

Abstract
Tumor neovascularization is considered to be a critical step in the development of a malignant tumor. Endothelin (ET)-1 is a powerful vasoconstrictor and mitogenic peptide that is produced by many cancer cell lines. The cellular distribution of the ET components was evaluated in human colon tumors and compared to normal colon. There was more of the ET components (preproET-1, endothelin-converting enzyme-1, and ETA and ETB receptors) in adenomas and adenocarcinomas than in the normal colon. There was overproduction of preproET-1 and endothelin-converting enzyme-1 in carcinoma cells and stromal vessels, suggesting that they are a local source of ET-1. ETA receptors were present in stromal myofibroblasts of neoplastic tissue, and there were large amounts of ETB receptors in the endothelium and myofibroblasts. There was also a redistribution of α-smooth muscle actin-positive cells in the vascular structures of tumors. An experimental rat model of induced colon cancer treated for 30 days with bosentan, a mixed antagonist of both ET receptors, confirmed the morphological changes observed during the tumor vascularization. Our data suggest that ET-1 and its receptor play a role in colon cancer progression, with ET-1 functioning as a negative modulator of the stromal response.
Endothelin (ET)-1, the most potent vasoconstrictor peptide known, was identified by Yanagisawa et al. 1 It belongs to a recently discovered family of 21-amino acid peptides, the ETs, which regulate vascular tone. 2 The ETs bind to two high-affinity receptor subtypes, ETA 3 and ETB, 4 which are seven-transmembrane G-protein-coupled receptors. ET-1 and ET-2 bind to ETA receptors at physiological concentrations, whereas ET-3 does not. But all three ETs bind to ETB receptors with similar affinities. The ETs are synthesized as large precursor polypeptides, called preproETs (PPETs), which are cleaved at two pairs of basic amino acids, generating intermediate peptides, the bigETs. The bigETs are then cleaved by an endothelin-converting enzyme (ECE) 5 to produce the mature ETs. ECE is a key enzyme in the biosynthesis of the ETs because the biological activity of bigETs is negligible. 6 Two ECE genes have been cloned, ECE-1 7 and ECE-2. 8 Their sequences are 59% identical, but only ECE-1, the most abundant, has been studied in detail (see Turner and Tanzawa 9 for a recent review).
ET-1 was initially described as a vasoconstrictor peptide but it has a variety of other effects in nonvascular tissues such as the stimulation of hormone release and the regulation of central nervous system activity. 10 ET-1 is also a potent mitogen in many cell types, including vascular smooth muscle cells, 11 playing a fundamental role in cardiovascular system development. 12 ET-1 has also been reported to stimulate the proliferation of various types of neoplastic cells. 13 Lastly, various human cancer cell lines derived from mammary, pancreatic, and colon carcinomas produce significant amounts of ET. 14
The growth of malignant tumors depends on neovascularization. Tumor angiogenesis requires angiogenic factors, such as vascular endothelial growth factor, provided by cancer cells and affecting the host tissue. 15 It was recently shown that vasoactive peptides modulate vascular endothelial growth factor production and endothelial cell proliferation and invasion. 16 The mechanisms involved in maturation of tumor vascularization are not well defined. Endothelial cells are a critical element responsible for new vessel formation but other cellular elements, like smooth muscle cells/pericytes are necessary. Maturation of the vascular system involves the recruitment of perivascular supporting cells that do not bear cell-specific markers, but which do contain α-smooth muscle actin (α-SMA). 17
A report suggests that migration of endothelial cells is promoted by ET-1 via the ETB receptor. 18 Raised ET-1 levels have been found in patients with liver metastases of colorectal cancer 19 and ET binding sites have been found in human colon cancer tissue. 20 In a previous study, we showed that there is ECE-1 mRNA and ECE-1 protein in the adult human colon. 21 And the whole ET-1 system has recently been identified in the human normal colon; its distribution suggests that it is secreted as a neuropeptide and a vasopeptide in this tissue. 22 However, the distribution of the ET system in various grades of colon cancer has not been evaluated.
The present study was therefore done to precisely determine and compare the distributions of all of the components of the ET-1 system in endothelial, smooth muscle cells, and macrophages. We examined mRNAs and proteins in the human normal colon, adenoma, and adenocarcinoma colon, to assess their potential role in tumor vascularization. We used an experimental rat model of colon cancer, with or without bosentan (a mixed antagonist of ETA and ETB receptors) treatment to further evaluate the influence of ET-1 receptors and α-SMA-positive cells in stromal angiogenic responses.
Materials and Methods
Human Tissues
Human colon tissues were obtained at the Institute of Pathology (Lausanne, Switzerland) at surgery from patients (n = 18) undergoing colectomy for cancer. Samples from 18 patients (41 to 84 years old, eight women and 10 men) were examined. Nine samples were from the cecum and nine from the sigmoid colon, all at the T3 or T4 stage. Tissues were either snap-frozen in liquid nitrogen and stored at −80°C (eight samples), or fixed in 4% buffered paraformaldehyde for at least 24 hours, processed, and embedded in paraffin (10 samples).
mRNA Analysis
Total RNA was isolated from frozen adenocarcinoma colon sections and control regions, excised at least 1 cm from the lesion, using the protocol of Chomczinski et al. 23 cDNA was prepared with 0.5 μg total RNA, 10 pmol oligo-dT using murine Moloney leukemia virus reverse transcriptase (Life Technologies, Inc., Rockville, MD) according to the manufacturer’s instructions. Polymerase chain reaction was performed using 3 μl of cDNA solution and 1.25 U of Taq polymerase (Roche Diagnostics, Deylan, France) according to the manufacturer’s instructions. Control reactions for reverse transcriptase-polymerase chain reaction analysis were performed from nonreverse-transcribed RNA samples. No amplification was observed for any of the RNA samples tested (not shown). Specific primers (10 pmol) for ECE-1, 5 PPET-1, 24 ETA 3 and ETB 4 receptors, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 25 were added as described. 22 The primers used were designed to avoid false-positive reactions from genomic DNA contamination. Thirty cycles were performed, consisting of denaturation at 94°C (30 seconds), annealing at 58°C (PPET-1, ETA, and ETB) or 55°C (ECE-1 and GAPDH) (30 seconds) and extension at 72°C (30 seconds) with a final extension step of 10 minutes at 72°C. Amplified products were analyzed on 2% agarose gel.
Preparation of Radiolabeled Riboprobes
The human PPET-1 partial cDNA, corresponding to the nucleotide sequence 70 to 630 was subcloned into pBK-CMV (Stratagene, La Jolla, CA). The recombinant plasmid was linearized by digestion with SacI to obtain the antisense or with KpnI to obtain the sense RNA probe. Probes for human ECE-1 5 were prepared as described in Korth et al. 21 Briefly, the ECE-1 partial cDNA corresponding to the nucleotides 304 to 1666 was linearized by digestion with HindIII to obtain the antisense or with XbaI to obtain the sense RNA. Probes for human ETA 3 and ETB 4 receptors, subcloned into pcDNA3, were prepared as described by Brand et al. 26 ETA and ETB cDNA were linearized by digestion with XhoI and KpnI, respectively, to obtain the antisense probes. ETA and ETB cDNA were linearized by digestion with XbaI to obtain the sense probe. In vitro transcription and labeling with 35S-UTP (Amersham, Les Vlis, France) were performed with T7 or SP6 RNA polymerase (Roche Diagnostics, Meylan, France). Probes were precipitated with ammonium acetate and ethanol, dried by centrifugation-evaporation (Speed-Vac) and dissolved in10 mmol/L Tris, 1 mmol/L ethylenediaminetetraacetic acid, 20 mmol/L dithiothreitol.
