

Abstract
Genetic causes of hereditary hemochromatosis (HH) include mutations in the HFE gene, a β2-microglobulin (β2m)-associated major histocompatibility complex class I-like protein. Accordingly, mutant β2m−/− mice have increased intestinal iron absorption and develop parenchymal iron overload in the liver. In humans, other genetic and environmental factors have been suggested to influence the pathology and severity of HH. Previously, an association has been reported between low numbers of lymphocytes and the severity of clinical expression of the iron overload in HH. In the present study, the effect of a total absence of lymphocytes on iron overload was investigated by crossing β2m−/− mice (which develop iron overload resembling human disease) with mice deficient in recombinase activator gene 1 (Rag1), which is required for normal B and T lymphocyte development. Iron overload was more severe in β2mRag1 double-deficient mice than in each of the single deficient mice, with iron accumulation in parenchymal cells of the liver, in acinar cells of the pancreas, and in heart myocytes. With increasing age β2mRag1−/− mice develop extensive heart fibrosis, which could be prevented by reconstitution with normal hematopoietic cells. Thus, the development of iron-mediated cellular damage is substantially enhanced when a Rag1 mutation, which causes a lack of mature lymphocytes, is introduced into β2m−/− mice. Mice deficient in β2m and Rag1 thus offer a new experimental model of iron-related cardiomyopathy.
The most relevant iron overload diseases in humans are primary, genetically determined, for example, hereditary hemochromatosis (HH) and secondary, transfusional and hemolysis related siderosis (eg, β-thalassemia). HH is an autosomal recessive disease, characterized by a defect in regulation of iron absorption, an increase of transferrin saturation, and progressive iron deposition predominantly in parenchymal cells of several organs. 1 Toxicity resulting from iron accumulation in selective target organs leads to the development of liver cirrhosis, cardiomyopathy, diabetes mellitus, hypogonadism, and arthritis. 1,2 The study of the mechanisms of selective tissue accumulation and damage in which iron excess is believed to play a role has been difficult in part as a result of the lack of adequate experimental models of iron overload.
Recently, a novel gene of the major histocompatibility complex class I family, HFE, has been found to be mutated in a large proportion of HH patients. 3 Previously, we characterized iron metabolism in major histocompatibility complex class I-deficient, β2-microglobulin knockout mice (β2m−/−), an animal model of HH. 4,5 Intestinal absorption of iron in β2m−/− mice is inappropriately increased, and transferrin saturation is abnormally high. 6 Pathological iron depositions occur predominantly in liver parenchymal cells, indicating defective iron storage in Kupffer cells. 5,7
In hemochromatosis patients, defective numbers of peripheral blood and liver lymphocyte populations are associated with a more severe clinical expression of iron overload. 8-10 Correction of the iron overload does not correct the reported anomalies in lymphocyte numbers, and patients with abnormally low numbers of lymphocytes reach high transferrin saturations at a faster rate than those with normal lymphocyte numbers after completion of the phlebotomy treatment. 9 Together these observations indicate that the lymphocyte abnormalities precede and are not the consequence of the iron overload.
To investigate the hypothesis that lymphocytes influence the development of iron overload, we introduced a deficiency in the recombinase activator gene 1 (Rag1) onto a β2m−/− genetic background. Rag1 deficiency results in total deficiency of B and T lymphocytes. 11 We report here the generation of double-deficient β2mRag1−/− mice, which develop spontaneous iron overload. Challenge with dietary iron loading was obtained by placing mice on an iron-enriched diet containing 2.5% (w/w) carbonyl iron. Iron burden was substantially aggravated by the additional absence of Rag1, with massive iron accumulation in liver parenchymal cells, acinar cells of the pancreas, and heart myocytes. Surprisingly, β2mRag1 double-knockout mice develop heart fibrosis, which could be prevented by reconstitution with normal hematopoietic cells. The β2m- and Rag1-deficient mice provide an interesting model to define the modifying influence of lymphocytes in iron homeostasis. In addition, this mouse model will facilitate investigation into the pathogenesis of iron-mediated myocardial failure.
Materials and Methods
Mice
C57BL/6 mice aged 6 to 8 weeks were purchased from the IFFA Credo (Brussels, Belgium) and used as controls. The β2-microglobulin knockout (β2m−/−) mice were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA), and Rag1−/− 11 were obtained from Dr. S. Tonegawa (Massachusetts Institute of Technology, Cambridge, MA). Both mutant mice have been back-crossed onto the C57BL/6 background. β2m−/− mice were bred to Rag1−/− to generate F1 offspring that were heterozygous for both genes. Because the β2m and Rag1 genes are closely linked, homozygous double knockout could only be obtained through recombination by breeding. Recombinants were detected as follows: the F2 offspring of the F1 interbreeding were screened by flow cytometry analysis (FACS) for the absence of T and B lymphocytes in peripheral blood samples. Mice identified as Rag1−/− were screened for recombination events by Southern blotting, using a β2m-specific probe, as described. 5 Identified Rag1−/−β2m+/−mice were further intercrossed, and the F3 offspring were screened by FACS and Southern blotting. Double-deficient Rag1−/−β2m−/− mice were further bred in our animal facility. For all strains, both males and females were studied. All animals were 8 weeks old at the beginning of the experiments.
All animals were given a commercial diet (RMH-B; Hope Farms, Woerden, The Netherlands), or, when indicated, an iron supplemented diet containing 2.5% (w/w) carbonyl iron (Sigma Immunochemicals, St. Louis, MO).
For all animal experiments, written approval was obtained from the local Animal Experiments Committee of Utrecht University (Utrecht, The Netherlands).
