

Abstract
The Cdc25 phosphatases play key roles in cell-cycle progression by activating cyclin-dependent kinases. The latter are absent from neurons that are terminally differentiated in adult brain. However, accumulation of mitotic phosphoepitopes, and re-expression and activation of the M phase regulator, Cdc2/cyclin B, have been described in neurons undergoing degeneration in Alzheimer’s disease (AD). To explain this atypical mitotic activation in neurons we investigated the Cdc2-activating Cdc25A phosphatase in human brain. The structural hallmarks of AD neurodegeneration, neurofibrillary tangles and neuritic plaques, were prominently immunolabeled with Cdc25A antibodies. In addition numerous neurons without visible structural alterations were also intensely stained, whereas control brain was very weakly positive. After immunoprecipitation from control and AD tissue, we found that the tyrosine dephosphorylating activity of Cdc25A against exogenous Cdc2 substrate was elevated in AD. Accordingly, Cdc25A from AD tissue displayed increased immunoreactivity with the mitotic phosphoepitope-specific antibody, MPM-2, and co-localized with MPM-2 immunoreactivity in AD neurons. These data suggest that Cdc25A participates in mitotic activation during neurodegeneration. The involvement of Cdc25A in cellular transformation, modulation of the DNA damage checkpoint, and linkage of mitogenic signaling to cell cycle machinery, also implicates one of these cell-cycle pathways in AD pathogenesis.
The unusual appearance of certain mitotic indices in neurons undergoing degeneration in Alzheimer’s disease (AD) 1-6 has prompted further studies of cell-cycle regulatory proteins in human brain. The earliest mitotic change seems to be the expression of the Cdc2 catalytic subunit and the regulatory cyclin B subunit of the mitotic kinase complex in affected neurons. 2 In terms of the predictable progressive spread of neuronal death in AD, 7,8 neurons vulnerable to degeneration also stain positive for these mitotic proteins. 3 Thus, we hypothesized that inappropriate activation of Cdc2/cyclin B in AD neurons is a preliminary step leading to accumulation of mitotic phosphoepitopes and eventual neuronal death. To better understand this process, we have studied regulatory enzymes upstream of Cdc2 in the M-phase signal transduction cascade. A prerequisite for activation of Cdc2 is removal of phosphates 9,10 previously introduced by the wee1 and mik1 kinases 11 in Thr14 and Tyr15 within the ATP-binding domain of Cdc2. Dephosphorylation of these residues is achieved by the dual specificity of Cdc25 phosphatase. 12 Although the B and C isoforms of the mammalian enzyme control the timing of entry into mitosis, 13,14 Cdc25A is expressed specifically in late G1, and functions in the start of the cell cycle. 15,16 However, Cdc25A remains activated from G1 through M phase, and could conceivably participate in activation of Cdc2/cyclin B during mitosis. 16 Other evidence has implicated Cdc25A in the oncogenic and apoptotic pathways switched on by c-myc. 17,18 In light of the mitotic changes, and the apoptotic changes described in AD neurons, 19-22 we undertook an investigation of the Cdc25A phosphatase in AD brain.
We have found that the enzyme is concentrated and activated in neurons containing accumulations of mitotic phosphoepitopes in AD. However we have also found, unexpectedly, that the enzyme is present at readily detectable levels in normal adult brain, and displays constitutive activity against exogenous cdc2. In AD, Cdc25A is hyperphosphorylated at the MPM-2 epitope and has higher activity than in control tissue. Our data suggest that Cdc25A participates in the neurodegenerative process in AD.
Materials and Methods
Antibodies
Two antibodies specific for human Cdc25A were used: a polyclonal antibody raised against the full-length human isoform obtained from Upstate Biotechnologies Incorporated (Lake Placid, NY), and a monoclonal antibody recognizing the C-terminal sequence unique to the human enzyme, from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). In support of the specificity of these antibodies for Cdc25A, we have demonstrated that treatment of cultured cells with antisense Cdc25A oligonucleotides eliminates immunoreactivity for Cdc25A with both antibodies. The MPM-2 monoclonal antibody specific for mitotic phosphoepitopes, and antiphosphotyrosine antibody, 4G10, were also from UBI, and the Cdc2-specific mouse monoclonal antibody was from Santa Cruz Biotechnology.
Tissues
A total of 14 controls and 18 AD cases were used for immunocytochemical analysis. The assignment of cases into control or AD groups was based on the plaque and neurofibrillary tangle count and distribution as determined by detailed neuropathological examination. Thirty-μm vibratome sections from nine control and 12 AD cases fixed with 4% paraformaldehyde were supplied by Dr. Dennis Dickson (Albert Einstein College of Medicine, Bronx, NY) and 10-μm formalin-fixed paraffin-embedded sections were obtained from the tissue data base of the Alzheimer Disease Research Center at the University of Washington, Seattle. Immunocytochemistry was performed as by the method of Vincent and colleagues. 3 Some frozen tissue of 21 control and 23 AD cases was provided by Dennis Dickson and the remainder was from the Alzheimer Disease Research Center at the University of Washington, Seattle. The average postmortem interval for the fixed and frozen tissues was 10 hours. Human biopsy brain tissue retrieved by resection of epileptogenic foci from the temporal cortex was supplied by Dr. Diana Casper of the Montefiore Medical Center, Bronx, NY. From each case, a piece of tissue was frozen at −70°C for biochemical analyses and another piece fixed in 4% paraformaldehyde for immunocytochemical studies, and the postmortem processing time was <1 hour. The age range for these cases was 19 to 44 years, and histopathological assessment showed no AD-type pathology.
