

Abstract
Dendritic cells (DCs) are thought to be key elements in the initiation and maintenance of autoimmune diseases. In this study, we sought evidence that DCs recruited to the central nervous system (CNS), a site that is primarily devoid of resident DCs, play a role in the effector phase and propagation of the immune response in experimental autoimmune encephalomyelitis (EAE). After immunization of SJL mice with proteolipid protein 139-151 peptide, process-bearing cells expressing the DC markers DEC-205 and CD11c appeared early in the spinal cord. During acute, chronic, and relapsing EAE, DEC-205+ DCs expressing a lymphostimulatory phenotype (including the mature DC marker MIDC-8, major histocompatibility complex class II, CD40, and CD86 molecules) accumulated within the CNS inflammatory cell infiltrates. More prominent infiltration of the spinal cord parenchyma by mature DCs was observed in mice with relapsing disease. Macrophage inflammatory protein 3α, a chemokine active on DCs and lymphocytes, and its receptor CCR6 were up-regulated in the CNS during EAE. These findings suggest that intracerebral recruitment and maturation of DCs may be crucial in the local stimulation and maintenance of autoreactive immune responses, and that therapeutic strategies aimed at manipulating DC migration could be useful in the treatment of CNS autoimmune disorders.
Dendritic cells (DCs) are a subset of bone marrow-derived leukocytes with the unique ability to activate resting T cells and to initiate immune responses. 1 DC progenitors can traffick from the blood to tissues, where they become immature DCs specialized for antigen capture. 2 Within the tissues, inflammatory signals (pathogens, cytokines) induce the maturation of DCs into potent antigen presenting cells (APCs) that express high levels of major histocompatibility complex (MHC) class II and adhesion/co-stimulatory molecules, ie, CD40, CD54 (intercellular adhesion molecule 1), and CD86 (B7–2), and migrate to peripheral lymphoid organs to initiate immune responses. 2 The constitutive and inflammation-induced trafficking of DCs to nonlymphoid tissues and lymphoid organs is selectively regulated by chemokines, small proteins involved in cell migration. 3,4
DCs have been implicated both in the initiation and maintenance of autoimmune diseases. 5 DCs capturing self-antigens in the target organ and migrating to regional lymph nodes are thought to initiate the activation of autoreactive T cells and to support chronic inflammation by sustaining successive waves of priming of naïve T cells. DCs recruited to the target tissue may participate in the maintenance of the inflammatory milieu by local activation of T cells and formation of organized lymphoid structures. 6,7
Experimental autoimmune encephalomyelitis (EAE) is a T-cell- and antibody-mediated autoimmune disease of the central nervous system (CNS) widely used to study the pathogenesis of the inflammatory disease multiple sclerosis. EAE can be induced in genetically susceptible animals by active immunization with myelin antigen(s), or by adoptive transfer of myelin-reactive CD4+ T cells. 8 Evidence for the involvement of DCs in initiating EAE comes from the observation that DCs efficiently activate encephalitogenic T cells for transfer of EAE, 9 and from a recent study showing that DCs pulsed with an encephalitogenic myelin basic protein peptide (Ac1-11) interact with Ac1-11-specific T cell receptor transgenic naïve T cells in the peripheral lymph nodes of recipient mice leading to induction of EAE. 10 After migration through the cerebrovascular endothelium, encephalitogenic CD4+ T cells recognize their target antigen on local APCs (reportedly perivascular macrophages and intraparenchymal microglia) and are activated to perform effector functions. 11,12 Intracerebral antigen presentation promotes secretion of T helper 1 (Th1) cytokines and the cascade of inflammatory events leading to massive recruitment of macrophages and of additional antigen-specific and nonspecific T cells to the CNS. 13
Progression to chronicity in EAE has been associated with the spreading of T cell-mediated autoreactivity to myelin determinants distinct from the initiating ones 14,15 and with humoral autoimmunity. 16,17 These findings raise two important issues: where the stimulation of new CNS autoreactive T and B cells takes place and whether the CNS harbors or recruits APCs capable of T cell priming locally or after migration to CNS-draining lymph nodes or spleen. Among resident CNS APCs, microglia are less effective than DCs in T cell priming 18 and are unlikely to migrate out of the CNS. In the normal CNS of humans and rodents, DCs are strategically located in potential sites of antigen entry such as the choroid plexuses and the meninges, but are not present within the CNS parenchyma. 19-22 In a rat model of acute EAE, small numbers of DCs were shown to localize within some spinal cord perivascular inflammatory infiltrates. 22 More recently Suter and colleagues 23 have demonstrated intracerebral expression of the MHC class II transactivator form I, specific for DCs, and the presence of CD11c+ DCs within CNS perivascular and meningeal inflammatory infiltrates during mouse EAE development. Although these findings suggest that DCs may contribute to intracerebral antigen presentation, it remains to be determined whether DCs are actively recruited to the CNS and play a role in sustaining autoreactive immune responses at the effector site.
The present study provides a detailed immunohistochemical description of the temporal appearance, spatial distribution, and functional phenotype of cells expressing DC markers (DEC-205, CD11c, MIDC-8) in the CNS of SJL mice with proteolipid protein (PLP) 139-151 peptide-induced EAE. It also shows that macrophage inflammatory protein 3α (MIP-3α), a chemokine involved in the trafficking of DCs and lymphocytes to peripheral tissues, and its receptor CCR6 are up-regulated in the CNS of EAE-affected mice. Collectively, our data suggest that DC recruitment and maturation within the CNS may be pivotal in local T cell activation and in maintaining cellular and humoral autoimmune responses leading to EAE progression.
Materials and Methods
Animals
Female adult SJL mice (Charles River, Calco, Italy) were used at 8 to 12 weeks of age and housed in a controlled environment in accordance with the guidelines of the European Community Council of the Welfare of Experimental Animals (86/609/EEC). All experimental procedures were approved by the Italian Ministry of Health.
EAE Induction
The peptide corresponding to amino acids 139 to 151 of mouse PLP 24 was purchased from Primm, Milano, Italy. Female SJL mice were injected subcutaneously in the flank with 0.2 mg of PLP 139-151 peptide in complete Freund’s adjuvant (CFA; Difco Laboratories, Detroit, MI) on days 0 and 7, and with pertussis toxin (0.2 μg; Sigma Chemical Co., St. Louis, MO) intraperitoneally on days 0, 1, 7, and 8. 25 Control mice were injected with phosphate-buffered saline (PBS) in CFA and pertussis toxin, according to the same schedule. Mice were weighted and examined daily for clinical signs of EAE, which was scored on the following scale: grade 0, no abnormality; grade 1, reduced tail tonus or slightly clumsy gait; grade 2, tail atony, moderately clumsy gait, impaired righting ability, or any combination of these signs; grade 3, additional hind limb weakness; grade 4, hind limb paralysis and fore limb weakness; grade 5, tetraplegia or moribund state. Mice were sacrificed for immunohistochemical and molecular studies at different times after disease induction: during preclinical EAE (day 10 or 13 after immunization; for this time point, we selected mice that already exhibited marked weight loss); during acute EAE (day 14 to 16 after immunization); during the early remission phase (day 20 to 22 after immunization, 24 hours after disappearance of clinical signs); 48 hours after the onset of relapses that followed an almost complete remission phase (day 25 to 30 after immunization); during chronic EAE (day 30 to 32 after immunization). At least two animals were examined for each time point.
