70.Wang J, Lenardo MJ: Molecules involved in cell death and peripheral tolerance. Curr Opin Immunol 1997, 9:818-825 [DOI] [PubMed] [Google Scholar]
- 71.Landowski TH, Wu N, Buyuksal I, Painter JS, Dalton WS: Mutations in the Fas antigen in patients with multiple myeloma. Blood 1997, 90:4266-4270 [PubMed] [Google Scholar]
- 72.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD: Protection from fas-mediated apoptosis by a soluble form of the fas molecule. Science 1994, 263:1759-1762 [DOI] [PubMed] [Google Scholar]
- 73.Hohlbaum AM, Moe S, Marshak-Rothstein A: Opposing effects of transmembrane and soluble fas ligand expression on inflammation and tumor cell survival. J Exp Med 2000, 191:1209-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ashkenazi A, Dixit VM: Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 1999, 11:255-260 [DOI] [PubMed] [Google Scholar]
- 75.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM: An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277:815-818 [DOI] [PubMed] [Google Scholar]
- 76.Pan G, Ni J, Yu G, Wei YF, Dixit VM: TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett 1998, 424:41-45 [DOI] [PubMed] [Google Scholar]
- 77.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A: Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors (see comments). Science 1997, 277:818-821 [DOI] [PubMed] [Google Scholar]
- 78.Gura T: How TRAIL kills cancer cells, but not normal cells (news comment). Science 1997, 277:768. [DOI] [PubMed] [Google Scholar]
- 79.Nagata S: Steering anti-cancer drugs away from the TRAIL (news comment). Nat Med 2000, 6:502-503 [DOI] [PubMed] [Google Scholar]
- 80.Kastan M: On the TRAIL from p53 to apoptosis? Nat Genet 1997, 17:130-131 [DOI] [PubMed] [Google Scholar]
- 81.Eberstadt M, Huang B, Chen Z, Meadows R, Ng S, Zheng L, Lenardo M, Fesik S: NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature 1998, 392:941-945 [DOI] [PubMed] [Google Scholar]
- 82.Stegh AH, Schickling O, Ehret A, Scaffidi C, Peterhansel C, Hofmann TG, Grummt I, Krammer PH, Peter ME: DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. EMBO J 1998, 17:5974-5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leo CP, Hsu SY, McGee EA, Salanova M, Hsueh AJ: DEFT, a novel death effector domain-containing molecule predominantly expressed in testicular germ cells. Endocrinology 1998, 139:4839-4848 [DOI] [PubMed] [Google Scholar]
- 84.Wallach D: Placing death under control. Nature 1997, 388:123-126 [DOI] [PubMed] [Google Scholar]
- 85.Tschopp J, Irmler M, Thome M: Inhibition of Fas death signals by Flips. Curr Opin Immunol 1998, 10:552-558 [DOI] [PubMed] [Google Scholar]
- 86.French L, Tschopp J: Inhibition of death receptor signaling by FLICE-inhibitory protein as a mechanism for immune escape of tumors. J Exp Med 1999, 190:891-893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O’Connell J, O’Sullivan GC, Collins JK, Shanahan F: The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med 1996, 184:1075-1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bennett M, O’Connell J, O’Sullivan G, Brady C, Roche D, Collins J, Shanahan F: The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J Immunol 1998, 160:5669-5675 [PubMed] [Google Scholar]
- 89.O’Connell J, Bennett M, O’Sullivan G, Roche D, Kelly J, Collins J, Shanahan F: Fas ligand expression in primary colon adenocarcinomas: evidence that the Fas counterattack is a prevalent mechanism of immune evasion in human colon cancer. J Pathol 1998, 186:240-246 [DOI] [PubMed] [Google Scholar]
- 90.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer J-L, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J: Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386:517-521 [DOI] [PubMed] [Google Scholar]
- 91.Tschopp J, Thome M, Hofmann K, Meinl E: The fight of viruses against apoptosis. Curr Opin Genet Dev 1998, 8:82-87 [DOI] [PubMed] [Google Scholar]