Immunocytochemistry, double immunofluorescence, and confocal microscopy were performed as previously described. 2,3
Preparation of Brain and Cell Extracts
Human brain tissue was homogenized with a polytron in 10 volumes of Triton-X containing lysis buffer. 2 The homogenates were centrifuged at 12,000 × g for 10 minutes at 4°C and the soluble fraction was used as extract for immunoblotting, or for immunoprecipitation, as described below. For immunoblot analysis, 100 μg of protein was loaded per lane for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Human M17 neuroblastoma cells (provided by Dr. Robert Ross, Fordham University, NY) were grown in Dulbecco’s modified medium containing 10% fetal calf serum. Nonsynchronous cultures having ∼8% of the cells in mitosis were used for preparation of interphase extracts, and cultures synchronized with colchicine were used for preparation of mitotic extracts. 1 Harvested cells were homogenized and detergent-soluble extracts were generated as by the method of Vincent and colleagues. 3 For comparison with human cells of nonneuronal origin, we used A431 epidermoid carcinoma cell lysates from UBI.
Immunoprecipitation
For assays of enzyme activity, immunoprecipitation was performed with 100 μg of protein from either brain or cultured cell extract and 0.5 to 1 μg of precipitating antibody. For immunoblot analyses of immune complexes, 500 μg of protein from brain or cell extract was incubated with 3 to 5 μg of appropriate primary antibody on a shaker for 2 hours at 4°C. To bring down the antigen-antibody immune complex, samples were then mixed with 30 μl (for activity assay) or 100 μl (for immunoblotting) of a 50% slurry of protein A Sepharose CL-4B (Amersham Pharmacia Biotech, Piscataway, NJ) for 1 hour, and centrifuged at a speed of 2,500 × g for 5 minutes. 2 The beads were washed and reconstituted to original volume either with phosphorylation buffer for activity assays, or with 1× sample buffer for electrophoresis. For immunoblot analyses, the immunoprecipitate (IP) was divided into two equivalent parts for loading into replicate lanes, and staining with different antibodies.
Assay of Cdc25A Activity
Tyr-15-phosphorylated Cdc2 substrate from nonsynchronized neuroblastoma cell extracts was isolated with an agarose-conjugate of Cdc2 mouse monoclonal antibody (Santa Cruz Biotechnology). Cdc25AIP from colchicine-treated mitotic cultures or from biopsy, control, or AD brain was incubated together with the Cdc2 substrate in the presence of phosphatase buffer for 40 minutes at 37°C, with vortexing every 10 minutes. At the end of the incubation, samples were subjected to SDS-PAGE and immunoblot analysis with antibody 4G10 to examine dephosphorylation of Tyr-15 in Cdc2.
Densitometric Analysis
Scanned images of electrochemiluminescence or autoradiographic data were quantitated using NIH image software. The scanned data were imported into Microsoft Excel spread sheets (Microsoft, Redmond, WA), and were statistically analyzed using EasyStat software (Hiroshi Tomonari, Tokyo, Japan).
Results
Cdc25A Immunoreactivity Demarcates Degenerating Neurons of AD Brain
The specificity of the Cdc25A antibodies was first confirmed in immunoblotting studies (shown in Figure 2 ▶ ). In paraffin-embedded hippocampal sections of AD brain, the monoclonal (Figure 1A ▶ , top row) and polyclonal (bottom row) Cdc25A antibodies displayed overt immunoreactivity with neurons containing neurofibrillary tangles (NFT) as shown in Figure 1A ▶ , arrows, b (see inset), c, e, and f. Neuritic components of plaques were also immunoreactive with both antibodies as shown in Figure 1A, b ▶ (see inset), c, and e. In the CA1, CA2, and CA4 hippocampal subfields of the AD cases, large numbers of neurons, with no apparent fibrillar inclusions, had prominent Cdc25A immunoreactivity in the cytoplasm of the somatodendritic compartment (Figure 1A, c and f ▶ , arrowheads). Based on the anatomical progression of disease in AD, it has been suggested that such neurons are engaged in initial stages of pathology. 7,8 Because Cdc25A staining was not observed in similar neurons of control brain, the positive neurons of AD most likely represent group 1 neurons with early pathological involvement, but no evidence yet of fibril formation. 23 The cytoplasmic distribution in these degenerating neurons is however, in contrast to the predominantly nuclear localization of Cdc25A in dividing cells. 15,16 In control hippocampal (Figure 1A, a and d ▶ , and corresponding insets) and temporal cortical (not shown) tissues, neurons appeared devoid of Cdc25A immunoreactivity. Surprisingly, dividing cells of the brain such as glia or endothelial cells, that might be expected to contain Cdc25A, were unstained in control and AD tissues.
Figure 2.