Immunohistochemistry
For specimen collection, mice were anesthetized with xylazine chloride hydrate and ketamine and perfused intracardiacally with PBS followed by cold 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4). Brains and spinal cords were removed, kept overnight in 4% paraformaldehyde at 4°C, passed in 15% and 30% sucrose, frozen in dry ice-chilled isopentane, entirely cut in serial sections with cryostat, and stored at −20°C. For immunohistochemistry, 10 μm-thick sections were air-dried, passed in 70%, 95%, and 100% ethanol, and then dried again. After rehydration with PBS and pre-incubation with 10% normal rabbit or goat serum, sections were incubated at 4°C overnight with monoclonal or polyclonal antibodies optimally diluted in PBS/1% BSA. To identify DCs, three widely used markers for mouse DCs were used: hamster monoclonal antibody (mAb) N418, which binds the CD11c integrin; 26 rat mAb NLDC-145, which binds the multilectin receptor DEC-205; 27 and rat mAb MIDC-8, which binds a still unidentified antigen within intracellular granules of mature, interdigitating DCs in secondary lymphoid organs 28,29 (all from Serotec, Oxford, UK). Anti-CD4 (RM4–5; PharMingen, San Diego, CA), anti-CD8 (53-6-7; PharMingen), anti-CD45R/B220 (RA3–6B2; PharMingen) and anti-CD11b/Mac-1 (5C6; Serotec) rat mAbs were used to identify CD4+ T cells, CD8+ T cells, B cells, and macrophages/microglia, respectively. To investigate the distribution of molecules which are typically expressed on APCs, we used mouse mAb anti-rat/mouse MHC class II (I-Ak,s) (MRC OX-6; Serotec), rat mAb anti-mouse CD40 (3/23; PharMingen) and rat mAb anti-mouse CD86 (B7-2) (GL1; PharMingen). Sections were also stained with a goat polyclonal antiserum recognizing rat/mouse MIP-3α (R&D Systems, Minneapolis, MN). After extensive washings with PBS, sections were incubated with the corresponding biotinylated secondary antibodies (rabbit anti-goat IgG, rabbit anti-rat IgG, goat anti-hamster IgG, goat anti-mouse IgG, all from Vector Laboratories, Burlingame, CA) and avidin:biotinylated peroxidase complex (ABC), using the ABC Vectastain Elite kit (Vector Laboratories), according to the manufacturer’s instructions. Staining reactions were performed with 3,3 diaminobenzidine (DAB; Sigma) as substrate. DEC-205, MIDC-8, and CD86 immunostainings were also performed using the amplification procedure described by Adams, 30 with minor modifications. After incubation with ABC, sections were incubated with 0.01% H2O2 and 28 mmol/L biotinylated tyramine, prepared as described, 30 for 10 minutes at room temperature. After extensive washings, sections were incubated with ABC for 20 minutes, PBS for 30 minutes, and DAB for another 5 minutes. To eliminate endogenous peroxidase activity, sections were incubated with 0.3% H2O2 in PBS, for 20 minutes, before secondary antibody addition. For negative controls, the primary antibody was omitted from the diluent. Sections were counterstained with hematoxylin and viewed with a Zeiss Axiophot photomicroscope (Oberkochen, Germany).
For double-immunohistochemical stainings with anti-CD11b and anti-CD11c mAbs, after an initial blocking with normal sera, sections were incubated with the two mAbs and then in sequence with goat biotinylated anti-hamster IgG (Vector Laboratories), ABC-alkaline phosphatase (Vector laboratories), and fuchsin (DAKO, Glostrup, Denmark) as chromogen (red color). After washing, sections were incubated with avidin-biotin (Vector Laboratories), rabbit anti-rat IgG, ABC-peroxidase, and DAB as chromogen (brown color).
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Mice were anesthetized as above and perfused intracardiacally with cold PBS. Total RNAs were extracted from the CNS, spleen, and small intestine of control (n = 2), presymptomatic (n = 2), EAE-affected (n = 5), and remitting (n = 2) SJL mice using the SV Total RNA Isolation System (Promega, Madison, WI), according to the manufacturer’s instructions. One μg of purified RNA was reverse-transcribed using the Reverse Transcription System (Promega), with 15 U of avian myeloblastosis virus reverse transcriptase. For detection of murine MIP-3α transcripts, 31 one-tenth of this reaction was added to 40 μl of PCR mix and amplified for 36 cycles (30 seconds at 94°C, 30 seconds at 60°C, 45 seconds at 72°C), with 2.5 U of Taq polymerase (Promega), using the following primers: 5′-TACAGACGCCTCTTCCTTCC-3′ (sense) and 5′-TCTTGACTCTTAGGCTGAGG-3′ (antisense) (size of the amplified product of MIP-3α cDNA was 174 bp). For detection of murine CCR6 transcripts, 31 equal amounts of cDNA were subjected to PCR amplification for 36 cycles (15 seconds at 94°C, 15 seconds at 59°C, 45 seconds at 72°C), using a forward primer specific to the untranslated 5′ region (5′-CTGCAGTTCGAAGTCATC-3′) and a reverse primer specific to the CCR6 open reading frame (5′-GTCATCACCACCATAATGTTG-3′) (sizes of the amplified products of CCR6 cDNA were 330 and 420 bp). As control, transcripts of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase were also amplified for 25 cycles (30 seconds at 94°C, 30 seconds at 60°C, 45 seconds at 72°C), using the following primers: sense 5′-CCATGGAGAAGGCCGGGG-3′, antisense 5′-CAAAGTTGTCATGGATGACC-3′ (size of the amplified product of glyceraldehyde-3-phosphate dehydrogenase cDNA was 194 bp). Negative controls lacking template RNA or RT were included in each experiment. The PCR products were fractionated on a 2% agarose gel and visualized by ethidium bromide staining. The data shown were obtained in individual, representative animals.
Results
Most of the SJL mice injected with PLP 139-151 peptide in CFA developed an acute form of EAE (grade 3 to 5) which peaked around days 14 to 19 after immunization and spontaneously resolved a few days later. In a minority of mice, the disease progressed to a chronic form (clinical grade 1 to 3) or the remission phase was followed by a relapse (clinical grade 1 to 4). Cryostat CNS sections of control SJL mice (either untreated or injected with PBS/CFA) and of SJL mice immunized with PLP 139-151 peptide in CFA were analyzed for the presence of CD11c+, DEC-205+ and MIDC-8+ DCs, CD4+ and CD8+ T cells, CD11b+ macrophages/microglia, and B220+ B cells. All immunohistochemical stainings were performed on sections of spinal cord, brain stem, and cerebellum, which are the CNS areas more severely affected in EAE.
Normal Mouse CNS: Localization of DCs in the Choroid Plexuses and Meninges
In agreement with previous studies on DC localization in the normal rat and human CNS, 19-22 the murine DC markers DEC-205 and CD11c were found to be expressed by process-bearing cells in the choroid plexus (fourth ventricle) and in the meninges of untreated SJL mice (Figure 1, A–C) ▶ , but not in the spinal cord, brain stem, or cerebellar parenchyma. Meningeal and choroid plexus DCs had the phenotype of immature, tissue resident DCs as they did not bind the mAb MIDC-8, reacting with an intracellular granule antigen of mature DCs 28,29 (not shown). In the CNS of control mice, CD11b, a marker for macrophages/microglia 32 and myeloid DCs, 33 stained round cells, most likely macrophages, in the meninges and choroid plexuses and only few intraparenchymal microglia (not shown). No CD4+, CD8+, or B220+ cells were detected (not shown).
Figure 1.