- 92.Bertin J, Armstrong R, Ottilie S, Martin D, Wang Y, Banks S, Wang G, Senkevich T, Alnemri E, Moss B, Lenardo M, Tomaselli K, Cohen J: Death effector domain-containing herpesvirus and poxvirus proteins inhibit Fas-and TNFR1-induced apoptosis. Proc Natl Acad Sci USA 1997, 94:1172-1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang G-H, Bertin J, Wang Y, Martin D, Wang J, Tomaselli K, Armstrong R, Cohen J: Bovine herpesvirus 4 BORFE2 protein inhibits Fas-and TNFR1-induced apoptosis and contains death effector domains shared with other gamma 2 herpesviruses. J Virol 1997, 71:8928-8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ng FWH, Nguyen M, Kwan T, Branton PE, Nicholson DW, Cromlish JA, Shore GC: p28 Bap31, a Bcl-2/Bcl-XL-and procaspase-8-associated protein in the endoplasmic reticulum. J Cell Biol 1997, 39:327-338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang H, Xu Q, Krajewski S, Krajewska M, Xie Z, Fuess S, Kitada S, Godzik A, Pawlowski K, Shabaik A, Konenen J, Bubendorf L, Reed J: BAR: an apoptosis regulator at the intersection of caspase and bcl-2 family proteins. Proc Natl Acad Sci USA 2000, 97:2597-2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, Thornberry NA, Huang J, MacPherson DP, Black SC, Hornung F, Lenardo MJ, Hayden MR, Roy S, Nicholson DW: Cell death attenuation by “Usurpin”, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ 1998, 5:271-288 [DOI] [PubMed] [Google Scholar]
- 97.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T: Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell 1995, 80:401-411 [DOI] [PubMed] [Google Scholar]
- 98.Bergeron L, Perez G, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham K, Flaws J, Salter J, Hara H, Moskowitz M, Li E, Greenberg A, Tilly J, Yuan J: Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 1998, 12:1304-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J: Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 1998, 92:501-509 [DOI] [PubMed] [Google Scholar]
- 100.Bergeron L, Yuan J: Sealing one’s fate: control of cell death in neurons. Curr Opin Neurobiol 1996, 8:55-63 [DOI] [PubMed] [Google Scholar]
- 101.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM: Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease (see comments). Nature 1999, 399:263-267 [DOI] [PubMed] [Google Scholar]
- 102.Nakagawa T, Zhu H, Morishima N, Ll E, Xu J, Yankner B, Yuan J: Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403:98-103 [DOI] [PubMed] [Google Scholar]
- 103.Chou J, Matsuo H, Duan H, Wagner G: Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell 1998, 94:171-180 [DOI] [PubMed] [Google Scholar]
- 104.Zhou P, Chou J, Olea RS, Yuan J, Wagner G: Solution structure of Apaf-1 CARD and its interation with caspase-9 CARD: a structural basis for specific adaptor/caspase interaction. Proc Natl Acad Sci USA 1999, 96:11265-11270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vaughn DE, Rodriguez J, Lazebnik Y, Joshua-Tor L: Crystal structure of Apaf-1 caspase recruitment domain: an alpha-helical Greek key fold for apoptotic signaling. J Mol Biol 1999, 293:439-447 [DOI] [PubMed] [Google Scholar]
- 106.Day CL, Dupont C, Lackmann M, Vaux DL, Hinds MG: Solution structure and mutagenesis of the caspase recruitment domain (CARD) from Apaf-1. Cell Death Differ 1999, 6:1125-1132 [DOI] [PubMed] [Google Scholar]
- 107.Koonin EV, Aravind L: The NACHT family: a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biol Sci 2000, 25:223-224 [DOI] [PubMed] [Google Scholar]
- 108.Chinnaiyan A, Chaudhary D, O’Rourke K, Koonin E, Dixit V: Role of CED-4 in the activation of CED-3. Nature 1997, 388:728-729 [DOI] [PubMed] [Google Scholar]
- 109.Seshagiri S, Miller L: Caenorhabditis elegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr Biol 1997, 7:455-460 [DOI] [PubMed] [Google Scholar]
- 110.Yang X, Chang H, Baltimore D: Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science 1998, 281:1355-1357 [DOI] [PubMed] [Google Scholar]