Measurement of Tissue Iron Levels
Organ samples were weighed wet, then dried overnight at 106°C and weighed again. The dried samples were ashed in an oven at 500°C for 17 hours, then fully solubilized in 6 mol/L HCl, and the final solution was adjusted with demineralized water to a final HCl concentration of 1.2 mol/L. Iron concentration of the samples was determined by flame atomic absorption spectrometry (Varian SpectrAA 250 Plus; Varian, Mulgrave, Victoria, Australia).
Transferrin Saturation and Hematological Measurements
Heparinized blood was obtained by orbital puncture under diethylether anesthesia. Hemoglobin, hematocrit, and mean corpuscular volume were determined using a Coulter-S counter (Coulter Electronics, Hialeah, FL). Plasma iron and total iron-binding capacity were determined by the ferrozine method (Iron FZ Test; Roche, Basel, Switzerland) with the COBAS-BIO autoanalyzer (Hoffman-La Roche BV, Mijdrecht, The Netherlands). Transferrin saturation was calculated from the total iron-binding capacity and plasma iron values.
Histology
Samples of liver, spleen, kidney, lung, heart, and pancreas were fixed in buffered 4% formaldehyde. After routine histology processing, the paraffin sections were stained with hematoxylin and eosin and with azan for demonstration of fibrosis. Ferric iron, Fe(III), was detected by Perl’s blue staining.
Electron Microscopy
Small pieces of pancreas and heart were fixed in a modified Karnovsky fixative consisting of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.8 mol/L Na-cacodylate buffer, supplemented with 0.25 mmol/L CaCl2, and 0.5 mmol/L MgCl2 for at least 24 hours at 4°C. The tissue was washed twice with the same buffer, postfixed in 1% OsO4 and embedded in Epon 812. Semithin sections (1 μm) were stained with methylene blue and pararosanilin. Ultrathin sections (60 nm) were cut and contrasted with 3% uranyl magnesium acetate for 45 minutes at 63°C followed by Reynolds’ lead citrate for 10 minutes. Stained and unstained sections were viewed in a Jeol JEM 1010 electron microscope (Joel LTD, Tokyo, Japan).
Gastrointestinal Iron Absorption
For iron absorption tests the mice were fasted for 6 hours and housed for 3 days in cages equipped with grates to minimize coprophagy. All test doses were freshly prepared and were administered in aqueous solution using demineralized water. Measurement of iron absorption was performed as previously described. 6 Ferric-citrate (Sigma Immunochemicals) was added to 59Fe(III) citrate to obtain a total of 5 μg per mouse, with a 20-fold molar excess of sodium citrate dihydrate (Sigma Immunochemicals) to maintain mononuclear ferric-citrate complexes and to prevent precipitation. Each mouse received ∼50 kBq of 59Fe.
The test dose was orally applied with the use of an olive-tipped oroesophageal needle. Total body radioactivity was measured with a whole-body γ counter (Automatic Scanner DS4/4S; Tracelab Ltd., Weybridge, Surrey, UK). The values were corrected for radioisotope decay and day-to-day fluctuations of the scanner with the use of a radium source. 59Fe absorption was determined by whole-body counting 7 days after administration of the test dose. When the animals were tested twice for iron absorption, background values of the first test dose were corrected for radioisotope decay.
Fetal Liver Cell Transfer
Recipient animals aged 8 weeks were lethally irradiated (9.5 Gy) and reconstituted with 5 × 10 6 fetal liver (embryonic day E13.5) cells by intravenous injection. Chimeras were sacrificed at 28 to 36 weeks after reconstitution and chimerism was monitored by flow cytometry analysis using αβTCR, B220, Mac-1, CD4, CD8, and H141.31.10 (anti-Kb) mAb (PharMingen, San Diego, CA).
Flow Cytometry
Expression of cell surface proteins was assayed by direct immunofluorescence. Samples of blood and spleen were stained with fluorescein isothiocyanate-conjugated or phycoerythrin-conjugated mAbs. Samples were then treated with FACS Lysing Solution (Becton Dickinson, Mountain View, CA) and washed in phosphate-buffered saline containing 2.5% fetal calf serum and 0.05% sodium azide. Fluorescence intensities were measured on a FACScan flow cytometer (Becton Dickinson).
Statistical Analysis
Results are presented as mean ± SEM. Student’s t-test was used for comparison between the control and knockout mouse groups. For individual comparisons between two measurements, the paired t-test was used. The level of significance was preset at P < 0.05.
Results
Altered Iron Storage and Distribution in β2mRag1 Double-Knockout Mice
β2mRag1−/− double-knockout mice obtained from β2m−/− and Rag1−/− crossings were screened by Southern blot analysis on DNA extracted from tail samples. The absence of T and B lymphocytes was confirmed by FACS analysis. The majority (>95%) of the peripheral blood mononuclear cells and spleen cells expressed Mac-1 (CD11b), which stains macrophages, natural killer cells, and granulocytes, and were negative for αβTCR (T lymphocytes), B220 (B lymphocytes), and Kb (β2m-dependent, major histocompatibility complex class I) (data not shown).
Determination of organ iron concentration, transferrin saturation, histochemical visualization of the cellular distribution of iron, and pathological examination of the extent of injury provide essential information about the type and degree of iron loading. To characterize iron homeostasis in β2mRag1 double-knockout mice these parameters were analyzed and compared to single-knockout and wild-type (B6) mice. The responses to iron overloading were studied by feeding animals with a carbonyl-iron-supplemented diet (2.5% w/w). No significant differences were found between males and females, and hence the results for both genders were pooled.