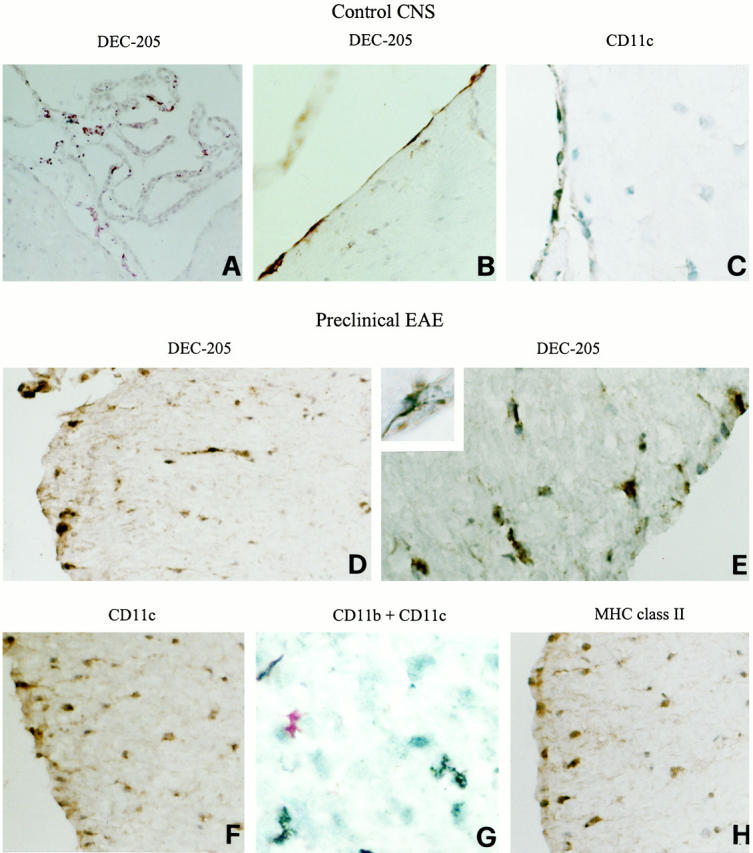
Localization of DC markers in the CNS of normal SJL mice (A–C) and of mice immunized with PLP 139-151 in CFA before the onset of EAE clinical signs (day 10 after immunization) (D–H). Representative cerebellar (A) and thoracic spinal cord (B–H) sections are shown. CNS sections were stained for the indicated markers and visualized with DAB, as described in Materials and Methods. Control CNS: DEC-205+ process-bearing cells in the choroid plexus of the fourth ventricle (A), DEC-205+ (B) and CD11c+ (C) process-bearing cells in the spinal cord meninges. Preclinical EAE: DEC-205+ (D and E), CD11c+ (F), and MHC class II+ (H) cells with thin cytoplasmic processes are present in the subpial white matter of the ventral and lateral spinal cord. Note the ramified morphology of a DEC-205+ cell immediately beneath the pial surface (inset in E). Double staining with anti-CD11b (brown color, DAB) and anti-CD11c (red color, fuchsin) mAbs reveals that CD11c mAb recognizes intraparenchymal cells that are distinct from CD11b+ microglia (G). Original magnifications: ×500 (A, D, F, and H), ×1000 (B, C, E, and G), and ×1575 (inset).
Preclinical EAE: Early Appearance of Immature DCs in the Spinal Cord
During preclinical EAE (day 10 after immunization), DEC-205+ and CD11c+ cells with thin cytoplasmic processes were detected not only in the meninges and choroid plexus but also in discrete areas of the spinal cord white matter, predominantly beneath the pial surface (Figure 1, D–F) ▶ . In most sections, the distribution of DEC-205+ and CD11c+ cells gave the clear impression that these were in the process of migrating from the meninges into the spinal cord. At this stage, no MIDC-8+ mature DCs were detected in the CNS (not shown). Double stainings with anti-CD11b and anti-CD11c mAbs revealed no overlapping of the two immunoreactivities, excluding that CD11c expression was induced on microglia (Figure 1G) ▶ . Using adjacent spinal cord sections, we found that process-bearing cells expressing MHC class II molecules were present in the meninges and in the same areas as DEC-205+ and CD11c+ cells (Figure 1H) ▶ . At 10 day after immunization, no CD4+, CD8+, or B cells infiltrated the CNS. Moreover, neither CD11b+ macrophages nor activated microglia were detected within the CNS parenchyma (not shown), rendering unlikely that these cell types contributed to early intracerebral expression of MHC class II molecules. No DEC-205+ or CD11c+ cells were present in the spinal cord of PBS/CFA-injected mice.
Acute EAE: Presence of Mature DCs in the CNS Inflammatory Cell Infiltrates
In the CNS of mice with acute EAE (grade 3 to 4), immune infiltrates were present in perivascular, meningeal, and submeningeal locations. CD11b+ macrophages and CD4+ T cells constituted the predominant infiltrating leukocytes and penetrated extensively into the CNS, particularly in the spinal cord (Figure 2, A and B) ▶ . Very rare, if any, CD8+ T cells and B220+ B cells were detected in acute EAE lesions (not shown). DEC-205+ cells with characteristic dendritic shape localized within the CNS infiltrates and only few were scattered in the spinal cord parenchyma (Figure 2, C–E) ▶ . CD11c did not seem to be a useful marker for DCs in EAE lesions as it was expressed on most of the infiltrating cells, likely macrophages (not shown). Notably, scattered cells within the infiltrates were labeled intracellularly by MIDC-8 mAb (Figure 2, F and G) ▶ , suggesting that DCs rapidly acquire a mature phenotype in the inflamed CNS.
Figure 2.
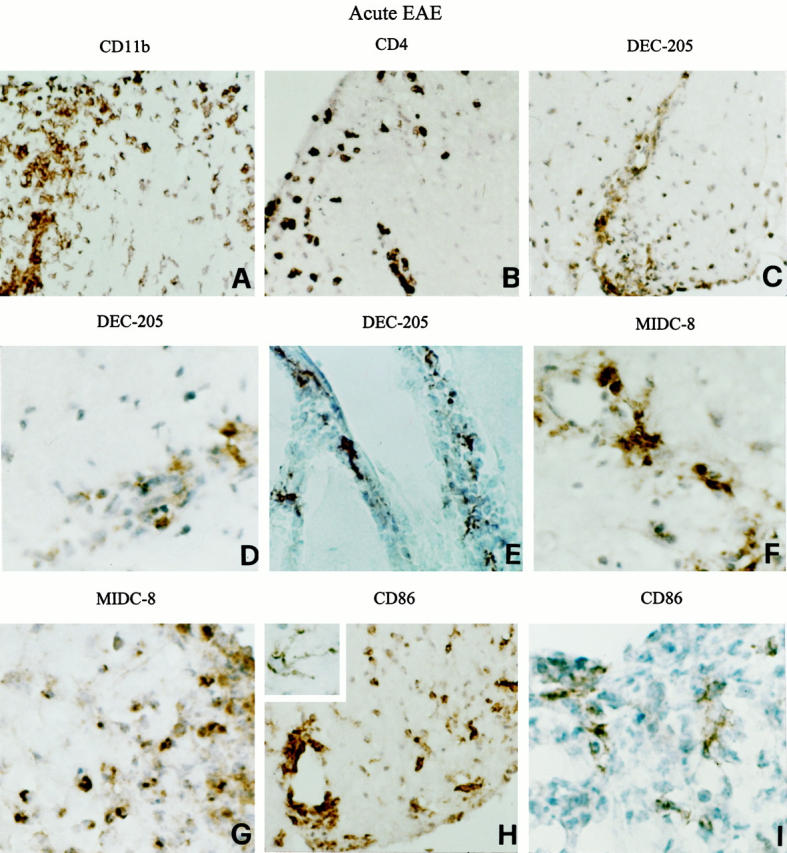
Cellular composition of the immune infiltrates and localization of DC markers in the CNS of mice with acute EAE. Representative lumbar (B, D, and I) and thoracic (A, C, F, G, and H) spinal cord and cerebellar (E) sections from a SJL mouse developing acute EAE (grade 4) are shown. CNS sections were stained for the indicated markers and visualized with DAB, as described in Materials and Methods. Infiltration of CD11b+ macrophages (A) and CD4+ T cells (B) into the spinal cord white matter is shown. Numerous DEC-205+ (C and D) and MIDC-8+ (F and G) cells are present within the perivascular and submeningeal inflammatory cell infiltrates in the spinal cord. Note the typical DC morphology of DEC-205+ cells accumulating within the cerebellar meningeal spaces (E). Staining of spinal cord sections for CD86 using the Adams’ amplification procedure (see Materials and Methods) evidenced the presence of CD86 immunoreactivity on most infiltrating cells, some activated microglia (H) and rare cells with irregularly shaped cell body and thin cytoplasmic processes typical of DCs (inset). Omission of the amplification step allowed to visualize only scattered CD86+ process-bearing cells, likely DCs, in the center of a submeningeal inflammatory cell infiltrate (I). Original magnifications: ×500 (A, B, C, E, and H) and ×1000 (D, F, G, I, and inset).