Immunoblot analyses of Cdc25A in human brain. Equivalent protein from detergent-soluble extracts of human epidermoid carcinoma cells (C), human neuroblastoma cells (N), human biopsy brain (B), and control and AD autopsy brain tissue were subjected to SDS-PAGE and transfer to nitrocellulose. The membranes were stained with Cdc25A polyclonal antibody, and detection of immunostaining was done using electrochemiluminescence. The exposure time was ∼2 minutes. A 70-kd band is detected with polyclonal and monoclonal antibody in all cases, and at similar levels in control and AD. The IP lane designates Cdc25A immunoprecipitate from biopsy brain extract, isolated with Cdc25A polyclonal antibody, and immunoblotted with the Cdc25A monoclonal antibody.
Figure 1.
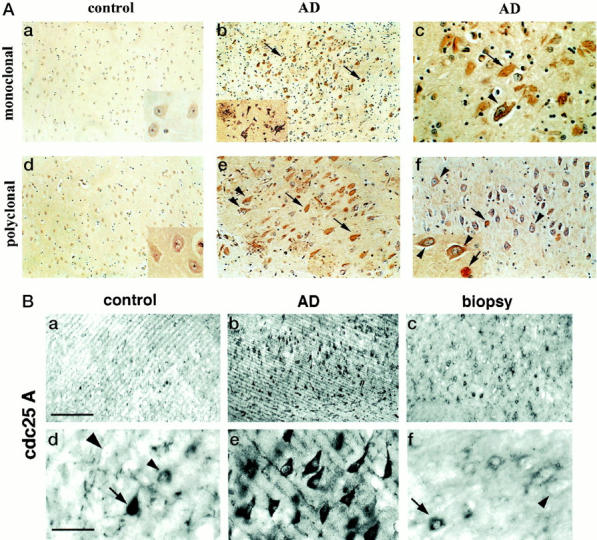
Immunocytochemical analyses of Cdc25A in paraffin-embedded (A) and vibratome-cut (B) sections of human brain. Paraffin-embedded sections from the indicated cases were immunostained with Cdc25A monoclonal (A, top row) and polyclonal antibody (A, bottom row), and then counterstained for cellular nuclei with hematoxylin (bluish purple). Neurons of control brain were very faintly stained, but degenerating AD neurons with or without NFT were intensely labeled with both antibodies. The light micrographs in a, b, and d are taken at an original magnification of ×10, e and the inset in b are at an original magnification of ×20, and all other images are at ×40 original magnification. The arrows point to NFT-containing neurons, and the arrowheads to immunolabeled neurons lacking apparent NFT. In B, paraformaldehyde-fixed vibratome sections of brain were immunostained with Cdc25A polyclonal antibody, using alkaline phosphatase for detection of antibody binding. a–c: Low-power magnifications (scale bar, 85 μm) showing the distribution of Cdc25A-positive neurons. d–f: Higher power micrographs (scale bar, 425 μm) of the same cases showing the perikaryal localization of Cdc25A in degenerating neurons in AD (e). The majority of normal neurons in control autopsy and biopsy tissue were unstained (large arrowheads), a few neurons displayed perinuclear staining (small arrowheads) and in three elderly controls small numbers of neurons apparently lacking fibrillar lesions (arrow) were detected.
The dramatic specificity of the Cdc25A antibodies for degenerating neurons in AD was replicated using 4% paraformaldehyde-fixed, vibratome-cut brain sections (Figure 1B) ▶ . In a few elderly controls, a rare neuron in the CA1 field was intensely stained (Figure 1B, a ▶ , arrow), perhaps reflecting incipient neurofibrillary disease. Biopsy-derived control cases having a postmortem processing time of less than an hour, showed a few neurons with intense Cdc25A immunostaining (Figure 1B, c) ▶ , some with a faint perinuclear ring of immunoreactivity in a few scattered neurons (Figure ▶ IB, f, small arrowhead), and several unstained neurons that were clearly demarcated (Figure 1B, d and f ▶ , large arrowheads). This pattern of staining was consistent in five biopsy cases ranging in age from 19 to 44 years, and imply that subtle differences in Cdc25A immunoreactivity are physiological and probably correlate with the function of Cdc25A in normal neurons. Additional studies will be required to explore this possibility, and are in progress in our lab. However, the major finding from the above studies was a marked increase in Cdc25A immunostaining correlating with AD-associated neuronal degeneration. Interestingly, this increase is evident at stages preceding cytoskeletal pathology.
Cdc25A Levels Are Unaltered in AD Brain
Immunoblot analyses were conducted with extracts from autopsy and biopsy human brain tissue and the two Cdc25A antibodies. The specificity of these antibodies was confirmed using similar extracts from A431 human epidermoid carcinoma cells (Figure 2 ▶ , lane C) and from dividing human neuroblastoma cultures (lane N). Both antibodies gave similar results (shown for the polyclonal antibody only), in that they recognized a 70-kd protein in each cell type. Extracts from human biopsy brain tissue (B) having a short postmortem processing period, also showed a prominent 70-kd band that co-migrated with the Cdc25A-immnuoreactive protein from cultured cells. This result suggested that the human brain isoform is similar to that of neuroblastoma and epidermoid carcinoma cells. The polyclonal Cdc25A antibody precipitated a 70-kd protein from biopsy brain extract, and this protein was strongly stained with the monoclonal antibody (Figure 2 ▶ , IP). Immunoblot analyses of extracts from control and AD autopsy brain tissue displayed a similar 70-kd protein in all cases (Figure 2) ▶ , but unlike the immunocytochemical data in Figure 1 ▶ , there was no apparent quantitative difference in the two groups. The 70-kd band was scanned densitometrically, and the amount of protein in the control and AD cases was compared. There was no statistical difference between these groups, and in addition, there was no difference between the amount of Cdc25A in biopsy brain as compared with the autopsy samples. These data suggest that the Cdc25A protein is stable postmortem, and that the levels are not different in control and AD. The absence of Cdc25A immunostaining in control brain may therefore be because of masking of epitopes or sensitivity of the antigen to fixation, and the marked increase in AD neurons may reflect a pathological change in conformation of the protein.