In Situ Hybridization
The in situ hybridization protocol used for paraffin sections involved microwave pretreatment to enhance the hybridization signal. 27 Paraffin-embedded sections (5 μm) were cut and two adjacent sections were mounted on each silane-coated slide. Deparaffinized sections were immersed in 0.01 mol/L citric acid, pH 6.0, and heated in a microwave oven for 12 minutes. The sections were then incubated with proteinase K (2 μg/ml; Roche Diagnostics) for 20 minutes and dehydrated. In situ hybridization performed on frozen sections used 7-μm sections fixed in paraformaldehyde/phosphate-buffered saline and dehydrated without microwaving. The following protocol was subsequently used for both frozen and paraffin-embedded sections. Sections were incubated overnight at 50°C with the respective antisense and sense riboprobes (3 to 4 × 10 5 cpm per section). The slides were washed with increasingly stringent solutions and treated with RNase A (20 μg/ml; Sigma, Saint-Quentin, France). The sections were then dehydrated and placed in contact with Biomax film (Kodak, Rochester, NY) for 1 to 3 days. They were subsequently dipped in NTB2 liquid emulsion (Kodak) and exposed for 2 weeks (ECE-1 or PPET-1 probes) or for 4 weeks (ETA and ETB probes). Sections were counterstained with toluidine blue. Figures shown are from in situ hybridization performed in paraffin-embedded tissue sections unless otherwise stated.
125I ET-1 Binding
Sections (7 μm) were cut using a cryostat, thaw-mounted on silane-coated slides, and stored overnight under vacuum at 4°C. Consecutive sections were fixed for 10 minutes in 4% formaldehyde/phosphate-buffered saline, and then incubated for 15 minutes in 50 mmol/L Tris-HCl buffer, pH 7.5, containing 120 mmol/L NaCl, 5 mmol/L MgCl2, and 40 mg/L bacitracin. Sections were then incubated with 100 pmol/L 125I ET-1 (2,125 Ci/mmol) in the previous buffer containing 1% bovine serum albumin (fraction V, protease-free; Sigma Chemical Co.) and 1 mmol/L phosphoramidon for 90 minutes at room temperature. Sections were given four 1-minute washes in ice-cold 50 mmol/L Tris-HCl, pH 7.4, dipped in ice-cold distilled water, air-dried, and placed in contact with Biomax MR films (Kodak). Nonspecific binding was determined in consecutive sections incubated as described above with 1 μmol/L unlabeled ET-1 (Bachem, Bubendorf, Switzerland). The receptor subtypes were identified by incubating consecutive sections as described above with 1 μmol/L BQ 123 (ETA antagonist), 10 nmol/L ET3 (natural ETB agonist), or 0.2 μmol/L sarafotoxin 6c (S6c) (selective ETB agonist). The sections were then fixed in paraformaldehyde at 80°C for 2 hours, dipped in NTB2 photographic emulsion (Kodak), exposed for 4 days, and counterstained with toluidine blue.
Immunohistochemistry
Paraffin-embedded sections (5 μm) were incubated with xylene (to remove paraffin), rehydrated in a graded ethanol series and their endogenous peroxidase inactivated with 3% hydrogen peroxide in methanol for 10 minutes. They were then washed in water and incubated with monoclonal antibodies to CD31, CD68 (both from DAKO, Hamburg, Germany), α-SMA (Sigma), and Ki-67 antigen (MIB-1, Dianova, Hamburg, Germany) according to the manufacturer’s instructions. The antiserum 473-17-A 28 was used to stain ECE-1. The bound anti-CD31 and anti-α-SMA antibodies were reacted with avidin-biotin complex (ABC; DAKO) and those for CD68 and ECE-1 were reacted with peroxidase-antiperoxidase (DAKO). Sections were treated with 0.035% diaminobenzidine (Fluka, Buch, Switzerland) for 30 minutes, counterstained with hematoxylin (according to Mayer), and mounted. Control reactions without first antibody showed no nonspecific staining (not shown).
Quantification
The tumors were scored semiquantitatively for mRNA expression in epithelial and stroma cells by assessing both the grade of labeling (low, moderate, high, and scattered) and the frequency of signal in each cell type considering 50 cells in the field of a ×40 objective. The distribution of markers (CD31, α-SMA, ETA and ETB mRNA) in stroma was evaluated in three different typical regions of normal (open bars), adenoma (gray bars), and adenocarcinoma (black bars) tissues. The field was chosen in longitudinal sections of crypts and polyps or in vascularized invasive areas. The paired t-test was used for statistical analysis.
Animal Experimentation
Bosentan Treatment
Peritoneal carcinomas (solid tumors) were induced in inbred BDIX rats (300 g males or females) purchased from IFFA Credo (l’Arbresle, France) by intraperitoneal injection of 10 6 syngeneic PROb cells. Animal use and handling was performed according to the French laws for animal experimentation. The PROb cells were derived from a colon adenocarcinoma chemically induced in BDIX rats. Control rats (n = 10) were fed normal rat chow (UAR, Epinay-sur-Oise, France), and an another group (n = 10) were fed bosentan (a gift from Dr. M. Clozel, Actelion, Switzerland) incorporated into the pellets of chow at 100 mg/kg/day, assuming that each animal ate 15 g chow per day. Bosentan treatment started the day before the injection of PROb cells. Under these conditions all rats developed peritoneal carcinomatosis and hemorrhagic ascitis. 29 Animals were examined at the time of their death or sacrifice, which was day 30 after implantation when tumors were evaluated according to: class 0, no nodules detected; class I, few 0.1- to 0.2-cm nodules; class II, numerous 0.1- to 0.5-cm nodules; class III, 1-cm nodules invaded peritoneal cavity; class IV, peritoneal cavity has been completely invaded. Nodules were characterized as viable tumor area. Treatment efficacy was evaluated by morphological analysis of the tumors in control and bosentan-treated rats.
Tumors Analysis
Sections (10 μm) of tumors were cut using a cryostat, thaw-mounted, and treated as human samples. 125I ET-1 binding was assessed as described for the human tissues. Immunohistochemistry was performed with antibodies against cytokeratin-18 (ICN, Costa Mesa, CA), α-SMA (Sigma), and rat von Willebrand factor (vWF) (Cedarlane, Hornby, Canada) as for human tissues. Collagen was visualized with hematoxylin-eosin-safran staining. The number of nodules (viable tumor areas) and necrotic areas were assessed in a ×X objective field of keratin and hematoxylin-eosin-safran staining slides. Quantification of markers (vWF and α-SMA) in tumors was evaluated in three different areas of each control (open bars) and bosentan-treated (black bars) animals considering all of the positive cells in the field of the ×40 objective. The impaired t-test was used for statistical analysis.