Sections of the spinal cord were also examined for the expression of APC molecules associated with enhanced lymphostimulatory activity. 2 In acute EAE, MHC class II and accessory/co-stimulatory (CD40, CD86) molecules were expressed on the majority of blood-derived cells and surrounding activated microglia as well as on rare intraparenchymal DC-like cells with thin processes and irregular cell body (only staining for CD86 is shown; Figure 2H ▶ ). In experiments in which the staining protocol did not include the amplification step with biotinylated tyramine, 30 CD86 mAb selectively stained few infiltrating process-bearing cells, most likely DCs (Figure 2I) ▶ . This finding is consistent with the fact that mature DCs express CD86 at higher levels than other blood-derived or tissue APCs. 2
Very rare DEC-205+ or MIDC-8+ cells were detected within the residual inflammatory infiltrates persisting in the perivascular and meningeal locations during the early remission phase of EAE and none were present at late remission stages (not shown).
Chronic EAE: Persistence of DCs in the Inflammatory Cell Infiltrates
Immunohistochemical analysis of the CNS of mice developing chronic EAE (grade 2 to 3) was performed at ∼2 weeks after the onset of clinical disease. Compared with acutely affected mice, mice developing chronic EAE had smaller and less invasive immune infiltrates in the spinal cord, brain stem, and cerebellum, whereas microglia activation was still evident, particularly in the spinal cord (Figure 3A) ▶ . The meningeal and perivascular infiltrates comprised CD4+ T cells as well as CD8+ T cells and B cells (Figure 3, B–D) ▶ . Within the infiltrates, many DEC-205+ and MIDC-8+ cells were detected (Figure 3, E and F) ▶ . Intriguingly, anti-DEC-205 (Figure 3G) ▶ , but not MIDC-8 (not shown), mAb also stained numerous process-bearing cells in discrete areas of the spinal cord white matter.
Figure 3.
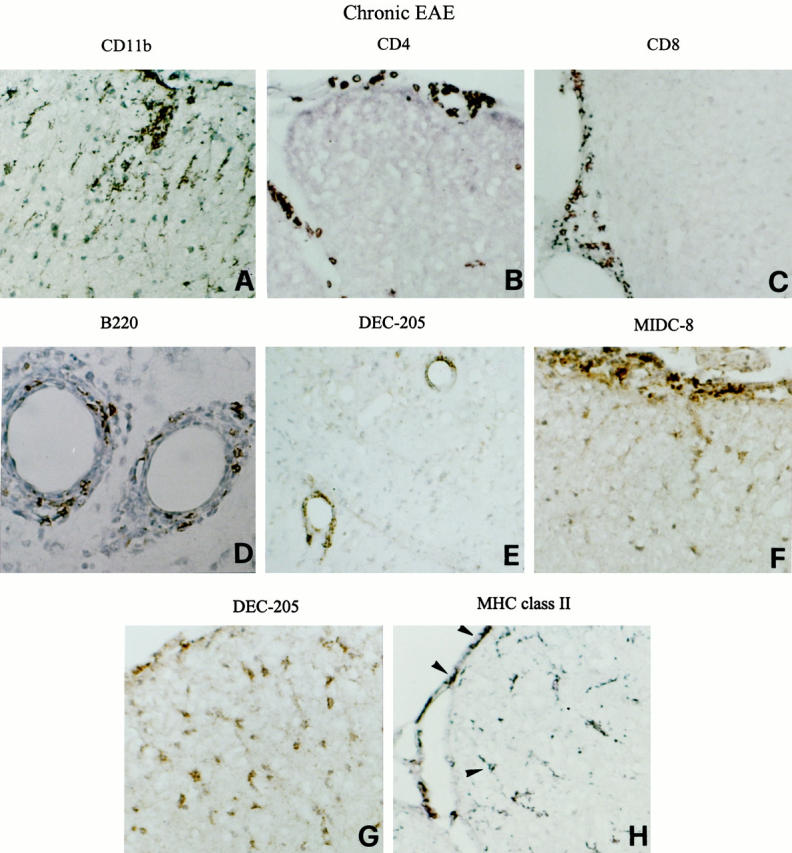
Persistence of mature DCs in the CNS of mice with chronic EAE. Representative thoracic (A, B, F, G, and H) and cervical (C) spinal cord and cerebellar (D and E) sections from a SJL mouse with chronic EAE (grade 3) are shown. CNS sections were stained for the indicated markers and visualized with DAB, as described in Materials and Methods. The immune infiltrates remain primarily confined to the meninges and around blood vessels and are composed of CD11b+ macrophages (A), CD4+ T cells (B), CD8+ T cells (C), as well as B220+ B cells (D). Cells positive for DEC-205 (E) and for the mature DC marker MIDC-8 (F) accumulate perivascularly and in the meninges. Anti-DEC-205 mAb stains process-bearing cells within the dorsal spinal cord white matter (G). Some of the MHC class II+ cells detected in the spinal cord meninges and white matter (H) have a DC-like morphology (arrowheads). Original magnifications: ×500 (A–C and E–H), and ×1000 (D).
In chronic EAE, expression of MHC class II, CD40, and CD86 molecules was mainly confined to cells (some with a DC-like morphology) in the meninges and perivascular cuffs. Within the spinal cord, APC molecules were detected on some activated microglia and rare process-bearing DC-like cells (only staining for MHC class II molecules is shown; Figure 3H ▶ ).
Relapsing EAE: Presence of Mature DCs within the Spinal Cord Parenchyma
For this set of experiments, we analyzed mice with an EAE relapse that had followed a complete remission. In relapsing EAE (grade 2 to 3), numerous CD4+ T cells were present in the CNS meninges, perivascular spaces and parenchyma (Figure 4A) ▶ , whereas few CD11b+ macrophages infiltrated the CNS (not shown). CD8+ T cells (Figure 4B) ▶ and B cells (Figure 4C) ▶ also accumulated within the CNS infiltrates.
Figure 4.