Phosphatase Activity of Cdc25A Is Elevated in AD
Because differences in levels of the Cdc25A phosphatase failed to explain the selective detection of the enzyme in AD neurons, we compared the activity of the enzyme in control and AD. We isolated Cdc25A immunoprecipitates (Cdc25AIPs) from these tissues, and analyzed their phosphatase activities against exogenous Cdc2 immune-complex (Cdc2IP) from nonsynchronously growing neuroblastoma cultures. In such cultures Cdc2 has a higher Tyr-15-phosphorylation content than Cdc2 from mitotic cells, and is therefore a suitable substrate for dephosphorylation by Cdc25A. Phosphorylation and dephosphorylation of Tyr-15 were monitored by immunoblotting with antiphosphotyrosine antibody, 4G10, and Cdc25A activity was estimated by comparative densitometric analysis of 4G10 immunoreactivity of Cdc2 before and after incubation with each IP sample. The specificity of 4G10 was verified by comparing its relative immunoreactivity with Cdc2IP from nonsynchronously growing (Figure 3c) ▶ and metaphase-arrested (m) cultures (Figure 3 ▶ , first panel). Cdc2IP from the nonsynchronous cultures showed markedly higher 4G10 immunoreactivity than the isolate from mitotic cell-enriched cultures. Moreover, the Tyr-15 phosphorylated Cdc2IPc had slower electrophoretic mobility than the dephosphorylated enzyme (Figure 3 ▶ , first panel). Cdc25AIP from M phase-enriched cultures (mitotic cells, Figure 3 ▶ ) effectively dephosphorylated Cdc2 as indicated by a 60% decrease in 4G10 staining (second panel). When preimmune serum was used to IP instead of Cdc25A antibody, phosphorylation of Cdc2 was unaffected (lane, PS). The Cdc2 substrate complex alone (second panel, last lane) did not contain appreciable Tyr-dephosphorylating activity. Incubation of Cdc2 with Cdc25AIP from biopsy brain resulted in a 37% decrease in 4G10 immunoreactivity (control panel, biop) compared with untreated Cdc2 (first panel, first lane), suggesting that Cdc25A from control brain possesses detectable levels of basal activity. This result is itself in opposition to the historic belief that cell cycle regulatory proteins are down-regulated and nonfunctional in postmitotic neurons. 24-27 When the activity of the enzyme from control and AD tissue was compared, it was found that the AD enzyme displayed activity levels equivalent to that from mitotic neuroblastoma cells, in that 4G10 immunoreactivity with Cdc2 was decreased by 57%. This decrease was significantly greater (P < 0.03) than that obtained with Cdc25A from control biopsy or control autopsy samples. There was no statistical difference between control biopsy and control cases. To better appreciate the increased activity in AD relative to control, the Cdc25AIPs were immunoblotted with Cdc25A monoclonal antibody and the results showed equivalent amounts of the enzyme immunoprecipitated in all cases (not shown).
Figure 3.

Tyr dephosphorylation and activation of Cdc2 by Cdc25A from brain. Cdc25A was IPed from mitotic neuroblastoma cultures (mitotic cells), or control biopsy (biop), control autopsy (autopsy), and AD brain (AD) as labeled in the top row, using the Cdc25A polyclonal antibody. These Cdc25AIPs were then incubated (+) with substrate for assaying Cdc25A activity, which consisted of Tyr-phosphorylated-Cdc2 IPed from nonsynchronous neuroblastoma cultures (c). The dephosphorylating effect of Cdc25A was subsequently analyzed by SDS-PAGE and immunoblotting with Cdc2-specific monoclonal antibody (middle row labeled Cdc2) to show total Cdc2, ie, phospho (upper band of doublet) and dephospho (lower band of doublet), and with 4G10 (bottom row), which is more selective for tyrosine-phosphorylated Cdc2 (upper band of the doublet). In the absence of any incubation with Cdc25A IP (first two columns labeled −) the Cdc2IP substrate from nonsynchronous cultures (c) had a lower electrophoretic mobility as seen with the Cdc2 antibody in the middle row and higher phosphotyrosine content as seen with 4G10 (bottom row), relative to Cdc2IP from colchicine-treated mitotic cultures (m). When incubated with active Cdc25AIP from mitotic neuroblastoma cultures (first column, mitotic cells) its 4G10 immunoreactivity was reduced. IP with preimmune serum (PS) or incubation of substrate in the absence of any phosphatase (−) had no effect. Cdc25AIP from AD had higher phosphatase activity, which resulted in significantly reduced 4G10 immunoreactivity than the controls.