Total Iron in Organs, Plasma Iron, and Plasma Transferrin Saturation
Spontaneous Iron Overload
To determine iron distribution in different organs from mice fed with a standard diet (n = 9 to 12 per group), iron content was measured by flame atomic absorption spectrometry. All mice were sacrificed at 5 months of age. β2m single and β2mRag1 double-knockout mice had significantly higher hepatic iron levels than B6 wild-type and Rag1−/− mice (Table 1 ▶ and Figure 1a ▶ ; P < 0.0001). In contrast, splenic total iron levels of β2m−/−, Rag1−/−, and double-knockout mice were lower than those seen in B6 wild-type mice (Figure 1b ▶ ; P < 0.01), a finding confirmed histologically. Noteworthy, β2mRag1 double-knockout mice fed the standard diet had significantly higher iron levels in the heart than Rag1 single, β2m single-knockout, and B6 wild-type mice (Table 1 ▶ and Figure 1c ▶ ; P < 0.0001). Plasma iron and transferrin saturation, as early markers of iron overload, were significantly higher in β2m-single and β2mRag1 double-knockout mice (plasma iron >40 μmol Fe/ml; transferrin saturation >80%) when compared to B6 control or Rag1−/− mice (plasma iron <25 μmol Fe/ml; transferrin saturation <60%; P < 0.001).
Table 1.
Tissue Iron Concentration in Mice Fed a Standard Diet
| Organ | μg Fe/g dry weight | P | |||||
|---|---|---|---|---|---|---|---|
| B6 | β2m−/− | Rag1−/− | β2mRag1−/− | versus B6 | versusβ2m−/− | versusRag1−/− | |
| Liver | 258 ± 61 | 552 ± 101 | 256 ± 52 | 682 ± 313 | <0.0001 | NS | <0.0001 |
| Heart | 331 ± 56 | 341 ± 74 | 383 ± 104 | 561 ± 138 | <0.0001 | <0.0001 | <0.0001 |
| Pancreas | 151 ± 33 | 168 ± 32 | 136 ± 26 | 604 ± 731 | NS | NS | NS |
| Spleen | 3688 ± 1299 | 1512 ± 500 | 2130 ± 1105 | 1458 ± 605 | <0.003 | NS | NS |
| Kidney | 287 ± 30 | 303 ± 68 | 254 ± 86 | 283 ± 70 | NS | NS | NS |
| Lungs | 393 ± 32 | 456 ± 68 | 309 ± 99 | 417 ± 59 | NS | NS | NS |
Data are presented as mean ± SD. Animals were analyzed at 5 months of age. P = Student’s t-test for comparison of β2mRag1−/− mice with B6 control, β2m−/−, and Rag1−/− mice.
Figure 1.

Distribution of iron storage in β2mRag1 double-knockout mice and controls kept on a standard diet (white bars; n = 9 to 12 per group) or iron-enriched diet (black bars; n = 12 to 16 per group). Animals were 2 months old at the start of the experiment and were sacrificed at 5 months of age. Total iron in livers (a); spleens (b); hearts (c); pancreas (d). Tissue samples from B6, β2m, Rag1−/−, and β2mRag1−/− (indicated as DKO, double knockout) mice were analyzed by flame atomic spectrometry for quantitative determination of iron. Values represent mean ± SEM. a: Total iron in livers of β2m and β2mRag1−/− (DKO) mice was significantly higher in animals kept on a standard diet (**, P < 0.001; ***, P < 0.0001 compared with B6 wild-type mice). b: Resistance to iron storage in spleen after dietary iron loading in β2m and β2mRag1−/− (DKO) mice (*, P < 0.01; **, P < 0.001; ***, P < 0.0001 compared with B6 wild-type mice). c: Persistent significantly higher iron storage in hearts of β2mRag1−/− (DKO) mice (***, P < 0.0001 compared with B6 wild-type mice). d: Increased iron accumulation in pancreas of dietary iron loaded β2mRag1−/− (DKO) mice (*, P < 0.01; **, P < 0.001 compared with B6 wild-type mice).
Overall, when comparing mice kept on a standard diet, body iron levels were the highest in β2mRag1 double-knockout mice, followed by β2m single-knockout mice, whereas no significant differences in body iron levels were found between Rag1 single-knockout and B6 wild-type mice.
Dietary Iron Overload
After feeding the animals an iron-enriched diet for 12 weeks (n = 12 to 16 per group), both β2m-single and β2mRag1 double-knockout mice were unable to increase iron levels in spleens (Figure 1b ▶ ). This inability to store excess iron in spleens was most evident in the β2mRag1 double-knockout mice, which had only half the total iron content (44 ± 8 μg Fe) of that in wild-type mice (96 ± 8 μg Fe). On the other hand significantly higher amounts of iron were found in the heart (Figure 1c ▶ ; P < 0.0001) and pancreas of double-knockout, but not single-knockout mice (Figure 1d ▶ ; P < 0.001) after dietary iron loading. Total iron levels in lungs and kidneys were not significantly different between mouse strains and treatments (data not shown).
Transferrin saturation after feeding the iron-enriched diet, increased in B6 control and Rag1−/− mice to >80%, reaching levels similar to those seen in β2m- and β2mRag1 double-knockout mice kept on a standard diet. Plasma iron concentration in iron-loaded animals was significantly lower in B6 control mice compared to all of the other strains (B6: plasma iron <29 versus >50 μmol Fe/ml in all other strains; P < 0.01).
Taken together, these results show that iron burden is accentuated in dietary iron-loaded β2mRag1 double-knockout mice when compared to the respective single knockout mice.
Cellular Distribution of Storage Iron
A typical feature of pathological iron overload in humans is the cellular distribution of storage iron, which has been particularly difficult to mimic in rodents. Therefore, we determined histologically the cellular distribution of storage iron in liver, pancreas, and heart in mice fed a standard diet and in dietary iron-loaded animals.