- 111.Abrams J: An emerging blueprint for apoptosis in drosophila. Trends Cell Biol 1999, 9:435-440 [DOI] [PubMed] [Google Scholar]
- 112.Srinivasula SM, Ahmad M, Guo Y, Zhan Y, Lazebnik Y, Fernandes-Alnemri T, Alnemri ES: Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res 1999, 59:999-1002 [PubMed] [Google Scholar]
- 113.Cardone M, Roy N, Stennicke H, Salvesen G, Franke T, Stanbridge E, Frisch S, Reed J: Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282:1318-1321 [DOI] [PubMed] [Google Scholar]
- 114.Saleh A, Nakazawa A, Kumar S, Srinivasula S, Kumar V, Weichselbaum R, Nalin C, Alnemri E, Kufe D, Kharbanda S: Negative regulation of cytochrome c-mediated oligomerization of apaf-1 and activation of procaspase-9 by heat shock protein 90β. Nat Cell Biol 2000, 2:476-483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR: Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000, 2:469-475 [DOI] [PubMed] [Google Scholar]
- 116.Matsuyama S, Llopi J, Deveraux Q, Tsien R, Reed J: Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol 2000, 2:318-325 [DOI] [PubMed] [Google Scholar]
- 117.Inohara N, Koseki T, Del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Nunez G: Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kB. J Biol Chem 1999, 274:14560-14567 [DOI] [PubMed] [Google Scholar]
- 118.Bertin J, Nir W-J, Fischer C, Tayber O, Errada P, Grant J, Keilty J, Gosselin M, Robison K, Wong G, Glucksmann M, DiStefano P: Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-κB. J Biol Chem 1999, 274:12955-12958 [DOI] [PubMed] [Google Scholar]
- 119.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW: Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998, 94:739-750 [DOI] [PubMed] [Google Scholar]
- 120.Willis T, Jadayel D, Peng H, Perry A, Rauf-Abdul M, Price H, Karran L, Majekodunmi O, Wlodarska I, Pan L, Crook T, Hamoudi R, Isaacson P, Dyer M: Bcl10 is involved in t(1;14) (p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96:35-45 [DOI] [PubMed] [Google Scholar]
- 121.Koseki T, Inohara N, Chen S, Carrio R, Merino J, Hottiger MO, Nabel GJ, Nunez G: CIPER, a novel NF kappaB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J Biol Chem 1999, 274:9955-9961 [DOI] [PubMed] [Google Scholar]
- 122.Yan M, Lee J, Schilbach S, Goddard A, Dixit V: mE10, a novel caspase recruitment domain-containing proapoptotic molecule. J Biol Chem 1999, 275:10287-10292 [DOI] [PubMed] [Google Scholar]
- 123.Zhang Q, Siebert R, Yan M, Hinzmann B, Cui X, Xue L, Rakestraw KM, Naeve CW, Beckmann G, Weisenburger DD, Sanger WG, Nowotny H, Vesely M, Callet-Bauchu E, Salles G, Dixit VM, Rosenthal A, Schlegelberger B, Morris SW: Inactivating mutations and overexpression of BCL-10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32). Nat Genet 1999, 22:63-68 [DOI] [PubMed] [Google Scholar]
- 124.Lee SH, Shin MS, Kim HS, Park WS, Kim SY, Lee HK, Park JY, Oh RR, Jang JJ, Park KM, Han JY, Kang CS, Lee JY, Yoo NJ: Point mutations and deletions of the Bcl-10 gene in solid tumors and malignant lymphomas. Cancer Res 1999, 59:5674-5677 [PubMed] [Google Scholar]
- 125.Inohara N, del Peso L, Koseki T, Chen S, Nunez G: RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J Biol Chem 1998, 273:12296-12300 [DOI] [PubMed] [Google Scholar]
- 126.Thome M, Hofmann K, Burns K, Martinon F, Bodmer J, Mattman C, Tschopp J: Identification of CARDIAK, a RIP-like kinase that assciates with caspase-1. Curr Biol 1998, 16:885-888 [DOI] [PubMed] [Google Scholar]
- 127.McCarthy J, Ni J, Dixit V: RIP2 is a novel NF-κB-activating and cell death-inducing kinase. J Biol Chem 1998, 273:16968-16975 [DOI] [PubMed] [Google Scholar]
- 128.Nolan G, Yu P, Huang C, Shen M, Quast J, Chan E, Xu X, Payan D, Luo Y: Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkB. Curr Biol 1999, 9:539-542 [DOI] [PubMed] [Google Scholar]
- 129.Deveraux Q, Reed J: IAP family proteins: suppressors of apoptosis. Genes Dev 1999, 13:239-252 [DOI] [PubMed] [Google Scholar]