MPM-2 Immunoreactivity with Cdc25A Is Increased in AD
It has been demonstrated that activation of Cdc25A is itself regulated by phosphorylation 16 and that increased phosphorylation of the enzyme is accompanied by an increase in immunoreactivity with the MPM-2 monoclonal antibody. 28 Activation of Cdc2 during M phase also culminates in a burst of MPM-2 immunoreactivity, 29 a situation recapitulated in degenerating neurons in AD. 1,3,4 We therefore analyzed MPM-2 immunoreactivity with Cdc25AIPs from control and AD brain tissue. A marked MPM-2 signal was observed with Cdc25AIP from AD (Figure 4A) ▶ , and controls only showed some weak electrochemiluminescence on more prolonged exposures (not shown). When replicate blots were immunostained with the precipitating Cdc25A antibody (Figure 4B) ▶ , it was clear that the increased MPM-2 immunoreactivity of the AD enzyme was not because of any increase in amount of enzyme precipitated, but represented an actual increase in phosphorylation that was consistent with activation of Cdc25A in this tissue.
Figure 4.

MPM-2 immunoreactivity with Cdc25A. Cdc25A was immunoprecipitated from brain using the Cdc25A polyclonal antibody, and the resulting IPs were immunoblotted with MPM-2 (A), or Cdc25A polyclonal antibody (B). Arrows point to the Cdc25A protein, which is more phosphorylated and more intensely MPM-2-immunoreactive in AD than in control.
Cdc25A and MPM-2 Immunoreactivities Co-Localize in Degenerating Neurons
Previously we had reported an accumulation of MPM-2 immunoreactivity in degenerating neurons of AD brain, whereas normal neurons appeared negative. 1,3 Although part of this increase may be accounted for by substrates of the Cdc2 kinase, 3 the data in Figure 4 ▶ above suggested that activation of Cdc25A might also contribute to increased MPM-2 immunoreactivity in AD. We therefore conducted double-staining studies of Cdc25A and MPM-2 immunofluorescence followed by confocal microscopic analysis of AD brain sections. Each antibody specifically labeled affected neurons in this tissue but not neurons free of pathological involvement. Cdc25A immunofluorescence was found in the same neurons immunoreactive with MPM-2 (Figure 5) ▶ . The subcellular overlap was partial in some cells, implying that the two antigens are not mutually exclusive. Nevertheless, the combined presence of Cdc25A and MPM-2 immunoreactive proteins in diseased neurons and their absence in normal neurons, suggests that Cdc25A might play a role in activation of mitotic kinase in these cells.
Figure 5.

Double-immunofluorescence staining of AD brain with Cdc25A and MPM-2. AD brain sections were double-immunofluorescence-stained with MPM-2 (red) and Cdc25A polyclonal antibody (green). The immunofluorescence staining was analyzed by confocal microscopy, and the micrographs show co-localization of the two antigens in diseased neurons, with fair amount of subcellular overlap. The arrow points to a neuritic plaque that contains Cdc25A and MPM-2 immunoreactivity. Scale bar, 20 μm.
Discussion
Consistent with the activation of mitotic Cdc2/cyclin B kinase and the accumulation of mitotic phosphoepitopes in degenerating neurons, this study describes an increase in activity of the Cdc2-activating Cdc25A tyrosine phosphatase in AD tissue. The enzyme is present and active, but not detected by immunocytochemistry in control brain, suggesting that some alteration of the protein in degenerating neurons favors its detection in these cells. Increased phosphorylation and MPM-2 immunoreactivity may account for this selective immunocytochemical visualization in AD, just as increased phosphorylation and altered conformation of the neuronal cytoskeletal tau protein result in its specific detection in AD tissue. 30 Alternatively, binding to Cdc2 or cyclin B which are absent in normal neurons 25,26 and down-regulated in dividing cells of the brain that have become quiescent, 31,32 may explain the selective presentation of the antigens in degenerating neurons. Nevertheless, the increase in activity of Cdc25A in AD neurons supports the hypothesis that neurodegeneration in this disease is preceded by inappropriate activation of a cell-cycle-driven process. The presence of a Cdc2-activating enzyme in control brain implies that induction of cdc2 and cyclin B alone, rather than the entire M phase signal transduction cascade, is sufficient for neurodegeneration to occur.
Evidence from the cell cycle literature suggests that Cdc25A is phosphorylated and active during M phase, 28 and causes arrest in mitosis when inhibited. 14 It has been shown that phosphorylation of Cdc25A in dividing cells is mediated by Cdc2/cyclin B as part of a positive feed-back regulatory loop. 16 In light of the co-localization of Cdc25A immunoreactivity and MPM-2 immunoreactivity in AD, it is possible that a similar potentiating effect of Cdc25A occurs in dying neurons, contributing to production of mitotic indices in these cells. Despite activation of Cdc2/cyclin B, cytokinesis of neurons in AD has never been detected in our studies, or in those of other research groups. Cdc2- or Cdc25A-positive cells do not display any signs of chromosomal condensation or spindle formation, implying that the structural phenomena associated with the onset of mitosis do not occur in AD. Thus, although affected postmitotic neurons of AD brain are capable of mitotic gene induction and activation, they fail to undergo mitosis.