Results
ET System mRNAs in Human Colon Adenocarcinoma
Reverse transcriptase-polymerase chain reaction analysis was performed on 0.5 μg of total RNA from seven samples to detect all of the components of the ET system. Despite the heterogeneity of the level of the amplified bands, the ET system was more abundant in adenocarcinoma than in normal colon tissue (Figure 1) ▶ . In particular, the concentrations of mRNA for PPET-1 and ECE-1 were higher in the cancer than in normal tissue, together with the expected greater concentration of GAPDH mRNA in cancer. 30
Figure 1.
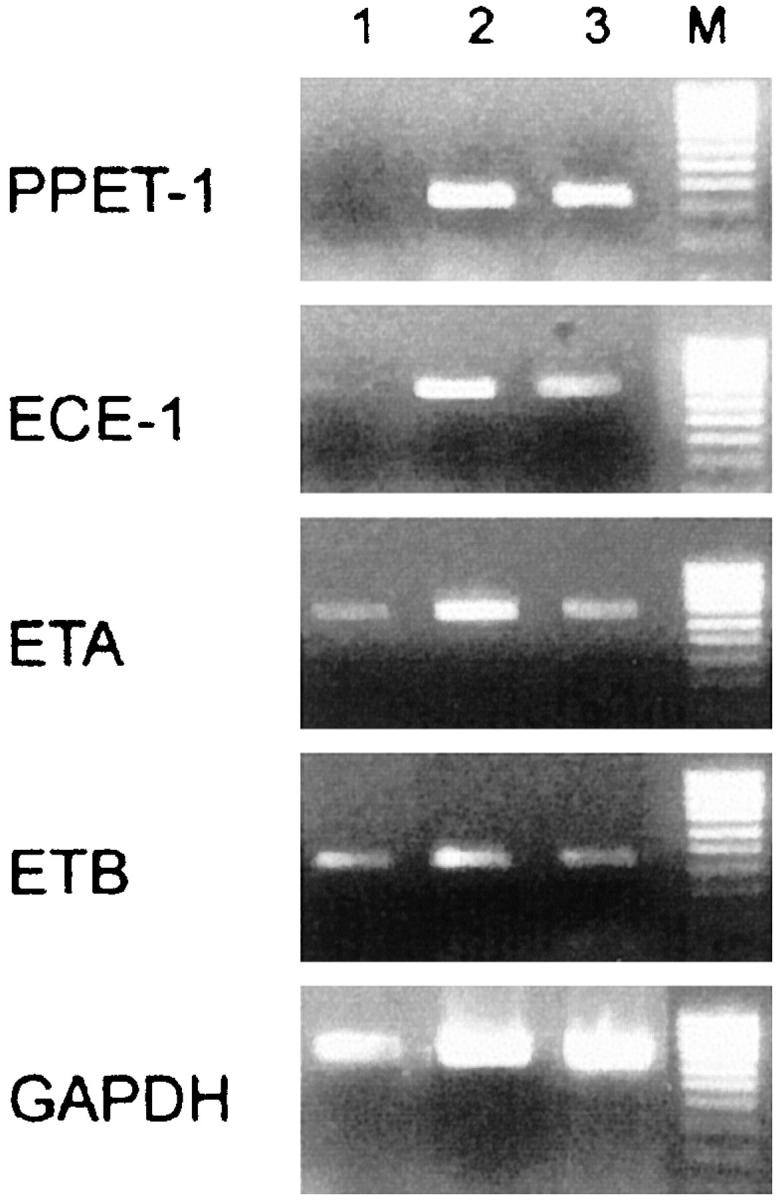
Components of the ET system in human colon adenocarcinoma. PPET-1, ECE-1, ETA, and ETB receptors and GAPDH were detected by reverse transcriptase-polymerase chain reaction using the specific primers described previously 22 and total RNA from normal (lane 1) or tumor tissue (lanes 2 and 3). One normal and two tumor samples out of seven of each are shown here. Thirty cycles of amplification gave each gene at the expected size, 341 bp (PPET-1), 622 bp (ECE-1), 675 bp (ETA), 400 bp (ETB), and 649 bp (GAPDH). Molecular weight markers (M) are λ/HindIII, φX174 DNA/HaeIII.
Tissue architecture of samples was analyzed by Ki-67 immunohistochemistry using MIB-1 antibody (Figure 2) ▶ . Figure 2a ▶ shows the typical immunostaining in the epithelial monolayer at the base of the crypts of normal colon mucosa whereas adenoma-disrupted crypts (Figure 2b) ▶ and adenocarcinoma (Figure 2c) ▶ had an anarchical MIB-1 staining pattern mainly in neoplastic epithelial cells.
Figure 2.
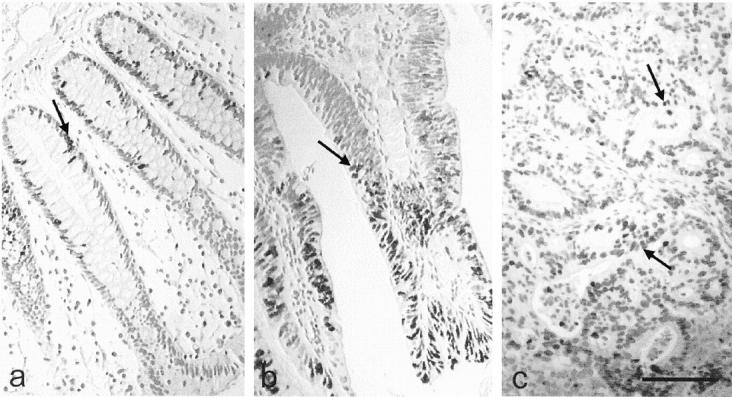
Histology of human colon samples. The proliferation state of each colon tissue sample, normal (a), adenoma (b), and adenocarcinoma (c) was evaluated by immunohistochemistry with MIB-1 antibody. This antibody detects Ki-67 antigen, a marker of cell proliferation. Tumor tissue had anarchic epithelial proliferation that is reflected in the MIB-1 immunostaining of the cells. Scale bar, 100 μm.
We located the cellular distribution of the ET system by in situ hybridization. Dark-field photographs of the labeling obtained with each riboprobe showed greater expression of the whole ET system in cancer tissue than in the normal colon (Figure 3) ▶ . The distribution of labeled PPET-1, ECE-1, and ETA receptor in neoplastic tissue and normal tissue were essentially the same. PPET-1 (Figure 3, a–c) ▶ and ECE-1 mRNA (Figure 3, d–f) ▶ were found mainly in the epithelium and ETA receptor mRNA (Figure 3, g–i) ▶ was found in the stroma. The distribution of ETA receptor mRNA was comparable to that of α-SMA immunoreactive cells along normal crypts and tumor vasculature (Figure 3, m–o) ▶ . ETB receptor mRNA was nearly undetectable in the normal colonic mucosa, but was abundant in the cancer stroma (Figure 3, j–l) ▶ , associated with the α-SMA stained cells (Figure 3, n and o) ▶ . ETA and ETB transcripts were also present in stromal vascular structures of adenocarcinoma. However, neither α-SMA signal (Figure 3o) ▶ , nor ETA or ETB mRNA was detected (Figure 3, i and l) ▶ where nests of tumor cells were invading the submucosa or the muscularis propria without a stromal reaction. Receptor mRNAs seemed to be much less abundant than mRNA for PPET-1 and ECE-1, with ETB mRNA being the least abundant, considering that ETA- and ETB-hybridized sections were exposed for twice as long as PPET-1 and ECE-1 probes. Sense probes yielded no labeling in any tissue (not shown).