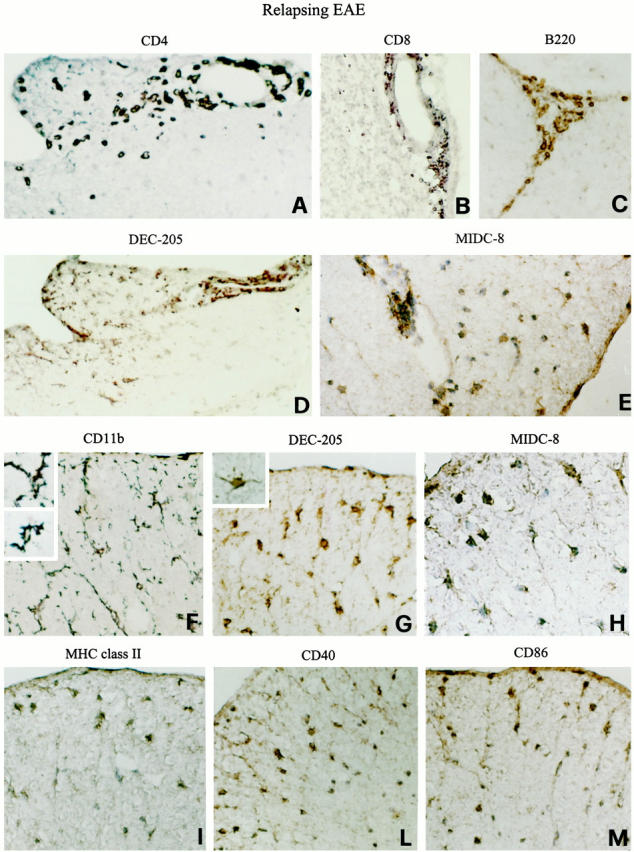
Diffuse infiltration of DCs with mature phenotype into the spinal cord white matter in EAE relapses. Representative thoracic (A and C–M) spinal cord and cerebellar (B) sections from a SJL mouse undergoing an EAE relapse (grade 2) are shown. CNS sections were stained for the indicated markers and visualized with DAB, as described in Materials and Methods. A–C: Shown are the distributions of CD4+ T cells, CD8+ T cells, and B220+ B cells recruited to the CNS during the EAE relapse phase. Numerous DEC-205+ (D) and MIDC-8+ (E) cells are present in the perivascular location as well as within the spinal cord parenchyma. DEC-205+ cells (D) co-localize with the CD4+ T cell infiltrates (A; adjacent spinal cord sections are shown in A and D). Note the different distribution and morphology of CD11b+-activated microglia (F), intraparenchymal DEC-205+ (G), and MIDC-8+ (H) DCs (dorsal part of adjacent spinal cord sections). CD11b+ microglia have elongated cell bodies and thick ramified processes (inset in F) whereas DEC-205+ DCs have a more rounded, irregular cell body with thin and long cytoplasmic processes (inset in G). I–M: Shown are the distributions of MHC class II, CD40, and CD86 molecules on intraparenchymal cells most of which exhibit a DC-like morphology. Original magnifications: ×500 (A–D, F, G, I, L, and M) and ×1000 (E, H, and insets).
Similarly to acute and chronic EAE, DEC-205+ and MIDC-8+ DCs were detected within the immune infiltrates of all CNS areas examined (Figure 4, D and E) ▶ . A striking feature of relapsing EAE was the diffuse infiltration of DEC-205+ and MIDC-8+ DCs in the spinal cord and caudal part of the medulla oblongata (Figure 4 ▶ ; D, E, G, and H). Intraparenchymal DCs were particularly enriched in areas of T cell infiltration (compare adjacent sections in panels A and D of Figure 4 ▶ ). Moreover, numerous DEC-205+ and MIDC-8+ cells were disseminated in vast areas of the spinal cord white matter (Figure 4, G and H) ▶ .
A prominent and diffuse activation of CD11b+ microglia was evident in the CNS of mice undergoing EAE relapses (Figure 4F) ▶ . In the spinal cord, both the localization and morphology of CD11b+-activated microglia clearly differed from those of intraparenchymal DEC-205+ cells ruling out the possibility that microglia had acquired positivity for DC markers (compare adjacent sections in panels F and G of Figure 4 ▶ ). MHC class II, CD40, and CD86 molecules were expressed on cells of the inflammatory infiltrates (not shown), some activated microglia, and intraparenchymal cells with a DC-like morphology (Figure 4, I–M) ▶ .
Up-Regulation of the β-Chemokine MIP-3α and its Receptor CCR6 in the CNS of EAE-Affected Animals
We then asked whether the CNS of EAE-affected mice would express chemokines involved in the recruitment of DCs at inflammatory sites. 3 We focused our attention on the β-chemokine MIP-3α (also termed liver and activation-regulated chemokine and Exodus-1), which is expressed constitutively in certain murine tissues and is up-regulated in inflammation. 31,34 CCR6, the MIP-3α-specific receptor, is expressed on murine DCs, T and B cells, 31 and on human immature DCs and memory T cells. 35,36 Analysis of MIP-3α and CCR6 transcripts in the CNS of control and EAE-affected mice was performed by RT-PCR. Small intestine and spleen tissues from normal mice served as positive controls for MIP-3α and CCR6, respectively. 31,34 Because of alternative splicing, 31 two amplified CCR6-specific products were obtained. MIP-3α mRNA was not detectable in the CNS of control mice (either untreated or injected with PBS/CFA) (Figure 5) ▶ . A very weak band for CCR6 mRNA was present in the cerebrum only. Both MIP-3α and CCR6 mRNAs became detectable during preclinical EAE (day 13 after immunization) and were further up-regulated in the acute disease phase in all CNS areas examined (Figure 5) ▶ . The induction of MIP-3α- and CCR6-specific transcripts in the cerebrum, an area which fails to develop typical perivascular and intraparenchymal EAE lesions, can be explained by the presence of abundant inflammatory infiltrates within the cerebral meninges and ventricular choroid plexuses (our unpublished observations). During EAE relapses, CCR6 mRNA was up-regulated in the cerebrum, cerebellum, and spinal cord, whereas expression of MIP-3α mRNA was particularly induced in the spinal cord. During the early remission phase, transcripts for MIP-3α and CCR6 became again undetectable, or poorly detectable in the case of cerebral CCR6 transcripts (Figure 5) ▶ .
Figure 5.

MIP-3α and CCR6 transcripts are up-regulated in the CNS of SJL mice with PLP 139-151-induced EAE. RNA was extracted from different CNS areas and from control tissues (spleen and small intestine), reverse-transcribed, and subjected to PCR amplification using MIP-3α- and CCR6-specific primers, as described in Materials and Methods. A RT-PCR using glyceraldehyde-3-phosphate dehydrogenase-specific primers is also shown as an internal control. RNA was extracted from control mice (PBS/CFA-injected) and from mice at the indicated EAE stages. The data shown were obtained in mice with grade 3 acute EAE and grade 4 relapsing EAE. The PCR products were visualized by ethidium bromide staining. Lane 1, cerebrum; lane 2, cerebellum; lane 3, spinal cord; lane 4, spleen; and lane 5, small intestine.
To identify the cellular sources of MIP-3α in the EAE-affected CNS, cryostat spinal cord sections were stained with a polyclonal anti-mouse/rat MIP-3α antibody. No MIP-3α immunoreactivity was detected in the spinal cord of control and presymptomatic SJL mice (not shown). During acute EAE, most MIP-3α immunoreactivity co-localized with the inflammatory cell infiltrates (Figure 6A) ▶ and only few MIP-3α+ cells were scattered in the surrounding CNS parenchyma. No MIP-3α immunoreactivity was detected in the CNS of mice undergoing disease remission (not shown). Notably, after EAE relapses (Figure 6, B and C) ▶ and to a lesser extent in chronic EAE (not shown), anti-MIP-3α antibody also stained numerous cells regularly distributed in the spinal cord white matter. Based on their morphological appearance (Figure 6, B and C) ▶ and localization of the astrocytic marker glial fibrillary acidic protein in adjacent sections (not shown), most of the MIP-3α+ cells were identified as astrocytes.