- 130.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE: The inhibitors of apoptosis (IAPs) and their emerging role in cancer Oncogene 1998, 17:3247-3259 [DOI] [PubMed] [Google Scholar]
- 131.Miller L: An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol 1999, 9:323-328 [DOI] [PubMed] [Google Scholar]
- 132.Sun C, Cal M, Gunasekera A, Meadows R, Wang H, Chen J, Zhang H, Wu W, Xu M, Ng S, Fesik S: NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 1999, 401:818-821 [DOI] [PubMed] [Google Scholar]
- 133.HInds M, Norton R, Vaux D, Day C: Solution structure of a baculoviral inhibitor of apoptosis (IAP) repeat. Nat Struct Biol 1999, 6:648-651 [DOI] [PubMed] [Google Scholar]
- 134.Verdecia MA, Huang, H-K, Dutil E, Kaiser DA, Hunter T, Noel JP: Structure of the Human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Biotechnol 2000, XX:1–25 [DOI] [PubMed]
- 135.Yang Y, Fang S, Jensen J, Weissman A, Ashwell J: Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288:874-877 [DOI] [PubMed] [Google Scholar]
- 136.Hauser H, Bardroff M, Pyrowolakis G, Jentsch S: A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J Cell Biol 1998, 141:1415-1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen Z, Naito M, Hori S, Mashima T, Yamori T, Tsuruo T: A human IAP family gene, apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun 1999, 264:847-854 [DOI] [PubMed] [Google Scholar]
- 138.Fraser A, James C, Evan G, Hengartner M: Caenorhabditis elegans inhibitor of apoptosis protein (IAP) homologue BIR-1 plays a conserved role in cytokinesis. Curr Biol 1999, 9:292-301 [DOI] [PubMed] [Google Scholar]
- 139.Vaux D, Uren A, Beilharz T, O’Connell M, Bugg S, VanDriel R, Lithgow T: Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc Natl Acad Sci Cell Biol 1999, 96:10170-10175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Deveraux QL, Takahashi R, Salvesen GS, Reed JC: X-linked IAP is a direct inhibitor of cell death proteases. Nature 1997, 388:300-304 [DOI] [PubMed] [Google Scholar]
- 141.Roy N, Deveraux QL, Takashashi R, Salvesen GS, Reed JC: The c-IAP-1 and c-IaP-2 proteins are direct inhibitors of specific caspases. EMBO J 1997, 16:6914-6925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Deveraux Q, Leo E, Stennicke H, Welsh K, Salvesen G, Reed J: Cleavage of human inhibitor of apoptosis protein xiap results in fragments with distinct specificities for caspases. EMBO J 1999, 18:5242-5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC: A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 1998, 273:7787-7790 [DOI] [PubMed] [Google Scholar]
- 144.Zhou Q, Krebs J, Snipas S, Price A, Alnemri E, Tomaselli K, Salvesen G: Interaction of the baculovirus antiapoptotic protein p35 with capases: specificity, kinetics, and characterization of the caspase/p35 complex. Biochemistry 1998, 37:10757-10765 [DOI] [PubMed] [Google Scholar]
- 145.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula M, Alnemri ES, Salvesen GS, Reed JC: IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 1998, 17:2215-2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Huang Q, Deveraux QL, Maeda S, Salvesen GS, Stennicke HR, Hammock BD, Reed JC: Evolutionary conservation of apoptosis mechanisms: Lepidopteran and baculoviral inhibitor of apoptosis proteins are inhibitors of mammalian caspase-9. Proc Natl Acad Sci USA 2000, 97:1427-1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Farahani R, Baird S, Besner-Johnson A, Lefebvre C, Kang X, Salih M, Aubry H, Tamai K, Guan X, Ioannou P, Crawford TO, de Jong PJ, Surh L, Ikeda J-E, Korneluk RG, MacKenzie A: The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80:167-178 [DOI] [PubMed] [Google Scholar]
- 148.Ambrosini G, Adida C, Altieri D: A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997, 3:917-921 [DOI] [PubMed] [Google Scholar]