Figure 3.
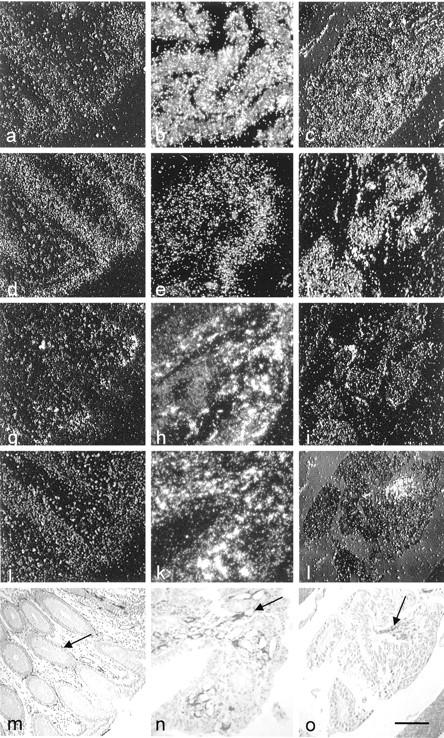
Components of the ET system in normal, adenoma, and adenocarcinoma human colon. In situ hybridization was performed with the antisense probe for PPET-1 (a–c), ECE-1 (d–f), ETA (g–i), and ETB (j–l). The photographs of consecutive sections of normal (a, d, g, j, and m), adenoma (b, e, h, k, and n), and adenocarcinoma (c, f, i, l, and o) human colon tissues are presented in dark-field illumination. Immunohistochemistry was performed to identify smooth muscle cells with anti-α-SMA (arrow in m–o) and define the regions studied. Scale bar, 100 μm.
PPET-1 mRNA was homogeneously detected in all epithelial cells of the adenoma (Figure 4a) ▶ and in adenocarcinomas at higher magnification (Figure 4b) ▶ . A PPET-1 signal was detected in the macrophages and myofibroblasts of the tumor stroma and also in endothelial cells in contrast to the normal tissue. There was ECE-1 mRNA in the epithelial and endothelial cells and in myofibroblasts of carcinomas (Figure 4, c and d) ▶ . ECE-1 immunoreactivity was found in endothelial cells from stromal vessels of the adenoma (Figure 4e) ▶ and also in cancer cells of the carcinoma in addition to vascular cells (Figure 4f) ▶ , showing similar distributions of the enzyme and its substrate in cancer.
Figure 4.

Cellular distribution of PPET-1 and ECE-1 mRNAs in human colon adenoma and adenocarcinoma. In situ hybridization with the antisense probe for PPET-1 (a and b) and ECE-1 (c and d), was performed in adenoma (a, c, and e) and carcinoma (b, d, and f). Photographs are presented in bright-field illumination. Immunohistochemistry was performed with the anti-ECE-1 antibody (e and f). There was a strong labeling in endothelial (arrowhead) and epithelial cells with both probes. Scale bar, 20 μm.
The cellular distributions of receptor transcripts in adenoma (Figure 5, a–f) ▶ and adenocarcinoma (Figure 5, g–l) ▶ confirmed the findings at low magnification, with receptor mRNA almost exclusively in the stroma. ETA mRNA was found in the adenomas and adenocarcinomas (Figure 5, a and g) ▶ , associated with a subpopulation of α-SMA-positive cells, probably subepithelial myofibroblasts (Figure 5, b and h) ▶ and also with CD31-positive cells (Figure 5i) ▶ . The vessels of adenomas were strongly labeled for ETB (Figure 5, c and f) ▶ which were also immunostained for α-SMA (Figure 5e) ▶ and the proliferation marker MIB-1 (Figure 5d) ▶ . ETB labeling was present in adenocarcinomas (Figure 5e) ▶ in the endothelial cells that were immunostained with CD31 antibody (Figure 5, i and j) ▶ and in myofibroblasts that were immunostained with α-SMA antibody (Figure 5k) ▶ . Thus, labeling for ETA and ETB mRNAs was more intense in regions with pronounced vascularization containing abundant α-SMA-positive cells.
Figure 5.
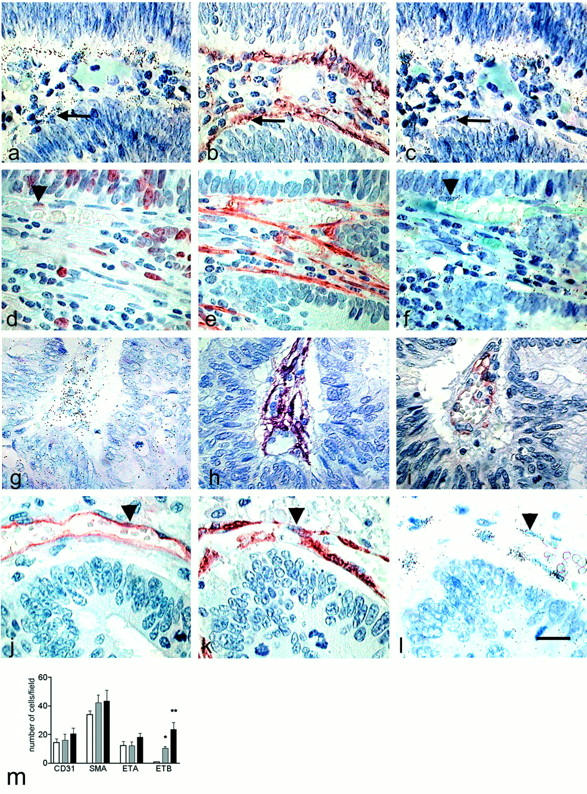
Cellular distribution of ETA and ETB receptors mRNAs in human colon adenoma and adenocarcinoma. In situ hybridization was performed in consecutive sections of adenoma (a–f) and adenocarcinoma (g–l) with the antisense probes for ETA (a and g) and ETB (c, f, and l) receptors. Photographs are presented in bright-field illumination. Immunohistochemistry was performed with anti-α-SMA (b, e, h, and k) (smooth muscle cell marker), anti-Ki-67 (d) (proliferation marker), and anti-CD31 (i and j) (endothelial cell marker) antibodies to identify the cells labeled with ETA and ETB probes. There was intense labeling in endothelial cells (arrowhead) and myofibroblasts (arrow) with both probes. Distribution of specific markers in the stroma (m). The cells labeled for CD31, α-SMA, ETA, and ETB mRNAs were counted in the field of the ×40 objective in normal (open bars), adenoma (gray bars), and adenocarcinoma (black bars). The field corresponded to the entire crypt in the normal colon. The results averaged values of seven different samples that are representative of all of the specimens analyzed. Significant differences are shown by * (P = 0.0162) and ** (P = 0.0022). Scale bar, 20 μm.