Figure 6.

Localization of MIP-3α immunoreactivity in the spinal cord of SJL mice with acute (A) and relapsing (B and C) EAE. Representative lumbar spinal cord sections are shown. Sections were stained for MIP-3α and visualized with DAB, as described in Materials and Methods. During acute EAE, MIP-3α immunoreactivity is localized in most cells within meningeal, submeningeal, and perivascular inflammatory infiltrates and in a few cells scattered in the spinal cord white matter (A). After an EAE relapse, numerous MIP-3α+ cells are also found within the spinal cord (B). At higher magnification, most intraparenchymal MIP-3α+ cells show the typical astrocytic morphology (C). Original magnifications: ×500 (A and B) and ×1000 (C).
Discussion
Our study demonstrates that DC appearance in the spinal cord is one of the first events detectable in the CNS after EAE induction. DCs accumulating in the inflamed CNS acquire a mature, lymphostimulatory phenotype and show a spatial and temporal distribution similar to that of MIP-3α, a chemokine implicated in DC and lymphocyte migration. Based on the present findings, we propose that intracerebral DC recruitment and maturation are key events in the local activation of encephalitogenic CD4+ T cells and in the maintenance of cytotoxic and antibody-mediated autoimmune responses leading to EAE progression and CNS damage.
At a stage immediately preceding EAE clinical signs and appearance of CNS immune infiltrates, cells exhibiting a typical DC morphology and expressing the DC markers DEC-205 and CD11c are found in subpial areas of the spinal cord white matter where the first intraparenchymal MHC class II+ cells are also detected. The present observations are in agreement with studies in other autoimmune pathologies showing that DC infiltration is one of the first abnormalities in the target organ. 5 We propose that the early appearance of DCs in the CNS provides an initial pool of intracerebral APCs which efficiently capture CNS antigens and present them to peripherally primed encephalitogenic CD4+ T cells. Matsumoto and colleagues 37 first described the presence of Ia+ cells with dendritic morphology in the spinal cord of rats at EAE preclinical stage (day 11 after immunization) but identified them as microglia. Using mice with myelin oligodendrocyte glycoprotein peptide 35-55-induced EAE, Suter and colleagues 23 have recently shown that CD11c+ DCs can be detected, together with CD4+ T cells and macrophages, within the spinal cord inflammatory infiltrates shortly before disease onset. These authors have also presented evidence that CD11b+ macrophages are the first hematogenous cell type to be recruited to the CNS. Discrepancies between the study of Suter and colleagues 23 and our findings could be due, at least in part, to the use of different mouse EAE models.
During acute EAE, DCs are predominantly distributed within the CNS immune infiltrates and express the mature DC markers MIDC-8 and CD86. We favor the hypothesis that DCs accumulating and maturing in the inflamed CNS play a pivotal role in Th1 effector activation, whereas the more numerous infiltrating macrophages and activated microglia contribute to the amplification and regulation of the immune response. Accordingly, several studies have shown that in the Th1 cytokine-rich microenvironment of EAE lesions the latter cell types are induced to express APC molecules and to secrete pro-inflammatory (interleukin-12, tumor necrosis factor) as well as anti-inflammatory (interleukin-10, transforming growth factor-β, prostaglandin E2) mediators. 11,12,38-40
The persistence of mature DCs in the CNS of mice with chronic and relapsing EAE suggests that DCs are also involved in disease progression. Most intriguingly, we have demonstrated enrichment of mature DCs in the spinal cord parenchyma of mice undergoing EAE relapses. EAE progression is associated with CNS infiltration by CD4+, CD8+, T cells, and B cells, suggesting that DCs may provide the signals necessary for sustaining local activation of these immune cell types. Consistent with this, CNS-infiltrating cells with a DC-like morphology express MHC class II, CD40, and CD86 molecules which are essential for optimal APC function. 2 Interaction of CD86 with CD28 on T cells is of critical importance for T cell activation, 41 whereas CD40 ligation by CD154 (CD40 ligand) on activated T cells induces DC maturation, cytokine secretion and expression of co-stimulatory molecules. 2,42 Encephalitogenic T cells accumulating in the CNS of EAE-affected mice express CD154, 43 suggesting that CD40-CD154 interactions are determinants for intracerebral DC maturation. This may be further promoted by cytokines, like granulocyte-macrophage colony-stimulating factor, tumor necrosis factor, and interleukin-1, produced intracerebrally during EAE. 44-46 The establishment of DC T and B cell interactions within the CNS parenchyma could sustain the intrathecal B-cell clonal expansion typically associated with multiple sclerosis 47 and the production of anti-myelin antibodies detected in EAE and multiple sclerosis lesions. 17 Mature intracerebral DCs are also likely to secrete chemokines contributing to the recruitment of additional DCs, CD4+ and CD8+ T cells to the inflamed CNS. 48,49
DCs were recently proposed to have a key role in the formation of ectopic lymphoid tissue occurring in target organs in a number of chronic inflammatory diseases. 6,7 The presence of organized lymphoid structures in the CNS of mice with adoptively transferred chronic-relapsing EAE 50 and in old multiple sclerosis plaques 51 suggests that DC might also orchestrate lymphoid neogenesis in the CNS and this in turn may be a key event in the maintenance of an autoimmune response against neural antigens. In addition to a role in sustaining immune responses within the CNS, intracerebral DCs could capture antigen generated during CNS damage and traffic to secondary lymphoid organs to stimulate new autoreactive T cells. This possibility is supported by a recent study showing that intracerebrally injected DCs home to cervical lymph nodes. 48
The present findings raise two important questions: where do intracerebral DCs come from, and how are they recruited to the CNS? During preclinical EAE, the subpial localization of DCs within the spinal cord white matter suggests that they could derive from DCs or DC precursors normally residing in the meninges. 21,22 The observation that DCs do not enter the parenchyma of the brain stem and cerebellum at any stage of EAE indicates a unique permissiveness of the spinal cord white matter to DC migration and/or differentiation. Appearance of intracerebral DCs early in the disease process occurs before detectable T cell infiltration into the CNS, suggesting that pro-inflammatory stimuli related to the immune response cause subtle alterations in the vascular endothelia or CNS tissue (eg, induction of adhesion molecules and/or chemokines), leading to selective DC recruitment. Because of the lack of a blood-brain barrier, the meninges are among the CNS compartments more readily affected by peripheral immune stimuli. It should be noted that peripheral immune activation in PLP 139-151 peptide-treated SJL mice is already very prominent at day 10 after immunization. 52 Although early DC mobilization is probably not restricted to the CNS, the CNS parenchyma represents a unique site to detect such event, as it normally does not contain a resident DC population.
After EAE development, the presence of DCs within the CNS inflammatory cell infiltrates is consistent with rapid recruitment of DCs or DC precursors from the blood circulation. Conversely, the sparse, intraparenchymal DC localization observed in the spinal cord of mice with chronic and relapsing EAE suggests that DC progenitors residing in the CNS parenchyma may be induced to differentiate into DCs. Monocytes/macrophages share a common myeloid precursor with DCs 53 and myeloid precursors continuously enter the CNS to differentiate into perivascular macrophages and microglia. 54,55 Chronic neuroinflammation and widespread glial activation could contribute to the establishment of a cytokine (granulocyte-macrophage colony-stimulating factor, interleukin-1, tumor necrosis factor)-rich milieu promoting intracerebral DC development and maturation. 53 This hypothesis is currently under investigation.