- 149.Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC: Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol 1998, 152:43-49 [PMC free article] [PubMed] [Google Scholar]
- 150.Madden SL, Zhang L, Lash AE, Yu J, Rago C, Lal A, Wang CJ, Beaudry GA, Ciriello KM, Cook BP, Dufault MR, Ferguson AT, Gao Y, He T-C, Hermeking H, Hiraldo SK, Hwang PM, Lopez MA, Luderer HF, Mathews B, Petroziello JM, Polyak K, Zawel L, Zhang W, Zhang X, Zhou W, Haluska FG, Jen J, Sukumar S, Landes GM, Riggins GJ, Vogelstein B, Kinzler KW, Velculescu VE: Analysis of human transcriptomes. Nat Genet 1999, 23:387-388 [DOI] [PubMed] [Google Scholar]
- 151.Li F, Ambrosini G, Chu EY, Marchisio PC, Altieri DC: Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396:580-584 [DOI] [PubMed] [Google Scholar]
- 152.Li F, Ackermann E, Bennett C, Rothermel A, Plescia J, Tognin S, Villa A, Marchisio P, Altieri D: Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol 1999, 1:461-466 [DOI] [PubMed] [Google Scholar]
- 153.Li F, Flanary PL, Altieri DC, Dohlman HG: Cell division regulation by BIR1, a member of the inhibitor of apoptosis family in yeast. J Biol Chem 2000, 275:6707-6711 [DOI] [PubMed] [Google Scholar]
- 154.Tamm I, Kornblau S, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero D, Tudor G, Qui Y, Monks A, Andreeff M, Reed J: Expression and prognostic significance of iap family genes in human cancers and myeloid leukemias. Clin Cancer Res 2000, 6:1796-1803 [PubMed] [Google Scholar]
- 155.Steller H: Mechanisms and genes of cellular suicide. Science 1995, 267:1445-1449 [DOI] [PubMed] [Google Scholar]
- 156.Vucic D, Kaiser WJ, Miller LK: Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol Cell Biol 1998, 18:3300-3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang S, Hawkins C, Yoo S, Muller H, Hay B: The drosophila caspase inhibitor diap1 is essential for cell survival and is negatively regulated by hid. Cell 1999, 98:453-463 [DOI] [PubMed] [Google Scholar]
- 158.Harvey AJ, Bidwai AP, Miller LK: Doom, a product of the Drosophila mod(mdg4) gene, induces apoptosis and binds to baculovirus inhibitor-of-apoptosis proteins. Mol Cell Biol 1997, 17:2835-2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goyal L, McCall K, Agapite J, Hartwieg E, Steller H: Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J 2000, 19:589-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Steller H: Drosophila p53: meeting the grim reaper. Nat Cell Biol 2000, 2:E100-E102 [DOI] [PubMed] [Google Scholar]
- 161.Haining WN, Carboy-Newcomb C, Wei CL, Steller H: The proapoptotic function of Drosophila Hid is conserved in mammalian cells. Proc Natl Acad Sci USA 1999, 96:4936-4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Du C, Fang M: Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102:33-42 [DOI] [PubMed] [Google Scholar]
- 163.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL: Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102:43-53 [DOI] [PubMed] [Google Scholar]
- 164.Thress K, Henzel W, Shillinglaw W, Kornbluth S: Scythe: a novel reaper-binding apoptotic regulator. EMBO J 1998, 17:6135-6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Reed JC: Double identity for proteins of the Bcl-2 family. Nature 1997, 387:773-776 [DOI] [PubMed] [Google Scholar]
- 166.Adams J, Cory S: The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281:1322-1326 [DOI] [PubMed] [Google Scholar]
- 167.Reed J: Bcl-2 family proteins. Oncogene 1998, 17:3225-3236 [DOI] [PubMed] [Google Scholar]
- 168.Gross A, McDonnell J, Korsmeyer S: BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999, 13:1899-1911 [DOI] [PubMed] [Google Scholar]
- 169.Kelekar A, Thompson CB: Bcl-2 family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol 1998, 8:324-330 [DOI] [PubMed] [Google Scholar]
- 170.Reed JC: Mechanisms of Bcl-2 family protein function and dysfunction in health and disease. Behring Inst Mitt 1996, 97:72-100 [PubMed] [Google Scholar]