Spontaneous Iron Overload
Perl’s blue-staining of liver sections from β2m-single and β2mRag1 double-knockout mice kept on a standard diet revealed the presence of excess iron, which was predominantly in parenchymal cells (data not shown). 4,5 Moderate deposits were also observed in the pancreas and the heart of 24- to 30-week-old β2mRag1 double-knockout mice, but not in the β2m-single and Rag1 single-knockout mice or B6 wild-type mice (data not shown).
Dietary Iron Overload
As previously reported for shorter loading periods, 5 iron deposition in the liver of B6 wild-type mice fed an iron-enriched diet up to 12 weeks was particularly prominent in Kupffer cells, and was also present in parenchymal cells (Figure 2a ▶ ). Surprisingly, Rag1 single-knockout mice, that supposedly have normal Kupffer cells, develop hepatic iron overload on dietary iron loading exclusively in parenchymal cells (data not shown), like HH patients and β2m−/− mice. 5 Dietary iron-loaded β2mRag1 double-knockout mice show heavy iron depositions in the livers that corresponded to the appearance of hepatocyte clusters (Figure 2b ▶ ). A remarkable iron loading was present in the pancreas and the heart of β2mRag1 double-knockout mice (Figure 2, d ▶ and f), which was not observed in control B6 (Figure 2, c ▶ and e), and β2m single-knockout mice (data not shown). Importantly, in the pancreas this prominent iron deposition was present in acinar cells (Figure 2d ▶ ), and in the heart it was present in myocytes and in the interstitial tissue (Figure 2f ▶ ).
Figure 2.
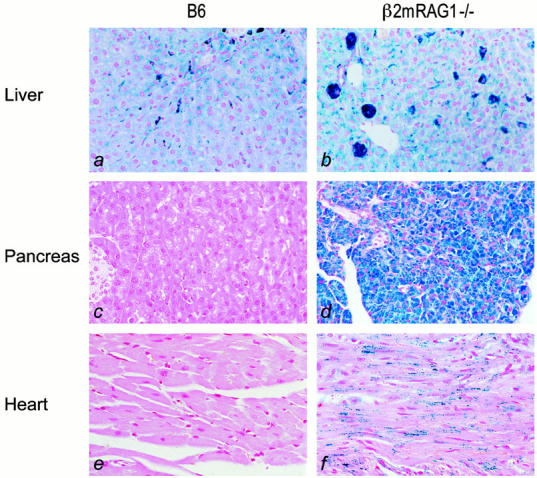
Storage of excess iron in organs of mice fed an iron-enriched diet (Perl’s blue staining). Animals were 2 months old at the start of the experiment and were sacrificed at 5 months of age. a: Light micrograph of a liver section from B6 showing heavy iron staining present in Kupffer cells; hepatic parenchymal cells have a low to moderate iron deposition. b: Liver section from β2mRag1−/− double-knockout mouse showing heavy iron deposition in hepatic parenchymal cells, forming hepatic cell clusters. c and e: Pancreas and heart of B6 mouse are devoid of iron. d: In the pancreas of β2mRag1−/− double-knockout mice prominent iron deposition is present in acinar cells. f: In the heart of β2mRag1−/− double-knockout mice iron deposition is present in myocytes and interstitial tissue. Original magnification, ×300.
Examination of hearts from β2mRag1 double-knockout mice by electron microscopy revealed frequent lysosomal structures containing granular electron-dense material in the cytoplasm of myocytes (Figure 3, a ▶ and b). Similar lysosomal iron deposition was observed in mesenchymal perivascular cells. In the pancreas, the acinar cells contained large lysosomes of moderate electron density (Figure 3c ▶ ). In these lysosomes, scattered ferritin particles were present (Figure 3d ▶ ). Ferritin accumulation was also evident in the cytoplasm of acinar cells.
Figure 3.
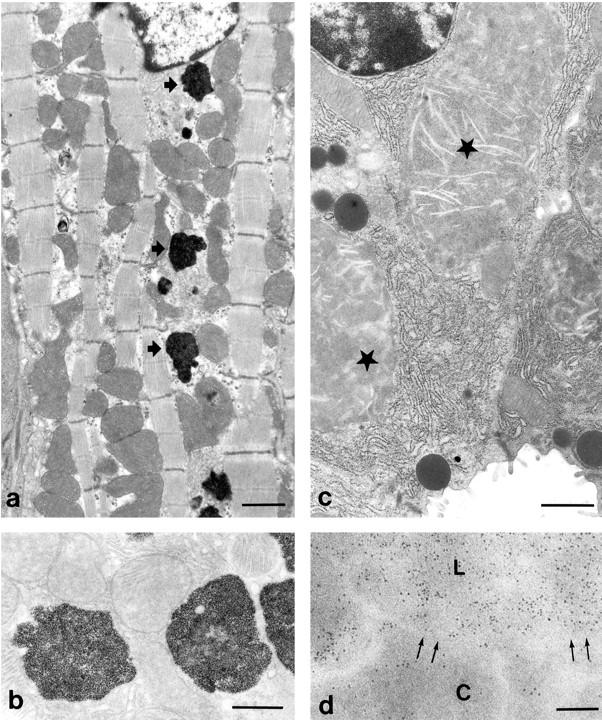
Intracellular iron deposition in heart and pancreas of a 5-month-old β2mRag1−/− mouse fed an iron-enriched diet. a: Electron micrograph of a myocyte showing electron-dense lysosomes containing iron (arrows). Scale bar, 1 μm. b: Unstained section showing lysosomes of myocytes (see a) with densely packed iron particles. Scale bar, 500 nm. c: Acinar cell of the pancreas containing large lysosomal structures of moderate electron density (asterisks). Scale bar, 1 μm. d: Unstained section of an acinar cell showing detail of a lysosome (L) and part of the cytoplasm (C). Arrows: membrane of the lysosome and cytoplasm. Electron-dense ferritin particles are present in a higher density in the lysosome than in the cytoplasm. Scale bar, 100 nm.