Quantification of these markers in the stroma of normal and abnormal regions showed slightly but not significantly more cells identified as endothelial cells between normal, adenoma, and adenocarcinoma tissues (Figure 5m) ▶ . The same was true for cells stained for α-SMA in adenomas than in the normal colon. Interestingly, two types of cells were stained by the α-SMA antibody: subepithelial myofibroblasts in normal tissues and smooth muscle cells strictly associated with vascular structures in adenocarcinomas, hence showing a progressive redistribution of the cells positive for α-SMA from adenoma to adenocarcinoma (Figure 5, e and h) ▶ . And counting the cells containing ETB mRNA confirmed the significantly increased mRNA in adenomas (P = 0.0162) and adenocarcinomas (P = 0.0022) over normal tissue observed by in situ hybridization, whereas there were no significant differences for ETA receptors.
It is worth noting that comparison of cells positive for α-SMA and ETB receptors in adjacent sections (Figure 5m) ▶ showed that not all α-SMA-positive cells (Figure 6a) ▶ were labeled for ETB mRNA (Figure 6b) ▶ .
Figure 6.
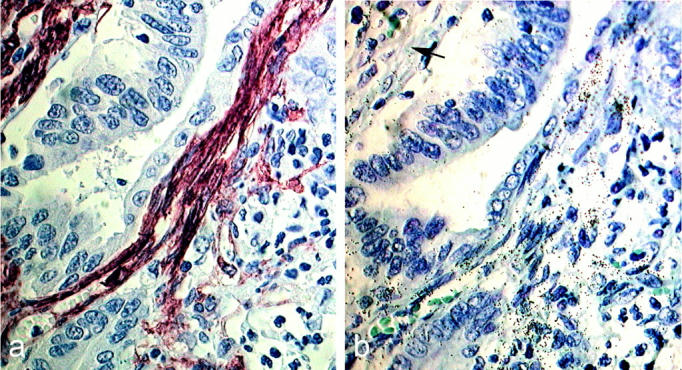
Cellular distribution of ETB mRNA in human colon adenoma. Immunohistochemistry for α-SMA (a) and in situ hybridization for ETB receptors (b) were performed in consecutive sections. Photographs are in bright-field illumination. Not all α-SMA-positive cells were labeled for ETB receptor mRNA (arrow). Scale bar, 20 μm.
Localization of ET Binding Sites in the Human Colon by Autoradiography
The distribution of ETA and ETB receptors in human colon adenocarcinomas were assessed by autoradiography of 125I ET-1 binding to frozen samples from eight patients (Figure 7) ▶ . Figure 7, a and b ▶ , shows total 125I ET-1 binding and Figure 7, c and d ▶ , shows the nonspecific binding that remains after displacement with ET-1, or BQ123 plus S6c, which was uniformly low. There was considerable specific binding in the lamina propria of the mucosa, but there was very little over the epithelium of the normal colon (Figure 7a) ▶ . The distribution of binding in tumor tissue was heterogeneous and was concentrated over clusters of fibroblasts adjacent to cancer cells (Figure 7b) ▶ . Receptor subtypes were identified in consecutive sections by 125I ET-1 binding in the presence of the specific ETA antagonist BQ123 and the specific ETB analogue S6c. Binding was displaced with S6c (Figure 7, e and f) ▶ and with BQ123 (Figure 7, g and h) ▶ in normal mucosa and adenocarcinomas indicating the presence of both receptor subtypes, with a higher proportion of ETB in the myofibroblasts adjacent to the cancer cell foci. The same pattern of expression was obtained by in situ hybridization in adjacent sections (not shown).
Figure 7.
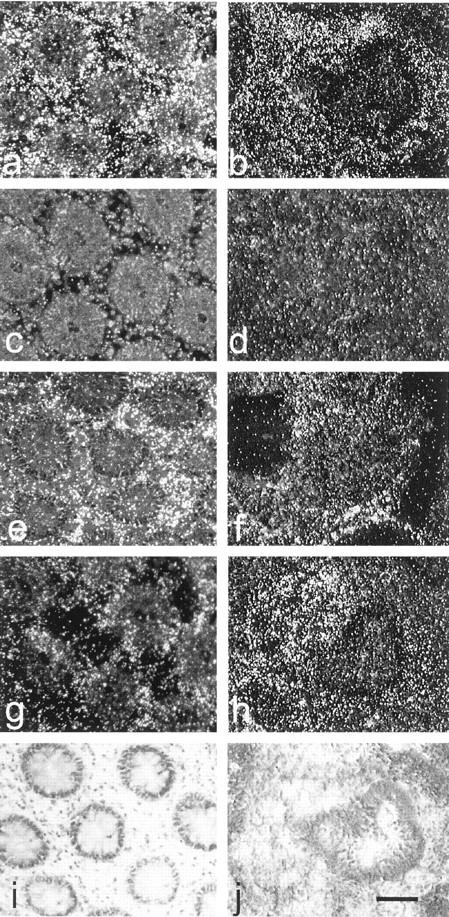
Autoradiographic 125I ET-1 binding sites in human colon adenocarcinoma. 125I ET-1 (100 pmol/L) binding was performed in frozen consecutive sections. Dark-field illuminations are shown of transversal sections of normal colon (a, c, e, g, and i) and adenocarcinoma (b, d, f, h, and j). a and b: Total binding. c and d: Nonspecific binding incubated as in a and b in the presence of 1 μmol/L ET-1 or both 1 μmol/L BQ123 and 0.2 μmol/L S6c. e and f: ETA binding, after incubation as in a and b in the presence of 0.2 μmol/L S6c as competitor. g and h: ETB binding, after incubation as in a and b in the presence of 1 μmol/L BQ123. i and j: Toluidine blue counterstaining. Scale bar, 100 μm.
In Vivo Effect of Bosentan Blockade in an Experimental Rat Model
Rats implanted with tumors and treated with 100 mg/kg−1 for 30 days of bosentan were killed and the tumors analyzed. PedutoEberl et al 31 have shown that bosentan-treated animals tend to have lower tumor grades than controls, but without any statistically significant differences and without complete control of tumor progression. Morphological analysis of the tumors (Figure 8) ▶ by hematoxylin-eosin-safran staining showed a decrease in collagen matrix around the nodules (insert) in bosentan-treated animals (Figure 8b) ▶ than in controls (Figure 8a) ▶ . In addition, tumors were less dense in bosentan-treated animals (Figure 8b) ▶ compared to controls (Figure 8a) ▶ , in agreement with the observation that the tumors in the treated rats were less cohesive at the time of sacrifice. Surprisingly, there was no differences in 125I ET-1 binding sites for both ET subtype receptors in tumors of treated and untreated rats despite bosentan treatment for 30 days (not shown).
Figure 8.