The events that precede mitotic activation in neurons are not well understood either. For instance, the possibility that DNA replication precedes M phase changes in neurons has not yet been addressed, although there is evidence for increased immunoreactivity of several G1/S phase markers such as Ki67, p105,and proliferating cell nuclear antigen, in tangle-bearing and pretangled neurons. 33,34 In the mammalian cell cycle, the timing of phosphorylation and activation of the Cdc25A phosphatase coincides with activation of the cdk2/cyclin E kinase in S phase, and microinjection of Cdc25A-specific antibodies into human or rat cells blocks cell division at the G1/S transition. 15,16,35 Thus, increased expression of Cdc25A in neurons of AD brain might be a more definitive indication of an attempt to restart a cell cycle. Knowledge of agents causing Cdc25A activation and entry into S phase might offer clues to possible effectors of this process in postmitotic neurons as well. For example, it has been shown that the adenovirus E1A oncoprotein and certain growth factors stimulate entry of quiescent cells into S phase, and that the Cdc25A phosphatase mediates this process. 15,16,36 In light of this discussion, Cdc25A could serve as a point of convergence for a variety of intrinsic (eg, inherited mutations, oxidative damage) and extrinsic (eg, mitogens, viruses) factors that influence neuronal death. Overall, the similarities between the neurodegenerative signal-transduction mechanism and cell cycle regulation are becoming more apparent, although evidence for the typical ordered progression of the cell division phases is lacking.
Cdc25A activity is also essential for c-myc-induced apoptosis, 17,18 although it is unclear how. Cdc2 activation has been implicated as a player in the apoptosis of a variety of cells 37-40 including neuronally differentiated PC12 cells. 29,41 The issue of whether neurons in AD die by apoptosis is shrouded in debate: one school of thought dwelling on the presence of apoptotic markers in AD, 19 and on cultured cell and animal models that show a causal relationship between certain etiological factors for AD and apoptosis 42-45 ; and the other focusing on the distinction between the protracted progressive death in AD and the rapid apoptotic process, and on the possibility that DNA fragmentation could result from postmortem autolysis, 20 or oxidative damage, 21,22 and would therefore not exclusively signify apoptosis. In our studies, the presence of active Cdc25A, Cdc2, and the resulting downstream phosphorylation of neuronal proteins, are observed not only before paired helical filaments accumulate, but also before any morphological abnormalities such as nuclear membrane dissolution or blebbing are evident. The mitotic changes persist until NFT are formed in neurons, which even at this stage do not display any obvious apoptotic changes. It is therefore unlikely that the appearance of mitotic indices in neurons with NFT is associated with an apoptotic mode of death. However, it has been postulated that a fair number of neurons in AD die without forming NFT. 46 Given the widespread distribution of Cdc25A and cdc2/cyclin B 3 in AD, it is possible that cdc2-mediated mechanisms drive such neurons into an apoptotic crisis.
A notable difference in the cell-cycle process of degenerating neurons is that the site of activation of cell-cycle regulators is in the neuronal cytoplasm, whereas the nucleus 47,48 is their site of activation in cycling cells. This difference in subcellular location alone might account for the different outcomes of mitotic activation in the two situations, ie, degeneration versus mitosis. There are instances where Cdc25 functions in the cytoplasm, and studies have implicated the family of 14-3-3 proteins in these instances. The DNA damage checkpoint that prevents mitosis while DNA repair is underway is one example where the Rad24 14-3-3 protein facilitates nuclear export and hence cytoplasmic build-up of inactive Cdc25A. 49,50 In AD neurons the high levels of DNA damage 19,51,52 and accumulation of Cdc25A in the neuronal cytoplasm resemble this situation, but Cdc25A is activated, rather than inhibited, making it unlikely that neuronal death in AD involves DNA damage-induced arrest. Another instance where Cdc25A functions in a cytoplasmic location is in association with the Raf-1 protein kinase after mitogenic stimulation of quiescent cells. 53 14-3-3 promotes Cdc25A phosphorylation and activation by Raf-1, an interaction that links mitogenic signaling with the cell cycle machinery. 53 Interestingly, various isoforms of 14-3-3 are particularly abundant in brain, functioning in a variety of neuronal processes such as regulation of protein kinase C, exocytosis, and synaptic plasticity, 54 and their presence in neurofibrillary tangles has also been reported. 55 Thus 14-3-3 may act to sequester activated Cdc25A in the cytoplasm of neurons, eventually causing degeneration. A closer look at the 14-3-3 proteins in AD brain might help uncover the signaling pathways leading to mitotic activation in neurons and to neurodegeneration.
Acknowledgments
We thank D. Dickson and D. Casper, for supplying us with the necessary tissue specimens for this study; and Dr. Robert Ross for the M17 human neuroblastoma cell line.
Footnotes
Address reprint requests to Inez Vincent, University of Washington, Department of Pathology, Box 357705, 1959 NE Pacific Ave., Seattle, WA 98195. E-mail: ivincent@u.washington.edu.
Supported by grants AG12721, and P50 AG 05136–16 (ADRC, Murray Raskind, PI) from the National Institute on Aging.