- 171.Tsujimoto Y, Cossman J, Jaffe E, Croce C: Involvement of the Bcl-2 gene in human follicular lymphoma. Science 1985, 228:1440-1443 [DOI] [PubMed] [Google Scholar]
- 172.Weiss LM, Warnke RA, Sklar J, Cleary ML: Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med 1987, 317:1185-1189 [DOI] [PubMed] [Google Scholar]
- 173.Reed JC, Miyashita T, Takayama S, Wang H-G, Sato T, Krajewski S, Aimé-Sempé C, Bodrug S, Kitada S, Hanada M: Bcl-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem 1996, 60:23-32 [DOI] [PubMed] [Google Scholar]
- 174.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M: Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275:967-969 [DOI] [PubMed] [Google Scholar]
- 175.Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T: Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 1997, 385:637-640 [DOI] [PubMed] [Google Scholar]
- 176.Miyashita T, Reed JC: Tumor suppressor p53 is a direct transcriptional activator of human Bax gene. Cell 1995, 80:293-299 [DOI] [PubMed] [Google Scholar]
- 177.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC: Upregulation of Bax protein levels in neurons following cerebral ischemia. J Neurosci 1995, 15:6364-6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ: BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 1996, 17:401-411 [DOI] [PubMed] [Google Scholar]
- 179.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM, Jr: Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol 1997, 139:205-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.MacGibbon GA, Lawlor PA, Sirimanne ES, Walton MR, Connor B, Young D, Williams C, Gluckman P, Faull RLM, Hughes P, Dragunow M: Bax expression in mammalian neurons undergoing apoptosis, and in Alzheimers disease hippocampus. Brain Res 1997, 750:223-234 [DOI] [PubMed] [Google Scholar]
- 181.Chen J, Flannery JG, LaVail MM, Steinberg RH, Xu J, Simon MI: bcl-2 overexpression reduces apoptotic photoreceptor cell death in three different retinal degenerations. Proc Natl Acad Sci USA 1996, 93:7042-7047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Middleton G, Nunez G, Davies AM: Bax promotes neuronal survival and antagonises the survival effects of neurotrophic factors. Development 1996, 122:695-701 [DOI] [PubMed] [Google Scholar]
- 183.Song Q, Kuang Y, Dixit VM, Vincenz C: Boo, a novel negative regulator of cell death, interacts with Apaf-1. EMBO J 1999, 18:167-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Inohara N, Gourley TS, Carrio R, Muniz M, Merino J, Garcia I, Koseki T, Hu Y, Chen S, Nunez G: Diva, a Bcl-2 homologue that binds directly to Apaf-1 and induces BH3-independent cell death. J Biol Chem 1998, 273:32479-32486 [DOI] [PubMed] [Google Scholar]
- 185.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Changs BS, Thompson CB, Wong S, Ng S, Fesik SW: X-ray and NMR structure of human Bcl-XL, an inhibitor of programmed cell death. Nature 1996, 381:335-341 [DOI] [PubMed] [Google Scholar]
- 186.Chou J, Li H, Salvesen G, Yuan J, Wagner G: Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 1999, 96:615-624 [DOI] [PubMed] [Google Scholar]
- 187.McDonnell J, Fushman D, Milliman C, Korsmeyer S, Cowburn D: Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell 1999, 96:625-634 [DOI] [PubMed] [Google Scholar]
- 188.Schendel S, Montal M, Reed JC: Bcl-2 family proteins as ion-channels. Cell Death Differ 1998, 5:372-380 [DOI] [PubMed] [Google Scholar]
- 189.Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB: Bcl-xL forms an ion channel in synthetic lipid membranes. Nature 1997, 385:353-357 [DOI] [PubMed] [Google Scholar]
- 190.Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC: Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA 1997, 94:5113-5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J-J, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou J-C: Inhibition of Bax channel-forming activity by Bcl-2. Science 1997, 277:370-372 [DOI] [PubMed] [Google Scholar]