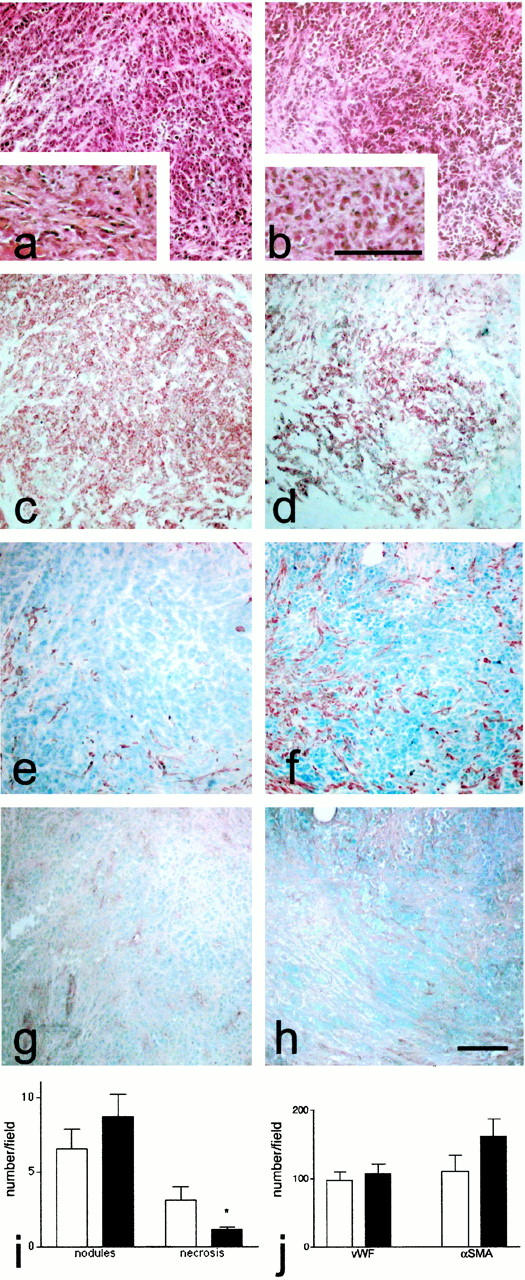
Morphology of the colon tumors in the experimental rat model. Immunohistochemistry was performed in consecutive sections of tumors from control (a, c, e, and g) and bosentan-treated rats (b, d, f, and h). Photographs are presented in bright-field illumination. Eosin-hematoxylin-safran staining (a and b) was performed for assessing the tumor appearance. Collagen characterized with the safran staining (orange) was shown at higher magnification (insert in a and b). Tumor epithelial cells bound anti-keratin antibody (c and d). Smooth muscle cells were identified with anti-α-SMA (e and f) and endothelial cells with an anti-vWF antibody (g and h). Histological scores (i) were obtained by counting the number of necrotic areas and nodules in the field of the ×4 objective in control (white bars) and bosentan-treated animals (black bars). j: Distribution of specific markers in rat nodules. The cells stained for vWF and α-SMA were counted in the field of the ×40 objective in control (open bars) and bosentan-treated animals (black bars). The results averaged values of seven different samples that are representative of all of the specimens analyzed. Significant differences (P ≤ 0.05) are indicated by an asterisk. Scale bar, 100 μm.
Analysis of keratin-positive cells showed that the tumor cells in bosentan-treated animals (Figure 8d) ▶ were more dispersed than in control rats (Figure 8c) ▶ . Staining for α-SMA demonstrated a few smooth muscle cells in the tumors of untreated rats (Figure 8e) ▶ . In contrast, the tumors of bosentan-treated animals contained α-SMA-positive cells (Figure 8f) ▶ . Endothelial cells identified by staining for vWF antigen were present in tumors of both groups (Figure 8, g and h) ▶ . The histological score on hematoxylin-eosin-safran-stained sections performed on seven different samples for each group of rats showed that the tumors in bosentan-treated rats had significantly less necrotic areas than the controls (P ≥ 0.05) (Figure 8i) ▶ . Quantification of vascular markers showed no significant difference on vWF-positive cells between the two groups and a strong but not significant difference on α-SMA-positive cells (Figure 8j) ▶ . In untreated animals, there was a correlation between vWF- and α-SMA-positive cells suggesting an exclusive vascular presence of these markers whereas in bosentan-treated rats α-SMA cells outnumbered vWF cells indicating the presence of nonvascular myofibroblasts.
Discussion
Tumor development depends on neovascularization with the recruitment and proliferation of endothelial and mural cell (pericytes, smooth muscle cells, myofibroblasts) and tissue remodeling. 32,33 ET-1 is produced by human cancer cell lines 14 and may be a paracrine tumor-derived growth promoting and survival factor for vascular cells. 16 We recently demonstrated the presence of the complete ET-1 system in the human normal colon 22 and ET-1 is an autocrine survival factor for rat colon cancer cell lines. 31 We therefore postulated that ET-1 could be involved in tumor progression, either through an autocrine effect on tumor cells or a paracrine effect on cancer-associated stromal cells. We tested this notion by comparing the cell-specific distribution of the ET system, including PPET-1, ECE-1, and ETA and ETB receptors, in human colon cancers. We also evaluated the role of the ET system in tumor vascularization in vivo using an experimental model of colon cancer in rats treated (or not) with bosentan, a mixed antagonist of ET receptors.
Reverse transcriptase-polymerase chain reaction revealed more PPET-1 and ECE-1 in human colon adenocarcinomas than in normal tissue. In situ hybridization showed that the stroma surrounding the cancer had highly vascularized regions with larger concentrations of PPET-1 and ECE-1 than the stroma in normal tissue. PPET-1 and ECE-1 mRNAs were mainly found in epithelial and endothelial cells in the adenomas, with labeling being 4.5-fold to fivefold more frequent in adenoma endothelial cells than in the normal colon (Table 1) ▶ . Thus, both the substrate, PPET-1, and its converting enzyme, ECE-1, have the same distribution, so that active ET-1 can be produced in tumors. ET-1 is also present in the endothelial cells, tumor cells, and myofibroblasts of colorectal liver metastasis. 19 To our knowledge no previous reports have studied the expression of cellular distribution of ECE-1 in human tumors.
Table 1.
Cellular Distribution of Endothelin Components in Human Normal, Adenoma, and Adenocarcinoma Colon Tissue
| PPET-1 | ECE-1 | ETA | ETB | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | Adenoma | Adenocarcinoma | Normal | Adenoma | Adenocarcinoma | Normal | Adenoma | Adenocarcinoma | Normal | Adenoma | Adenocarcinoma | |
| Whole tissue | + | +++ | + | +++ | ++ | +++/++ | ++ | +++/++ | ||||
| Epithelial cell | + | +++ | +++ (1.2) | + | ++ | +++ | − | s | − (3) | − | s | + (2.3) |
| Endothelial cell | − | ++ | + (4.5) | + | +++ | +++ | − | + | − (2.3) | + | +++ | +++ (1.3) |
| Myofibroblast* | − | + | + (2.5) | − | − | + (5) | + | ++ | ++ | − | +++ | +++ (4) |
| Macrophage | + | ++ | ++ (1.4) | + | + | + (1.4) | + | ++ | ++ (1.5) | − | + | − |
Amounts of mRNA were evaluated from reverse transcriptase-polymerase chain reaction studies (whole tissue) and from in situ hybridization studies (cell type). For each riboprobe, − corresponds to no labeling; +, to low labeling; ++, to moderate labeling; +++, to high labeling in the vast majority of cells in the designated population; and s corresponds to labeling in only scattered cells. Numbers in brackets indicate the fold increase in the frequency of labeled cells outnumbered 50 cells in adenoma/adenocarcinoma regions compared to normal tissue. No number indicates no difference in the frequency of the labeled cells. The average values are representative of at least 10 different specimens examined. No significant differences were found in the amounts of ET system in the cecum and distal colon. Evaluation was performed by different investigators (GE, LJ, FP), each blind to the other assessments.
*We did not discriminate between subepithelial and perivascular myofibroblasts.