References
- 1.Vincent I, Rosado M, Davies P: Mitotic mechanisms in Alzheimer’s disease? J Cell Biol 1996, 132:413-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent I, Jicha G, Rosado M, Dickson DW: Aberrant expression of mitotic cdc2/cyclin B 1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci 1997, 17:3588-3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P: Mitotic phosphoepitopes precede paired helical filaments in Alzheimer’s disease. Neurobiol Aging 1998, 19:287-296 [DOI] [PubMed] [Google Scholar]
- 4.Kondratick CM, Vandre DD: Alzheimer’s disease neurofibrillary tangles contain mitosis-specific phosphoepitopes. J Neurochem 1996, 67:2405-2416 [DOI] [PubMed] [Google Scholar]
- 5.Busser J, Geldmacher DS, Herrup K: Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci 1998, 18:2801-2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagy Z, Esiri MM, Smith AD: The cell division cycle and the pathophysiology of Alzheimer’s disease. Neuroscience 1998, 87:731-739 [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Braak E: Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991, 82:239-259 [DOI] [PubMed] [Google Scholar]
- 8.Hyman BT, Gomez-Isla T: Alzheimer’s disease is a laminar, regional, and neural system specific disease, not a global brain disease. Neurobiol Aging 1994, 15:353-354 [DOI] [PubMed] [Google Scholar]
- 9.Kumagai A, Dunphy WG: Molecular mechanism of the final steps in the activation of MPF. Cold Spring Harb Symp Quant Biol 1991, 56:585-589 [DOI] [PubMed] [Google Scholar]
- 10.Millar J, McGowan C, Jones R, Sadhu K, Bueno A, Richardson H, Russell P: cdc25 M-phase inducer. Cold Spring Harb Symp Quant Biol 1991, 56:577-584 [DOI] [PubMed] [Google Scholar]
- 11.Parker LL, Piwnica-Worms H: Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257:1955-1957 [DOI] [PubMed] [Google Scholar]
- 12.Moreno S, Nurse P, Russell P: Regulation of mitosis by cyclic accumulation of p80cdc25 mitotic inducer in fission yeast. Nature 1990, 344:549-552 [DOI] [PubMed] [Google Scholar]
- 13.Sadhu K, Reed SI, Richardson H, Russell P: Human homolog of fission yeast cdc25 mitotic inducer is predominantly expressed in G2. Proc Natl Acad Sci USA 1990, 87:5139-5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galaktionov K, Beach D: Specific activation of cdc25 tyrosine phosphatases by B-type cyclins: evidence for multiple roles of mitotic cyclins. Cell 1991, 67:1181-1194 [DOI] [PubMed] [Google Scholar]
- 15.Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H: Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J 1994, 13:1549-1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffmann I, Draetta G, Karsenti E: Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J 1994, 13:4302-4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galaktionov K, Chen X, Beach D: Cdc25 cell-cycle phosphatase as a target of c-myc. Nature 1996, 382:511-517 [DOI] [PubMed] [Google Scholar]
- 18.Macdonald K, Bennett MR: cdc25A is necessary but not sufficient for optimal c-myc-induced apoptosis and cell proliferation of vascular smooth muscle cells. Circ Res 1999, 84:820-830 [DOI] [PubMed] [Google Scholar]
- 19.Cotman CW: Apoptosis decision cascades and neuronal degeneration in Alzheimer’s disease. Neurobiol Aging 1998, 19:S29-S32 [DOI] [PubMed] [Google Scholar]
- 20.Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H: Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 1998, 57:456-464 [DOI] [PubMed] [Google Scholar]
- 21.Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N: Oxidative damage in Alzheimer’s [letter]. Nature 1996, 382:120-121 [DOI] [PubMed] [Google Scholar]
- 22.Tsang SY, Tam SC, Bremner I, Burkitt MJ: Research communication copper-1,10-phenanthroline induces internucleosomal DNA fragmentation in HepG2 cells, resulting from direct oxidation by the hydroxyl radical. Biochem J 1996, 317:13-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braak E, Braak H, Mandelkow EM: A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 1994, 87:554-567 [DOI] [PubMed] [Google Scholar]
- 24.Rakic P: DNA synthesis and cell division in the adult primate brain. Ann NY Acad Sci 1985, 457:193-211 [DOI] [PubMed] [Google Scholar]
- 25.Hayes TE, Valtz NL, McKay RD: Downregulation of CDC2 upon terminal differentiation of neurons. New Biol 1991, 3:259-269 [PubMed] [Google Scholar]
- 26.Okano HJ, Pfaff DW, Gibbs RB: RB and Cdc2 expression in brain: correlations with 3H-thymidine incorporation and neurogenesis. J Neurosci 1993, 13:2930-2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchkovich KJ, Ziff EB: Nerve growth factor regulates the expression and activity of p33cdk2 and p34cdc2 kinases in PC12 pheochromocytoma cells. Mol Biol Cell 1994, 5:1225-1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuang J, Ashorn CL, Gonzalez-Kuyvenhoven M, Penkala JE. cdc25 is one of the MPM-2 antigens involved in the activation of maturation-promoting factor [published erratum appears in Mol Biol Cell: May;5(5):611]. Mol Biol Cell 1994, 1994:5:135–145 [DOI] [PMC free article] [PubMed]