- 192.Schlesinger P, Gross A, Yin X-M, Yamamoto K, Saito M, Waksman G, Korsmeyer S: Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci USA 1997, 94:11357-11362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Schendel S, Azimov R, Pawlowski K, Godzik A, Kagan B, Reed J: Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem 1999, 274:21932-21936 [DOI] [PubMed] [Google Scholar]
- 194.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW: Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 1997, 275:983-986 [DOI] [PubMed] [Google Scholar]
- 195.Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley R, Saxena S, Gietz R, Greenberg A: The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med 1997, 186:1975-1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wang K, Yin W-M, Chao DT, Milliman CL, Korsmeyer SJ: BID: a novel BH3 domain-only death agonist. Genes Dev 1996, 10:2859-2869 [DOI] [PubMed] [Google Scholar]
- 197.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Maundrell K, Antonsson B, Martinou J-C: Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c depletion during apoptosis. J Cell Biol 1999, 144:891-901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li H, Zhu H, Xu C, Yuan J: Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94:491-501 [DOI] [PubMed] [Google Scholar]
- 199.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94:481-490 [DOI] [PubMed] [Google Scholar]
- 200.Yin X-M, Wang K, Gross A, Klocke B, Roth K, Korsmeyer S: Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999, 400:886-891 [DOI] [PubMed] [Google Scholar]
- 201.Puthalakath H, Huang D, O’Reilly L, King S, Strasser A: The proapoptotic activity of the Bcl-2 family member bim is regulated by interaction with the dynein motor complex. Mol Cell 1999, 3:287-296 [DOI] [PubMed] [Google Scholar]
- 202.Franke TF, Cantley L: C. A bad kinase makes good. Nature 1997, 390:116-117 [DOI] [PubMed] [Google Scholar]
- 203.Datta S, Brunet A, Greenberg M: Cellular survival: a play in three Akts. Genes Dev 1999, 13:2905-2927 [DOI] [PubMed] [Google Scholar]
- 204.Wang H-G, Pathan N, Ethell I, Krajewski S, Yamaguchi Y, Shibasaki F, Mckeon F, Bobo T, Franke T, Reed J: Calcineurin promotes apoptosis by dephosphorylating BAD. Science 1998, 284:339-343 [DOI] [PubMed] [Google Scholar]
- 205.Nechushtan A, Smith C, Hsu Y-T, Youle R: Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 1999, 18:2330-2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Khaled AR, Kim K, Hofmeister R, Muegge K, Durum SK: Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc Nat Acad Sci USA 1999, 96:14476-14481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kroemer G, Reed JC: Mitochondrial control of cell death. Nat Med 2000, 6:513-519 [DOI] [PubMed] [Google Scholar]
- 208.Mancini M, Nicholson D, Roy S, Thornberry N, Peterson E, Casciola-Rosen L, Rosen A: The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J Cell Biol 1998, 140:1485-1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Krajewski S, Krajewska M, Ellerby LM, Welsch K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC: Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci USA 1999, 96:5752-5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Susin SA, Lornzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost M-C, Alzari PM, Kroemer G: Mitochondrial release of caspases-2 and -9 during the apoptotic process. J Exp Med 1999, 189:382-394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Susin S, Lorenzo H, Zamzami N, Marzo I, Snow B, Brothers G, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett D, Aebersold R, Siderovski D, Penninger J, Kroemer G: Molecular characterisation of mitochondrial apoptosis-inducing factor (AIF). Nature. 1999, 397:441-446 [DOI] [PubMed] [Google Scholar]
- 212.Nicholson DW: Apoptosis-based therapeutics: from bench to clinic, how far have we come? Nature 2000 (in press) [DOI] [PubMed]