There were also more of targets for ET-1 and the ETA and ETB receptors in colorectal cancer than in normal tissue, both the mRNA and the protein, as defined by reverse transcriptase-polymerase chain reaction and in situ hybridization and ET-1 binding, respectively. ETA receptors were increased in myofibroblasts. ETB receptors were almost undetectable in normal colon mucosa, but they were abundant in all vascularized areas of the cancer stroma. Our results are in agreement with earlier reports describing more ET-1 binding sites in tumor vessels than in epithelium. 20 We found ETB mRNA in endothelial cells and in vessel-surrounding myofibroblasts in the vessels of cancer stromas. The myofibroblasts contain four times more labeled cells than in the normal colon (Table 1) ▶ . Consistent with these findings, both ETA and ETB receptor mRNA were detected in human colon fibroblasts. 22 Counting of α-SMA-positive cells demonstrated a redistribution of α-SMA-stained cells below the epithelium to around the vasculature in adenocarcinomas. Benjamin et al 34 recently showed the importance of α-SMA-positive cells as an index of vessel maturation in human brain and prostate tumors. Thus, our data suggest that the ET system is important as modulator of the colon tumor vascularization causing interaction of myofibroblasts with endothelial cells and ETB receptor induction. Tumor-infiltrating myofibroblasts are α-SMA-positive fibroblasts that are believed to be involved in tumor invasiveness 35 and the remodeling of the extracellular matrix. 36 They are different from the subepithelial α-SMA-positive cells in the human normal colon.
This raises the question of the role of the ET system and myofibroblasts in colon cancer. Tumor cells are an important source of factors that can promote stroma remodeling, including neovascularization and myofibroblast differentiation. The appearance of a high concentration of ETB receptor in endothelial and myofibroblast cells in cancer could be a key step in the process of tumor vascularization, involving these two cell types. The proliferation and migration of endothelial cells are promoted by ET-1 via the ETB receptor. 18 ET-1 may also exert an anti-apoptotic effect on endothelial cells, 37 smooth muscle cells, 38 and fibroblasts, 39 in addition to colon adenocarcinoma cells. 31
We have attempted to elucidate the phenotypic changes that occur during the colon tumor vascularization. We looked to see whether the induction of ET receptors, in particular ETB, and the redistribution of α-SMA-positive cells were simultaneous or successive events using an experimental rat model of induced colon carcinoma. We blocked ET receptors using bosentan, a mixed antagonist of both ET receptors, shown to incompletely control tumor progression in vivo using the same experimental model. 31 There were structural modifications within the tumors in bosentan-treated animals; the tumors were less dense with less collagen around the nodules. Bosentan also reduced deposition of collagen I and III in the extracellular matrix in a murine model of glomerulonephritis. 40 We found a tendency of an increased ratio of α-SMA-positive cells in the tumors of bosentan-treated animals suggesting that ET-1 acts negatively on α-SMA myofibroblasts. The treatment modified cell phenotypes and the cohesion of the tumor nodules, and necrosis was decreased. Bosentan has been recently shown to also decrease necrosis in a murine model of myocarditis. 41 But we found no apparent difference in the amount of ETA and ETB receptors in treated and control animals after 30 days on bosentan, suggesting a dissociation in time between myofibroblast recruitment and ETB induction or the existence of at least two populations of α-SMA-positive cells. In accordance with this, we did not find ETB receptors in all of the α-SMA-stained cells in human colon tumors.
Thus, our data indicate that ET-1 is one of the factors released by endothelial and tumor cells, putatively modulating the recruitment, differentiation, and/or proliferation of undifferentiated fibroblasts into α-SMA-positive cells in a negative manner.
Our findings of various components of the ET system in specific cells of colon cancers, suggest that ET-1 and its receptors could play a role in colon cancer progression. Increased ECE-1 and PPET-1 in endothelial and tumor cells provide a local source of ET-1, which might act in an autocrine role in tumor cell survival and most likely in a paracrine role on the proliferation of tumor stroma cells. ET-1 seems to be functioning as a negative modulator of the stromal angiogenic response, which may be primarily directed through the repression of fibroblast differentiation and may in turn or concomitantly induce the appearance of ETB receptors.
Acknowledgments
We thank Drs. E. Saraga, L. Guillou, J. Benhattar, and P. Chaubert for their help in selecting colon specimen, routine histological and immunohistological examination, and for very helpful discussion; V. Saint-Giorgio for her help in animal handling; Dr. M Clozel (Actelion, Basel) for donating the bosentan; and Dr. Owen Parkes for editing the English text.
Footnotes
Address reprint requests to Dr. Florence Pinet, INSERM Unit 36, Collège de France, 3 rue d’Ulm, 75005 Paris, France. E-mail: florence.pinet@college-de-france.fr.
Supported in part by the Ministère de la Recherche et de l’Enseignement (ACC-SV9), the Ministère des Affaires Etrangères and the French Embassy in Switzerland, and by grants from the Swiss National Science Foundation (grant No 32.045908.95) and the Swiss League against Cancer (grant No SKL 353-9-1996). Drs. G. Egidy and P. Korth received funding from La Fondation pour la Recherche Medicale.
Present address for Petra Korth is Hoechst Roussel Vet, Wiesbaden, Germany.
References
- 1.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayishi Y, Mitsui Y, Yazaki Y, Goto K, Masaki T: A novel potent, vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332:411-415 [DOI] [PubMed] [Google Scholar]
- 2.Kennedy R, Haynes W, Webb D: Endothelins as regulators of growth and function in endocrine tissues. Clin Endocrinol (Oxf) 1993, 39:259-265 [DOI] [PubMed] [Google Scholar]
- 3.Hosoda K, Nakao K, Hiroshi S, Suga S, Ogawa Y, Mukoyama M, Shirami G, Saito Y, Nakanishi S, Imura H: Cloning and expression of human endothelin-1 receptor cDNA. FEBS Lett 1991, 287:23-26 [DOI] [PubMed] [Google Scholar]
- 4.Sakamoto A, Yanagisawa T, Sakurai T, Takuwa Y, Yanagisawa H, Masaki T: Cloning and functional expression of human cDNA for the ETB endothelin receptor. Biochem Biophys Res Commun 1991, 178:656-663 [DOI] [PubMed] [Google Scholar]
- 5.Shimada K, Matsushita Y, Wakabayashi K, Takahashi M, Matsubar A, Iijima Y, Tanzawa K: Cloning and functional expression of human endothelin-converting enzyme cDNA. Biochem Biophys Res Commun 1995, 207:807-812 [DOI] [PubMed] [Google Scholar]