Overall, dietary iron-loaded β2mRag1 double-knockout mice develop a more severe iron burden in multiple organs than each of the single-knockout mice, indicating an additive effect of the two mutations.
Erythroid Parameters
To exclude the possibility that anemia could account for the abnormal iron storage defect in β2mRag1 double-knockout mice, several erythroid parameters were determined. The results demonstrated that hemoglobin, hematocrit, and mean corpuscular volume were even higher in β2m-single and in β2mRag1 double-knockout mice when compared to B6 and Rag1−/− mice fed a standard diet (Table 2) ▶ . We observed an increase of hemoglobin, hematocrit, and mean corpuscular volume values to a similar extent when B6 and Rag1−/− mice were fed the iron-enriched diet for 12 weeks. Thus, the excess storage iron found in β2mRag1 double-knockout mice could not be attributed to defective erythropoiesis or hemoglobin synthesis.
Table 2.
Erythroid Parameters
| Strain | Treatment | n | RBC, ×1012/L | Hb, mmol/L | HCT, % | MCV, fl |
|---|---|---|---|---|---|---|
| B6 | − | 8 | 9.0 ± 0.5 | 8.7 ± 0.5 | 39 ± 3 | 43 ± 2 |
| Carbonyl-iron | 8 | 9.4 ± 0.7 | 10.4 ± 0.3 | 44 ± 4 | 46 ± 1 | |
| βm−/− | − | 6 | 9.2 ± 0.6 | 9.9 ± 0.9 | 44 ± 4 | 48 ± 2 |
| Carbonyl-iron | 8 | 9.9 ± 0.1 | 10.8 ± 0.2 | 49 ± 2 | 49 ± 2 | |
| RAG1−/− | − | 7 | 8.9 ± 0.8 | 8.9 ± 0.5 | 39 ± 3 | 42 ± 2 |
| Carbonyl-iron | 8 | 9.4 ± 0.4 | 9.9 ± 0.5 | 43 ± 3 | 46 ± 2 | |
| β2mRAG1−/− | − | 8 | 9.6 ± 0.5 | 10.2 ± 0.7 | 44 ± 4 | 46 ± 1 |
| Carbonyl-iron | 6 | 9.4 ± 0.1 | 10.5 ± 0.2 | 47 ± 2 | 47 ± 2 |
Data are presented as mean ± SD. n, number of animals. Animals were 5 months old. RBC, red blood cell; Hb, hemoglobin; HCT, hematocrit; MCv, mean corpuscular volume.
Iron Absorption
To investigate the effect of the Rag1 mutation on the absorption of iron, ferric iron, Fe(III), absorption 6 was measured before and after feeding an iron-enriched diet for 14 days (Figure 4) ▶ . Ferric iron absorption after this treatment significantly decreased in all mouse strains (P < 0.0001). However, iron absorption in β2m-single and β2mRag1 double-knockout mice was persistently higher, before and after treatment, when compared to wild-type (B6) or Rag1 single-knockout mice (P < 0.0001, Figure 4 ▶ ). No significant differences were found between iron absorption in β2m-single and β2mRag1 double-knockout mice, indicating that the Rag1 mutation has no further influence on iron absorption in the gut.
Figure 4.

Increased iron absorption in β2m-single and β2mRag1 double-knockout (indicated as DKO) mice. All animals were 8 weeks old at the start of the experiment. Iron absorption was measured before (black circles) and after (white circles) feeding an experimental diet containing 2.5% w/w carbonyl iron for 14 days. Each group of mice (n = 11 to 14 per group) was given a radioactive test dose solution containing 59Fe. Iron absorption was determined at day 7 in a whole-body counter. Individual values for each mouse are shown.
Heart Fibrosis in β2mRag1 Double-Knockout Mice
Iron deposition in the heart deserves special interest, because heart failure is a frequent cause of death in untreated HH and posttransfusional secondary hemochromatosis. 12-16 Remarkably, 17 out of 21 β2mRag1 double-knockout mice aged between 20 and 28 weeks and kept on a standard diet developed heart fibrosis, as detected by azan staining, which was never seen in β2m- and Rag1-single-knockout mice or control mice of the same age and kept on the standard diet (Figure 5, a ▶ and b). Only after feeding an iron-enriched diet for 3 months, heart fibrosis was additionally observed in Rag1 single-knockout mice, but not in β2m single-knockout or B6 wild-type mice (data not shown).
Figure 5.

Heart fibrosis in β2mRag1−/− mice kept on a standard diet (azan staining). a: The hearts of B6 control mice show a normal histology. b: Extensive fibrotic lesions (blue) are present in the heart of a 5-month-old β2mRag1−/− mouse. c and d: Heart histology in radiation chimeras; animals were 2 months old at the start of the experiment. c: Prevention of heart fibrosis in a β2mRag1−/− mouse reconstituted with fetal liver cells from B6 donor mice (9 months old). d: Fibrotic lesions in a β2mRag1−/− mouse reconstituted with fetal liver cells from β2mRag1−/− donor mice (7 months old). Original magnification, ×3.2.
Previously we have demonstrated that reconstitution of β2m−/− mice with normal hematopoietic cells, redistributes the iron from parenchymal to Kupffer cells in the liver. 5 To further investigate the influence of hematopoietic cells in the development of iron-related heart fibrosis, we reconstituted lethally irradiated 8-week-old β2mRag1 double-knockout mice with fetal liver-derived hematopoietic progenitor cells from normal mice. All reconstituted β2mRag1 double-knockout mice (n = 4) showed a normal histology up to 36 weeks of age (Figure 5 ▶ c). The control β2mRag1 double-knockout mice reconstituted with β2mRag1−/−-derived cells (n = 5) were sacrificed between 20 to 28 weeks when they became ill and had developed extensive fibrosis in the heart (Figure 5d ▶ ). Thus, wild-type hematopoietic cell transfer prevents the development of heart fibrosis in β2mRag1 double-knockout mice.