Chemokines are small proteins that regulate leukocyte migration to lymphoid organs and nonlymphoid tissues. They are produced by activated leukocytes themselves as well as by endothelial and parenchymal cells during inflammation. 56 Depending on their maturation stage, DCs respond to specific chemokines and express the corresponding subsets of chemokine receptors. 35,57,58 MIP-3α has been proposed to mediate constitutive DC trafficking to certain peripheral tissues and to be involved in the rapid recruitment of DCs during inflammation. 31,34,35
We have shown that transcripts specific for MIP-3α and its receptor CCR6 are up-regulated in the CNS during EAE development and that the spatial distribution of MIP-3α immunoreactivity matches that of DCs, suggesting that this chemokine may have a role in intracerebral DC recruitment. Although the localization of MIP-3α in acute EAE lesions indicates that the first chemotactic signals are provided by blood-derived cells, the presence of numerous intraparenchymal MIP-3α+ cells, predominantly astrocytes, in the CNS of mice with chronic and relapsing EAE supports the idea that the injured CNS becomes the major source of MIP-3α. Because CCR6 receptors are expressed on DCs as well as on T and B lymphocytes, 31,35,36 it is likely that intracerebral MIP-3α production is important for coordinating interactions among these cell types, resulting in sustained immune activation. In addition to MIP-3α, several other chemokines (eg, MIP-1α, MIP-1β, RANTES, and MCP-1) are chemotactic for immature murine DCs in vitro 59 and are produced in the CNS during acute and chronic EAE. 60,61 Further studies need to be performed to understand whether and which chemokine(s) are critically involved in DC recruitment to the inflamed CNS. Given the presence of numerous DCs with a mature phenotype in EAE lesions, it will be interesting to verify whether chemokines acting on mature DCs, such as MIP-3β 35 and SLC, 62 are also produced in the CNS of EAE-affected mice and contribute to intracerebral DC homing.
In conclusion, our study strongly suggests that DCs recruited to and maturing within the CNS parenchyma may have a pivotal role both in the onset and progression of EAE. Manipulation of DC recruitment and function within the CNS may represent a new strategy to treat experimental and human neuroinflammatory diseases.
Acknowledgments
We thank Luciano Adorini, Giulio Levi, and Luisa Minghetti for critical reading of the manuscript.
Footnotes
Address reprint requests to Francesca Aloisi, Laboratory of Organ and System Pathophysiology, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy. E-mail: fos4@iss.it.
Supported by Research Project on Multiple Sclerosis and Project “Inflammatory, Oxidative and Autoimmune Mechanisms in CNS Diseases” of the Istituto Superiore di Sanità/Italian Ministry of Health.
References
- 1.Steinman RM: The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991, 9:271-296 [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM: Dendritic cells and the control of immunity. Nature 1998, 392:245-252 [DOI] [PubMed] [Google Scholar]
- 3.Dieu-Nosjean M-C, Vicari A, Lebecque S, Caux C: Regulation of dendritic cell trafficking: a process that involves the participation of selective chemokines. J Leukoc Biol 1999, 66:252-262 [DOI] [PubMed] [Google Scholar]
- 4.Cyster JG: Chemokines and the homing of dendritic cells to the T cell areas of lymphoid organs. J Exp Med 1999, 189:447-450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas R, Lipsky P: Could endogenous self-peptides presented by dendritic cells initiate rheumatoid arthritis? Immunol Today 1996, 17:559-564 [DOI] [PubMed] [Google Scholar]
- 6.Ludewig B, Odermatt B, Ochsenbein AF, Zinkernagel RM, Hengartner H: Role of dendritic cells in the induction and maintenance of autoimmune diseases. Immunol Rev 1999, 169:45-54 [DOI] [PubMed] [Google Scholar]
- 7.Hjelmström P, Fjell J, Nakagawa T, Sacca R, Cuff AC, Ruddle NH: Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol 2000, 156:1133-1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradl M, Linington C: Animal models of demyelination. Brain Pathol 1996, 6:303-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gautam AM, Glynn P: Lewis rat lymphoid dendritic cells can efficiently present homologous myelin basic protein to encephalitogenic lymphocytes. J Neuroimmunol 1989, 22:113-121 [DOI] [PubMed] [Google Scholar]
- 10.Dittel BN, Visintin I, Merchant RM, Janeway CA, Jr: Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J Immunol 1999, 163:32-39 [PubMed] [Google Scholar]
- 11.Hickey WF, Kimura H: Perivascular microglial cells of the CNS are bone-marrow derived and present antigen in vivo. Science 1988, 239:290-292 [DOI] [PubMed] [Google Scholar]
- 12.Aloisi F, Ria F, Adorini A: Regulation of T cell responses by central nervous system antigen presenting cells: different roles for microglia and astrocytes. Immunol Today 2000, 21:141-147 [DOI] [PubMed] [Google Scholar]
- 13.Steinman L: A few autoreactive cells in an autoimmune infiltrate control a vast population of nonspecific cells: a tale of smart bombs and the infantry. Proc Natl Acad Sci USA 1996, 93:2253-2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehmann PV, Forsthuber T, Miller A, Sercarz EE: Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358:155-157 [DOI] [PubMed] [Google Scholar]
- 15.McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD: Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med 1995, 182:75-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linington C, Lassman H: Antibody responses in chronic relapsing experimental allergic encephalomyelitis: correlation of serum demyelinating activity with antibody titre to the myelin/oligodendrocyte glycoprotein (MOG). J Neuroimmunol 1987, 17:61-69 [DOI] [PubMed] [Google Scholar]
- 17.Genain CP, Cannella B, Hauser SL, Raine CS: Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med 1999, 5:170-175 [DOI] [PubMed] [Google Scholar]
- 18.Aloisi F, Ria F, Columba-Cabezas S, Hess H, Penna G, Adorini L: Relative efficiency of microglia, astrocytes, dendritic cells and B cells in naive CD4+ T cell priming and Th1/Th2 restimulation. Eur J Immunol 1999, 29:2705-2714 [DOI] [PubMed] [Google Scholar]
- 19.Serot JM, Foliquet B, Béné MC, Faure GC: Ultrastructural and immunohistological evidence of dendritic-like cells within human choroid plexus epithelium. Neuroreport 1997, 8:1995-1998 [DOI] [PubMed] [Google Scholar]
- 20.Hanly A, Petito CK: HLA-DR positive dendritic cells of the human choroid plexus: a potential reservoir of HIV in the central nervous system. Hum Pathol 1998, 29:88-93 [DOI] [PubMed] [Google Scholar]
- 21.McMenamin PG: Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mather, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol 1999, 405:553-562 [PubMed] [Google Scholar]
- 22.Matyszak MK, Perry VH: The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience 1996, 74:599-608 [DOI] [PubMed] [Google Scholar]
- 23.Suter T, Malipiero U, Otten L, Ludewig B, Muelethaler-Mottet A, Mach B, Reith W, Fontana A: Dendritic cells and differential usage of the MHC class II transactivator promoters in the central nervous system in experimental autoimmune encephalitis. Eur J Immunol 2000, 30:794-802 [DOI] [PubMed] [Google Scholar]
- 24.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB: Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol 1989, 142:1523-1527 [PubMed] [Google Scholar]