- 6.Kashiwabara T, Inagaki Y, Ohta H, Iwamatsu A, Nomizu M, Nishikori K: Putative precursors of endothelin have less vasoconstrictor activity in vitro but a potent pressor effect. FEBS Lett 1989, 247:73-76 [DOI] [PubMed] [Google Scholar]
- 7.Valdenaire O, Rohrbacher E, Mattei M: Organization of the gene encoding the human endothelin-converting enzyme (ECE-1). J Biol Chem 1995, 270:29794-29798 [DOI] [PubMed] [Google Scholar]
- 8.Emoto N, Yanagisawa M: Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem 1995, 270:15262-15268 [DOI] [PubMed] [Google Scholar]
- 9.Turner A, Tanzawa K: Mammalian membrane metallopeptidases: NEP, ECE, Kell and PEX. FASEB J 1997, 11:355-364 [DOI] [PubMed] [Google Scholar]
- 10.Masaki T: Possible role of endothelin in endothelial regulation of vascular tone. Annu Rev Pharmacol 1995, 35:235-255 [DOI] [PubMed] [Google Scholar]
- 11.Hirata Y, Takagi Y, Fukuda Y, Marumo F: Endothelin is a potent mitogen for rat vascular smooth muscle cells. Atherosclerosis 1989, 78:225-228 [DOI] [PubMed] [Google Scholar]
- 12.Kurihara Y, Kurihara H, Oda H, Maemura K, Nagai R, Ishikawa I, Yazaki Y: Aortic malformations and ventricular septal defect mice deficient in endothelin-1. J Clin Invest 1995, 96:293-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagnato A, Tecce R, DiCastro V, Catt K: Activation of mitogenic signaling by endothelin 1 in ovarian carcinoma cells. Cancer Res 1997, 57:1306-1311 [PubMed] [Google Scholar]
- 14.Kusuhara M, Yamaguchi K, Nagasaki K, Hayashi C, Suzaki A, Hori S, Handa S, Nakamura Y, Abe K: Production of endothelin in human cancer cell lines. Cancer Res 1990, 50:3257-3261 [PubMed] [Google Scholar]
- 15.Folkman J: Tumor angiogenesis: therapeutic implication. N Engl J Med 1971, 285:1182-1186 [DOI] [PubMed] [Google Scholar]
- 16.Pedram A, Razandi M, Hu R, Levin E: Vasoactive peptides modulate vascular endothelial cell growth factor production and endothelial cell proliferation and invasion. J Biol Chem 1997, 272:17097-17103 [DOI] [PubMed] [Google Scholar]
- 17.Darland D, D’Amore P: Blood vessel maturation: vascular development comes of age. J Clin Invest 1999, 103:157-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziche M, Morbidelli L, Donnini S, Ledda F: ETB receptors promote proliferation and migration of endothelial cells. J Cardiovasc Pharmacol 1995, 26:S284-S286 [PubMed] [Google Scholar]
- 19.Shankar A, Loizidou M, Aliev G, Fredericks S, Holt D, Boulos P, Burnstock G, Taylor I: Raised endothelin 1 levels in patients with colorectal liver metastases. Br J Surg 1998, 85:502-506 [DOI] [PubMed] [Google Scholar]
- 20.Inagaki H, Bishop A, Eimoto T, Polak J: Autoradiographic localization of endothelin-1 binding sites in human colonic cancer tissue. J Pathol 1992, 168:263-267 [DOI] [PubMed] [Google Scholar]
- 21.Korth P, Bohle R, Corvol P, Pinet F: Cellular distribution of endothelin-converting enzyme-1 in human tissues. J Histochem Cytochem 1999, 47:447-462 [DOI] [PubMed] [Google Scholar]
- 22.Egidy G, Juillerat-Jeanneret L, Korth P, Bosman F, Pinet F: The endothelin system in human normal colon. Am J Physiol 2000, 279:G211-G222 [DOI] [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N: Single step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroforme extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 24.Itoh Y, Yanagisawa M, Ohkubo S, Kimura C, Tosaka T, Inoue A, Ishida N, Mitsui Y, Onda H, Fujino M: Cloning and sequence analysis of cDNA encoding the precursor of a human endothelium-derived vasoconstrictor peptide, endothelin: identity of human and porcine endothelin. FEBS Lett 1988, 231:440-444 [DOI] [PubMed] [Google Scholar]
- 25.Tso J, Sun X, Kao T, Reece K, Wu R: Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res 1985, 13:2485-2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brand M, LeMoullec J, Corvol P, Gasc J: Ontogeny of endothelins-1 and -3, their receptors, and endothelin converting enzyme-1 in the early embryo. J Clin Invest 1998, 101:549-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sibony M, Commo F, Callard P, Gasc J: Enhancement of mRNA in situ hybridization signal by microwave heating. Lab Invest 1995, 73:586-591 [PubMed] [Google Scholar]
- 28.Korth P, Egidy G, Parnot C, LeMoullec J, Corvol P, Pinet F: Construction, expression and characterization of a soluble form of human endothelin-converting enzyme. FEBS Lett 1997, 417:365-370 [DOI] [PubMed] [Google Scholar]
- 29.Jeannin J, Onier N, Lagarde C, VonJeney N, Stutz P, Liehl E: Antitumor effect of synthetic derivatives of lipid A in an experimental model of colon cancer in the rat. Gastroenterology 1991, 101:726-733 [DOI] [PubMed] [Google Scholar]
- 30.Tokunaga K, Nakamura Y, Sakata K, Fujimori K, Ohkubo M, Sawada K, Sakiyama S: Enhanced expression of a glyceraldehyde-3-phosphate dehydrogenase gene in human lung cancers. Cancer Res 1987, 47:5616-5619 [PubMed] [Google Scholar]
- 31.PedutoEberl L, Valdenaire O, Saintgiorgio V, Jeannin J-F, Juillerat-Jeanneret L: Endothelin receptor blockade potentiates FasL-induced apoptosis in rat colon carcinoma cells. Int J Cancer 2000, 86:182-187 [DOI] [PubMed] [Google Scholar]
- 32.Folkman J: Addressing tumor blood vessels. Nat Biotechnol 1997, 15:510. [DOI] [PubMed] [Google Scholar]
- 33.Ohtani H: Stromal reaction in cancer tissue: pathophysiologic significance of the expression of matrix-degrading enzymes in relation to matrix turnover and immune/inflammatory reactions. Pathol Int 1998, 48:1-9 [DOI] [PubMed] [Google Scholar]
- 34.Benjamin L, Golijanin D, Itin A, Pode D, Keshet E: Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999, 103:159-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Powell D, Mifflin R, Valentich J, Crowe S, Saada J, West A: Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol 1999, 277:C1-C19 [DOI] [PubMed] [Google Scholar]
- 36.Pujuguet P, Hamman A, Moutet M, Samuel J, Martin F, Martin M: Expression of fibronectin ED-A+ and ED-B+ isoforms by human and experimental colorectal cancer. Contribution of cancer cells and tumor-associated myofibroblasts. Am J Pathol 1996, 148:579-592 [PMC free article] [PubMed] [Google Scholar]
- 37.Shichiri M, Kato H, Marumo F, Hirata Y: Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension 1997, 30:1198-1203 [DOI] [PubMed] [Google Scholar]
- 38.Wu-Wong J, Chiou W, Dickinson R, Opgenorth T: Endothelin attenuates apoptosis in human smooth muscle cells. Biochem J 1997, 328:733-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shichiri M, Sevidy J, Marumo F, Hirata Y: Endothelin-1 is a potent survival factor for c-myc-dependent apoptosis. Mol Endocrinol 1998, 12:172-180 [DOI] [PubMed] [Google Scholar]
- 40.Chatziantoniou C, Boffa J, Ardaillou R, Dussaule J: Nitric oxide inhibition induces early activation of type I collagen in renal resistance vessels and glomeruli in transgenic mice. J Clin Invest 1998, 101:2780-2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S: Contribution of endothelin-1 to myocardial injury in a murine model of myocarditis. Circulation 1999, 100:1823-1829 [DOI] [PubMed] [Google Scholar]