Discussion
The aim of this study was to investigate the modifying influence of lymphocytes in the pathology of iron overload. Such a modifying role has been suggested by the association between low numbers of T lymphocytes in patients with HH and a more severe clinical expression of iron overload. 8-10
In the β2m-deficient mice that develop a progressive iron overload similar to that seen in HH patients, 4-7 we introduced the Rag1 mutation, 11 to create a total absence of mature lymphocytes.
When kept on a standard diet, the double-knockout mice develop a more severe phenotype than the β2m-deficient mice, involving increased iron accumulation in the liver, heart, and pancreas. The β2mRag1 double-knockout mice have visible iron depositions specifically in parenchymal cells of the liver and significantly higher iron levels in the heart than single-knockout and control mice. This indicates that the additional absence of lymphocytes, in the β2m model of iron overload, exacerbates the accumulation of iron in target organs, especially the heart. Moreover, the double-deficient mice spontaneously develop fibrosis in the heart.
The observed phenotype in the double-deficient mice is also an accentuation of the phenotype of the Rag1 single-knockout mice, which can normally regulate iron absorption and storage, and do not develop heart fibrosis under standard conditions. Rag1 single-knockout mice will develop heart fibrosis after very long periods of dietary iron loading of at least 12 weeks. Thus, dietary iron loading in combination with the lack of lymphocytes leads to cardiomyopathy. Altered cellular distribution of the iron in the heart may be a contributing factor in the development of cardiomyopathy, and may change in the absence of lymphocytes, as was observed in the liver of Rag1 single-knockout mice after dietary overloading. In the β2mRag1 double-knockout mice, dietary iron loading is not necessary because the β2m mutation leads to iron overload already under normal conditions.
When fed an iron-supplemented diet, β2mRag1 double-knockout mice, like β2m-single and HFE−/− mice, 17 have a significantly lower capacity to store iron in the spleens when compared with B6 control mice on the same diet. This is partially because of the absence of a functional HFE-β2m complex, which could lead to defective storage of iron in reticuloendothelial cells. Importantly, HH patients have been reported to have a defect in iron storage in reticuloendothelial cells. 18-20 The lower capacity to store iron may be aggravated by the lack of lymphocytes. 21,22 The lack of lymphocytes alone in Rag1-deficient mice leads to an aberrant storage of iron exclusively in parenchymal cells on dietary iron overload, indicating that lymphocytes may influence the iron storage capacity of reticuloendothelial cells.
As a consequence of the deficient iron metabolism in the double-mutant mice, excess iron is progressively deposited in the liver, heart, and pancreas. Thus, dietary iron overload in double-mutant mice leads to an exacerbation of the pattern of tissue iron deposition observed when the mice are kept on a standard diet.
Iron deposition in the hearts of β2mRag1 double-knockout mice, presumably leading to fibrosis, deserves special attention because heart failure is the most important life-threatening situation in untreated HH and in secondary hemochromatosis. 12-16 To our knowledge, experimentally induced iron-related cardiomyopathy has never been reported before in mice.
Cardiac manifestations are apparent in ∼20% to 30% of patients presenting with clinical manifestations of HH. In younger patients they are often the presenting feature and almost always the cause of early death unless the iron is removed. 1 In both HH and secondary hemochromatosis the iron is found predominantly within myocytes, leading to degeneration and fibrosis, with disturbances of cardiac rhythm and eventually death. 12-16,23 The typical deposition of iron in myocytes and the associated tissue damage has been difficult to mimic in animal models. In rats, after regular feeding of carbonyl iron 24 or the more efficient trimethylhexanoyl-ferrocene, 25 modest iron deposits are found in endothelial cells and perivascular macrophages. In these animal models, no stainable iron is found in myocytes and cellular damage does not occur.
The mechanism by which excess iron in myocytes causes damage may involve oxidative stress and the consecutive alteration of myocyte functions, through the iron-catalyzed Fenton chemistry. 26,27 The reason why the heart is the first organ to be affected may relate to the fact that the anti-oxidant enzyme equipment varies among tissues. 28 It is interesting to note that in several other instances related to oxidative stress the heart also seems to be a major target organ involved. 29,30
A β2m-deficient mouse lacks appropriate surface expression of the HFE gene product. The introduction of the Rag1 mutation leads to the additional absence of T and B cells. In HH patients, a correlation between T lymphocytes and the severity of the iron overload has been reported. 8 In these patients the numbers of B lymphocytes are normal and do not change after phlebotomy treatment. 9 No direct influence of B lymphocytes on iron metabolism or iron-binding proteins has been suggested. Thus, it is unlikely that the absence of B cells leads to the reported effects on iron metabolism in β2mRag1 double-deficient mice.
On the other hand, T lymphocytes are major regulators of cytokine production, either directly or indirectly via regulation of macrophage function. The lack of iron storage in the Kupffer cells of dietary overloaded Rag1 mutant mice may be an illustration of such an indirect mechanism. Cytokines produced locally by T lymphocytes and macrophages, namely interleukins, tumor necrosis factor-α, and interferon-γ, are powerful modifiers of iron homeostasis. 31,32 For example, after dietary iron overload, wild-type mice respond with an increase in tumor necrosis factor-α production, which in turn down-regulates intestinal iron absorption via increase in ferritin expression in intestinal epithelial cell. 33 Such cytokine-induced alterations in iron metabolism are also clearly illustrated in the pathogenesis of anemia of chronic disease, the most frequent anemia found in hospitalized patients, often occurring in patients with chronic infectious, inflammatory, and neoplastic disorders. 34,35 In anemia of chronic disease, associated disturbances of iron homeostasis include withdrawal of the metal from the sites of erythropoiesis and the circulation to the storage compartment in the reticuloendothelial system.