- 25.Di Rosa F, Francesconi A, Di Virgilio A, Finocchi L, Santilio I, Barnaba V: Lack of Th2 cytokine increase during spontaneous remission of experimental allergic encephalomyelitis. Eur J Immunol 1998, 28:1-11 [DOI] [PubMed] [Google Scholar]
- 26.Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM: The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med 1990, 171:1753-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang W, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM, Nussenzweig MC: The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 1995, 375:151-155 [DOI] [PubMed] [Google Scholar]
- 28.Breel M, Mebius RE, Kraal G: Dendritic cells of the mouse recognized by two monoclonal antibodies. Eur J Immunol 1987, 17:1555-1559 [DOI] [PubMed] [Google Scholar]
- 29.Inaba K, Pack M, Inaba M, Sakuta H, Isdell F, Steinman RM: High levels of a major histocompatibility complex II-self peptide complex on dendritic cells from the T cell areas of lymph nodes. J Exp Med 1997, 186:665-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams JC: Biotin amplification of biotin and horseradish peroxidase signals in histochemical stains. J Histochem Cytochem 1992, 40:1457-1463 [DOI] [PubMed] [Google Scholar]
- 31.Varona R, Zaballos A, Gutiérrez J, Martín P, Roncal F, Albar JP, Ardavín C, Márquez G: Molecular cloning, functional characterization and mRNA expression analysis of the murine chemokine receptor CCR6 and its specific ligand MIP-3α. FEBS Lett 1998, 440:188-194 [DOI] [PubMed] [Google Scholar]
- 32.Perry VH, Hume DA, Gordon S: Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 1985, 15:313-326 [DOI] [PubMed] [Google Scholar]
- 33.Reid SD, Penna G, Adorini L: The control of T cell responses by dendritic cell subsets. Curr Opin Immunol 2000, 12:114-121 [DOI] [PubMed] [Google Scholar]
- 34.Tanaka Y, Imai T, Baba M, Ishikawa I, Uehira M, Nomiyama H, Yoshie O: Selective expression of liver and activation-regulated chemokine (LARC) in intestinal epithelium in mice and humans. Eur J Immunol 1999, 29:633-642 [DOI] [PubMed] [Google Scholar]
- 35.Dieu M-C, Vanbervliet B, Vicari A, Bridon J-M, Oldham E, Aït-Yahia S, Brière F, Zlotnik A, Lebecque S, Caux C: Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med 1998, 188:373-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao F, Rabin RL, Smith CS, Sharma G, Nutman TB, Farber JM: CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3α. J Immunol 1999, 162:186-194 [PubMed] [Google Scholar]
- 37.Matsumoto Y, Hara N, Tanaka R, Fujiwara M: Immunohistochemical analysis of the rat central nervous system during experimental allergic encephalomyelitis, with special reference to Ia-positive cells with dendritic morphology. J Immunol 1986, 136:3668-3676 [PubMed] [Google Scholar]
- 38.Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T: TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol 1995, 154:944-953 [PubMed] [Google Scholar]
- 39.Jander S, Pohl J, D’Urso D, Gillen C, Stoll G: Time course and cellular localization of interleukin-10 mRNA and protein expression in autoimmune inflammation of the rat central nervous system. Am J Pathol 1998, 152:975-982 [PMC free article] [PubMed] [Google Scholar]
- 40.Kiefer R, Schweitzer T, Jung S, Toyka KV, Hartung HP: Sequential expression of transforming growth factor-beta by T cells, macrophages and microglia in rat spinal cord during autoimmune inflammation. J Neuropathol Exp Neurol 1998, 57:385-395 [DOI] [PubMed] [Google Scholar]
- 41.June CH, Bluestone JA, Nadler LM, Thompson CB: The B7 and CD28 receptor families. Immunol Today 1994, 15:321-331 [DOI] [PubMed] [Google Scholar]
- 42.Stout RD, Suttles J: The many roles of CD40 in cell-mediated inflammatory responses. Immunol Today 1996, 17:487-492 [DOI] [PubMed] [Google Scholar]
- 43.Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJA, Claassen E: CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA 1996, 93:2499-2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyons JA, Zhao ML, Fritz RB: Pathogenesis of acute passive murine encephalomyelitis II. Th1 phenotype of the inducing population is not sufficient to cause disease. J Neuroimmunol 1999, 93:26-36 [DOI] [PubMed] [Google Scholar]
- 45.Kennedy MK, Torrance DS, Picha KS, Mohler KM: Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol 1992, 149:2496-2505 [PubMed] [Google Scholar]
- 46.Tanuma N, Kojima T, Shin T, Aikawa Y, Kohji T, Ishihara Y, Matsumoto Y: Competitive PCR quantification of pro- and anti-inflammatory cytokine mRNA in the central nervous system during autoimmune encephalomyelitis. J Neuroimmunol 1997, 73:197-206 [DOI] [PubMed] [Google Scholar]
- 47.Tourtellotte WW, Walsh MJ, Baumhefner RW, Staugaitis SM, Shapshak P: The current status of multiple sclerosis intra-blood-brain-barrier IgG synthesis. Ann NY Acad Sci 1984, 436:52-67 [DOI] [PubMed] [Google Scholar]
- 48.Carson MJ, Reilly CR, Sutcliffe JG, Lo D: Disproportionate recruitment of CD8+ T cells into the central nervous system by professional antigen-presenting cells. Am J Pathol 1999, 154:481-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang HL, Cyster JG: Chemokine up-regulation and activated T cell attraction by maturing dendritic cells. Science 1999, 284:819-822 [DOI] [PubMed] [Google Scholar]
- 50.Raine CS, Mokhtarian F, McFarlin DE: Adoptively transferred chronic relapsing experimental autoimmune encephalomyelitis in the mouse. Neuropathologic analysis. Lab Invest 1984, 51:534-546 [PubMed] [Google Scholar]
- 51.Prineas JW: Multiple sclerosis: presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science 1979, 203:1123-1125 [DOI] [PubMed] [Google Scholar]
- 52.Di Rosa F, Serafini B, Scognamiglio P, Di Virgilio A, Finocchi L, Aloisi F, Barnaba V: Short-lived immunization site inflammation in self-limited active experimental allergic encephalomyelitis. Int Immunol 2000, 12:711-719 [DOI] [PubMed] [Google Scholar]
- 53.Cella M, Sallusto F, Lanzavecchia A: Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol 1997, 9:10-16 [DOI] [PubMed] [Google Scholar]
- 54.Hickey WF, Vass K, Lassmann H: Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J Neuropathol Exp Neurol 1992, 51:246-256 [DOI] [PubMed] [Google Scholar]
- 55.Lawson LJ, Perry VH, Gordon S: Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48:405-415 [DOI] [PubMed] [Google Scholar]
- 56.Baggiolini M: Chemokines and leukocyte traffic. Nature 1998, 392:565-568 [DOI] [PubMed] [Google Scholar]
- 57.Sallusto F, Lanzavecchia A, Mackay CR: Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol Today 1998, 19:568-574 [DOI] [PubMed] [Google Scholar]
- 58.Sozzani S, Allavena P, D’Amico G, Luini W, Bianchi G, Kataura M, Imai T, Yoshie O, Bonecchi R, Mantovani A: Cutting edge: differential regulation of chemokine receptors during dendritic cell maturation: a model for their trafficking properties. J Immunol 1998, 161:1083-1086 [PubMed] [Google Scholar]
- 59.Vecchi A, Massimiliano L, Ramponi S, Luini W, Bernasconi S, Bonecchi R, Allavena P, Parmentier M, Mantovani A, Sozzani S: Differential responsiveness to constitutive vs. inducible chemokines of immature and mature mouse dendritic cells. J Leukoc Biol 1999, 66:489-494 [DOI] [PubMed] [Google Scholar]
- 60.Karpus WJ, Ransohoff RM: Cutting edge commentary: chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. J Immunol 1999, 161:2667-2671 [PubMed] [Google Scholar]
- 61.Asensio VC, Campbell IL: Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci 1999, 22:504-512 [DOI] [PubMed] [Google Scholar]
- 62.Saeki H, Moore AM, Brown MJ, Hwang ST: Secondary lymphoid tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol 2000, 162:2472-2475 [PubMed] [Google Scholar]