- 29.Davis PK, Dudek SM, Johnson GV: Select alterations in protein kinases and phosphatases during apoptosis of differentiated PC 12 cells. J Neurochem 1997, 68:2338-2347 [DOI] [PubMed] [Google Scholar]
- 30.Mandelkow EM, Mandelkow E: Tau in Alzheimer’s disease. Trends Cell Biol 1998, 8:425-427 [DOI] [PubMed] [Google Scholar]
- 31.Ducommun B: From growth to cell cycle control. Semin Cell Biol 1991, 2:233-241 [PubMed] [Google Scholar]
- 32.Doree M, Galas S: The cyclin-dependent protein kinases and the control of cell division. FASEB J 1994, 8:1114-1121 [DOI] [PubMed] [Google Scholar]
- 33.Nagy Z, Esiri MM, Cato AM, Smith AD: Cell cycle markers in the hippocampus in Alzheimer’s disease [see comments]. Acta Neuropathol (Berl) 1997, 94:6-15 [DOI] [PubMed] [Google Scholar]
- 34.Nagy Z, Esiri MM, Smith AD: Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions [see comments]. Acta Neuropathol (Berl) 1997, 93:294-300 [DOI] [PubMed] [Google Scholar]
- 35.Sexl V, Diehl JA, Sherr CJ, Ashmun R, Beach D, Roussel MF: A rate limiting function of cdc25A for S phase entry inversely correlates with tyrosine dephosphorylation of Cdk2. Oncogene 1999, 18:573-582 [DOI] [PubMed] [Google Scholar]
- 36.Spitkovsky D, Jansen-Durr P, Karsenti E, Hoffman I: S-phase induction by adenovirus E1A requires activation of cdc25a tyrosine phosphatase. Oncogene 1996, 12:2549-2554 [PubMed] [Google Scholar]
- 37.Steinmann KE, Belinsky GS, Lee D, Schlegel R: Chemically induced premature mitosis: differential response in rodent and human cells and the relationship to cyclin B synthesis and p34cdc2/cyclin B complex formation. Proc Natl Acad Sci USA 1991, 88:6843-6847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi L, Nishioka WK, Th’ng J, Bradbury EM, Litchfield DW, Greenberg AH: Premature p34cdc2 activation required for apoptosis [see comments]. Science 1994, 263:1143-1145 [DOI] [PubMed] [Google Scholar]
- 39.Pandey S, Wang E: Cells en route to apoptosis are characterized by the upregulation of c-fos, c-myc, c-jun, cdc2, and RB phosphorylation, resembling events of early cell-cycle traverse. J Cell Biochem 1995, 58:135-150 [DOI] [PubMed] [Google Scholar]
- 40.Shimizu T, O’Connor PM, Kohn KW, Pommier Y: Unscheduled activation of cyclin B1/Cdc2 kinase in human promyelocytic leukemia cell line HL60 cells undergoing apoptosis induced by DNA damage. Cancer Res 1995, 55:228-231 [PubMed] [Google Scholar]
- 41.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA: Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol 1998, 143:457-467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP: Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci 1997, 17:4212-4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP: Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer disease [see comments]. Nat Med 1998, 4:957-962 [DOI] [PubMed] [Google Scholar]
- 44.Ivins KJ, Bui ET, Cotman CW: Beta-amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol Dis 1998, 5:365-378 [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP, Partin J, Begley JG: Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res 1998, 807:167-176 [DOI] [PubMed] [Google Scholar]
- 46.Hyman BT: Neuronal loss in Alzheimer’s disease. Aging (Milano) 1998, 10:156. [PubMed] [Google Scholar]
- 47.Gallant P, Fry AM, Nigg EA: Protein kinases in the control of mitosis: focus on nucleocytoplasmic trafficking. J Cell Sci Suppl 1995, 19:21-28 [DOI] [PubMed] [Google Scholar]
- 48.Pines J: Cell cycle. Checkpoint on the nuclear frontier [news; comment]. Nature 1999, 397:104-105 [DOI] [PubMed] [Google Scholar]
- 49.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ: Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25 [see comments]. Science 1997, 277:1497-1501 [DOI] [PubMed] [Google Scholar]
- 50.Kumagai A, Dunphy WG: Binding of 14-3-3 proteins and nuclear export control the intracellular localization of the mitotic inducer Cdc25. Genes Dev 1999, 13:1067-1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabbita SP, Lovell MA, Markesbery WR: Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J Neurochem 1998, 71:2034-2040 [DOI] [PubMed] [Google Scholar]
- 52.Sheng JG, Mrak RE, Griffin WS: Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J Neuropathol Exp Neurol 1998, 57:323-328 [DOI] [PubMed] [Google Scholar]
- 53.Galaktionov K, Jessus C, Beach D: Raf1 interaction with Cdc25 phosphatase ties mitogenic signal transduction to cell cycle activation. Genes Dev 1995, 9:1046-1058 [DOI] [PubMed] [Google Scholar]
- 54.Aitken A, Jones D, Soneji Y, Howell S: 14-3-3 proteins: biological function and domain structure. Biochem Soc Trans 1995, 23:605-611 [DOI] [PubMed] [Google Scholar]
- 55.Layfield R, Fergusson J, Aitken A, Lowe J, Landon M, Mayer RJ: Neurofibrillary tangles of Alzheimer’s disease brains contain 14-3-3 proteins. Neurosci Lett 1996, 209:57-60 [DOI] [PubMed] [Google Scholar]