Cytokines have also been associated with cardiomyocyte loss in other studies. 36 Cytokines secreted by T-helper type 1 lymphocytes, such as interleukin-1, interleukin-2, and interferon-γ can induce tumor necrosis factor production from target cells, including myocytes. Tumor necrosis factor and several other cytokines are able to induce nitric oxide production, which depresses cardiac function and can induce apoptosis. 37
The effect of dietary iron overload on the heart of Rag1 single-mutant mice and the exacerbated phenotype in the double-mutant mice suggest that dysregulation of cytokine production may be responsible for the specific cellular iron storage in the heart and the loss of cardiomyocytes. The cytokines may either be released by lymphocytes or be locally produced and regulated by lymphocytes.
The cardiac phenotype was prevented by transfer of normal hematopoietic cells into double-deficient mice, indicating that the combined effect of both mutations on the heart during the first 8 weeks of life could be reversed by the combined introduction of normal reticuloendothelial cells expressing a functional HFE molecule and mature lymphocytes expressing a functional antigen receptor. This reversal may reflect a redistribution of iron under the influence of normal hematopoietic cells.
In conclusion, the present study shows that the development of iron overload pathology is substantially enhanced when a Rag1 mutation, which causes a lack of mature lymphocytes, is introduced into β2m−/− mice. The β2mRag1 double-knockout mouse model represents an ideal animal model of iron-mediated cardiomyopathy, and will be a useful model to evaluate therapeutic strategies not only for prevention and correction of iron overload, but also for the treatment of iron-related tissue damage. In addition, mice deficient in both β2m and Rag1 offer a new experimental model for defining in vivo which lymphocytes play a role in iron-related pathological processes and by what mechanism. This model may contribute to the understanding of the heterogeneity of the pathology of HH in man. 38,39
Acknowledgments
We thank Toon Hesp, Jan Smits, and Else Dorrestein for taking care of the animals; and Dr. F. Arosa for stimulating discussions.
Footnotes
Address reprint requests to Manuela Santos, Molecular Immunology Laboratory, Instituto de Biologia Molecular e Celular, Rua do Campo Alegre, 823, 4150-180 Porto, Portugal. E-mail: msantos@ibmc.up.pt.
Supported by a grant from Junta Nacional de Investigação Científica e Tecnológica-PRAXIS XXI (BD/2866/94).
References
- 1.Bothwell TH, MacPhail AP: Hereditary hemochromatosis: etiologic, pathologic and clinical aspects. Semin Hematol 1998, 35:55-71 [PubMed] [Google Scholar]
- 2.Bottomley SS: Secondary iron overload disorders. Semin Hematol 1998, 35:77-86 [PubMed] [Google Scholar]
- 3.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo Jr R, Ellis MC, Fullan A, Hinton LM, Jones NI, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morkang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayne DT, Risch NJ, Bacon BR, Wolff RK: A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996, 13:399–408 [DOI] [PubMed]
- 4.De Sousa M, Reimão R, Lacerda R, Hugo P, Kaufmann SEH, Porto G: Iron overload in β2-microglobulin-deficient mice. Immunol Lett 1994, 39:105-111 [DOI] [PubMed] [Google Scholar]
- 5.Santos M, Schilham MW, Rademakers LHPM, Marx JJM, De Sousa M, Clevers H: Defective iron homeostasis in β2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J Exp Med 1996, 184:1975-1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santos M, Clevers H, De Sousa M, Marx JJM: Adaptive response of iron absorption to anemia, increased erythropoiesis, iron deficiency, and iron loading in β2-microglobulin knockout mice. Blood 1998, 91:3059-3065 [PubMed] [Google Scholar]
- 7.Rothenberg BE, Volan JR: β2m knockout mice develop parenchymal iron overload: a putative role for class I genes of the major histocompatibility complex in iron metabolism. Proc Natl Acad Sci USA 1996, 93:1529-1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reimão R, Porto G, De Sousa M: Stability of CD4/CD8 ratios in man: new correlation between CD4/CD8 profiles and iron overload in idiopathic haemochromatosis patients. CR Acad Sci Paris 1991, 313:481-487 [PubMed] [Google Scholar]
- 9.Porto G, Vicente C, Teixeira MA, Martins O, Cabeda JM, Lacerda R, Gonçalves C, Fraga J, Macedo G, Silva BM, Alves H, Justiça B, De Sousa M: Relative impact of HLA phenotype and CD4/CD8 ratios on the clinical expression of hemochromatosis. Hepatology 1997, 25:397-402 [DOI] [PubMed] [Google Scholar]
- 10.Arosa FA, Oliveira L, Porto G, da Silva BM, Kruijer W, Veltman J, De Sousa M: Anomalies of the CD8+ T cell pool in haemochromatosis: HLA-A3-linked expansions of CD8+CD28- T cells. Clin Exp Immunol 1997, 107:548-554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE: Rag-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68:869-877 [DOI] [PubMed] [Google Scholar]
- 12.Finch SC, Finch CA: Idiopathic hemochromatosis, an iron storage disease. Medicine 1955, 34:381-430 [DOI] [PubMed] [Google Scholar]
- 13.Buja LM, Roberts WC: Iron in the heart. Etiology and clinical significance. Am J Med 1971, 51:209-221 [DOI] [PubMed] [Google Scholar]
- 14.Cutler DJ, Isner JM, Bracey AW, Hufnagel CA, Conrad PW, Roberts WC, Kerwin DM, Weintraub AM: Hemochromatosis heart disease: an unemphasized cause of potentially reversible restrictive cardiomyopathy. Am J Med 1980, 69:923-928 [DOI] [PubMed] [Google Scholar]
- 15.MacDonald RA, Mallory GK: Hemochromatosis and hemosiderosis: study of 211 autopsied cases. Arch Intern Med 1960, 105:686-697 [PubMed] [Google Scholar]
- 16.Tuomainen T-P, Punnonen K, Nyyssönen K, Salonen JT: Association between body iron stores and the risk of acute myocardial infarction in men. Circulation 1998, 97:1461-1466 [DOI] [PubMed] [Google Scholar]
- 17.Zhou XY, Tomatsu S, Fleming RE, Parkkila S, Waheed VA, Jiang J, Fei Y, Brunt EM, Ruddy DA, Prass CE, Schatzman RC, O’Neill R, Britton RS, Bacon BR, Sly WS: HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci USA 1998, 95:2492-2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fillet G, Marsaglia G: Idiopathic haemochromatosis abnormality in RBC transport of iron by the reticuloendothelial system. Blood 1975, 46:1007-1015 [Google Scholar]
- 19.Flanagan PR, Lam D, Banerjee D, Valberg LS: Ferritin release by mononuclear cells in hereditary hemochromatosis. J Lab Clin Med 1989, 113:145-150 [PubMed] [Google Scholar]
- 20.Düllmann J, Wulfhekel U, Mohr A, Riecken K, Hausmann K: Absence of macrophage and presence of plasmacellular iron storage in the terminal duodenum of patients with hereditary hemochromatosis. Virchows Arch A Pathol Anat 1991, 418:241-247 [DOI] [PubMed] [Google Scholar]
- 21.Doherty TM: T cell regulation of macrophage function. Curr Opin Immunol 1995, 7:400-404 [DOI] [PubMed] [Google Scholar]
- 22.Tormey VJ, Faul J, Leonard C, Burke CM, Dilmec A, Poulter LW: T-cell cytokines may control the balance of functionally distinct macrophage populations. Immunology 1997, 90:463-469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry WL, Nienhuis AW, Wiener M, Miller DR, Canale VC, Piomelli S: Echocardiographic abnormalities in patients with transfusion-dependent anemia and secondary myocardial iron deposition. Am J Med 1978, 64:547-555 [DOI] [PubMed] [Google Scholar]
- 24.Iancu TC, Ward RJ, Peters TJ: Ultrastructural observations in the carbonyl iron-fed rat, an animal model of hemochromatosis. Virchows Arch B Cell Pathol 1987, 53:208-217 [DOI] [PubMed] [Google Scholar]
- 25.Braumann A, Wulfhekel U, Nielsen P, Balkenhol B, Düllmann J: Pattern of iron storage in the rat heart following iron overloading with trimethylhexanoyl-ferrocene. Acta Anat (Basel) 1994, 150:45-54 [DOI] [PubMed] [Google Scholar]
- 26.Imlay JA, Chin SM, Linn S: Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 1988, 240:640-642 [DOI] [PubMed] [Google Scholar]
- 27.Halliwell B: Free radical, antioxidants, and human disease: curiosity, cause or consequence? Lancet 1994, 344:721-724 [DOI] [PubMed] [Google Scholar]
- 28.Fridovich I: Superoxide radical and superoxide dismutases. Annu Rev Biochem 1995, 64:97-112 [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Saarri JT, Kang YJ: Weak antioxidant defences make the heart a target for damage in copper-deficient rats. Free Radic Biol Med 1994, 17:529-536 [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Huang T-T, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ: Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995, 11:376-381 [DOI] [PubMed] [Google Scholar]
- 31.Fahmy M, Young SP: Modulation of iron metabolism in monocyte cell line U937 by inflammatory cytokines: changes in transferrin uptake, iron handling and ferritin mRNA. Biochem J 1993, 296:175-181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss G, Bogdan C, Hentze MW: Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL13. J Immunol 1997, 158:420-425 [PubMed] [Google Scholar]
- 33.Ten Elshof AE, Brittenham GM, Chorney KA, Page MJ, Gerhard G, Cable EE, Chorney MJ: Gamma delta intraepithelial lymphocytes drive tumor necrosis factor-alpha responsiveness to intestinal iron challenge: relevance to hemochromatosis. Immunol Rev 1999, 167:223-232 [DOI] [PubMed] [Google Scholar]
- 34.Means RT, Krantz SB: Progress in understanding the pathogenesis of the anemia of chronic disease. Blood 1992, 80:1639-1647 [PubMed] [Google Scholar]
- 35.Weiss G, Wachter H, Fuchs D: Linkage of cell-mediated immunity to iron metabolism. Immunol Today 1995, 16:495-500 [DOI] [PubMed] [Google Scholar]
- 36.Meldum DR: Tumor necrosis factor in the heart (review). Am J Physiol 1998, 274:R577-R595 [DOI] [PubMed] [Google Scholar]
- 37.Ing DJ, Zang J, Dzau VJ, Webster KA, Bishopric NH: modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, bak, and bcl-x. Circ Res 1999, 84:21-33 [DOI] [PubMed] [Google Scholar]
- 38.Piperno A, Arosio C, Fargion S, Roetto A, Nicoli C, Girelli D, Sbaiz L, Gasparini P, Boari G, Sampietro M, Camaschella C: The ancestral haemochromatosis haplotype is associated with a severe phenotype expression in Italian patients. Hepatology 1996, 24:43-46 [DOI] [PubMed] [Google Scholar]
- 39.Crawford DHG, Jazwinska EC, Cullen LM, Powell LW: Expression of HLA-linked hemochromatosis in subjects homozygous or heterozygous for the C282Y mutation. Gastroenterology 1998, 114:1003-1008 [DOI] [PubMed] [Google Scholar]