

Abstract
Functional inhibition of tissue factor (TF) has been shown to improve coronary blood flow after myocardial ischemia/reperfusion (I/R) injury. TF initiates the coagulation protease cascade, resulting in the generation of the serine protease thrombin and fibrin deposition. Thrombin can also contribute to an inflammatory response by activating various cell types, including vascular endothelial cells. We used a rabbit coronary ligation model to investigate the role of TF in acute myocardial I/R injury. At-risk areas of myocardium showed increased TF expression in the sarcolemma of cardiomyocytes, which was associated with a low level of extravascular fibrin deposition. Functional inhibition of TF activity with an anti-rabbit TF monoclonal antibody administered either 15 minutes before or 30 minutes after coronary ligation reduced infarct size by 61% (P = 0.004) and 44% (P = 0.014), respectively. Similarly, we found that inhibition of thrombin with hirudin reduced infarct size by 59% (P = 0.014). In contrast, defibrinogenating the rabbits with ancrod had no effect on infarct size, suggesting that fibrin deposition does not significantly contribute to infarct size. Functional inhibition of thrombin reduced chemokine expression and inhibition of either TF or thrombin reduced leukocyte infiltration. We propose that cardiomyocyte TF initiates extravascular thrombin generation, which enhances inflammation and injury during myocardial I/R.
Myocardial ischemia-reperfusion (I/R) injury exists as a continuum ranging from mild stunning, which is characterized by reversible postischemic organ dysfunction, to permanent tissue damage, which is characterized by irreversible myocellular necrosis. 1 I/R injury contributes to loss of myocardial tissue after restoration of blood flow after angioplasty, coronary artery bypass grafts, and reperfusion therapies, including thrombolytics. Although reperfusion of ischemic myocardium is essential for the survival of cardiomyocytes, the restoration of blood flow to ischemic myocardium is associated with an acute inflammatory response 2 Cytokines, chemokines, and adhesion molecules are induced during I/R injury. 3-5 These molecules promote the recruitment of polymorphonucleocytes (PMNs) and monocytes, 4,6 which secrete cytotoxic molecules that lead to damage of ischemic myocardium. Additionally, a no reflow effect leads to continued ischemia. 7,8
Tissue factor (TF) is the transmembrane receptor and cofactor for plasma factor VII/VIIa that functions as the primary cellular initiator of blood coagulation. 9 TF is constitutively expressed at extravascular sites, including the vascular adventitia, where it is proposed to play a hemostatic role to limit hemorrhage in the event of vessel damage. 10 In pathological settings, TF can initiate intravascular thrombosis. For instance, disruption of atherosclerotic plaques exposes TF-positive foam cells within the plaque to plasma-clotting factors, 11 leading to thrombosis, occlusion of coronary vessels, and myocardial infarction. Patients with unstable angina, myocardial infarction, and patients postangioplasty also exhibit elevated levels of circulating TF on the surface of monocytes and in vesicles in plasma, 12-14 which may contribute to the occlusion and re-occlusion of coronary vessels.
TF may contribute to inflammation observed in various disease states, such as sepsis, 15 trauma, 16 and glomerulonephritis. 17 The proinflammatory role of TF seems to require thrombin generation but may be independent of fibrin deposition. Thrombin can contribute to local inflammation and tissue damage by activation of a family of protease-activated receptors 18,19 that stimulate cells to express cytokines, such as interleukin (IL)-1, and IL-6; chemokines, such as IL-8 and monocyte chemotactic protein-1 (MCP-1); and adhesion molecules such as P-selectin, E-selectin, and ICAM-1. 5,20-22
A recent study demonstrated that TF activity was increased in the hearts of rabbits subjected to myocardial I/R injury. 23 Moreover, administration of an inhibitory anti-rabbit TF monoclonal antibody improved coronary blood flow. 23 In the present study, we used a similar rabbit model of myocardial I/R injury to identify the cells responsible for increased TF expression and to examine the mechanism by which the TF-thrombin pathway enhanced myocardial I/R injury.
Materials and Methods
In Situ Coronary Ligation Model
We used a well-characterized rabbit model of regional cardiac I/R injury. 24 In this model, adult New Zealand White rabbits weighing 3 to 4 kg were used in research protocols approved by the Animal Care Committee of the University of Washington, Seattle. All animals received humane care according to the “Guide for the Care and Use of Laboratory Animals” prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH Publication No. 86-23, revised 1985). Rabbits were anesthetized with an initial intramuscular injection of a ketamine (35 mg/kg) and xylazine (5 mg/kg). Rabbits were endotracheally intubated (3 mm ID, Aire-Cuff Veterinary endotracheal tube; Bivona, Gary, IN) and mechanically ventilated with 100% oxygen at a rate of 18 to 20 breaths/minute with a tidal volume of 48 ml using a small animal respirator (Harvard Apparatus Co., Cambridge, MA). Continuing anesthesia was provided by inhaled 4% halothane for 2 minutes followed by a 1% maintenance dose during the procedure. Intravenous Ringer’s lactate was administered at 5 ml/kg/hour and the temperature of the rabbit was maintained with a warming pad. A 4.0-Vicryl suture (Ethicon, Inc., Somerville, NJ) was passed twice around a large anterolateral branch of the left main coronary artery supplying most of the left ventricle (LV) and the ends of the suture were passed through a small length of polyethylene tubing to form a snare. After a 20- to 30-minute stabilization period, regional myocardial ischemia was produced by reversibly tightening the snare and occluding the artery for 45 minutes. The coronary snare was then released to allow 120 minutes of reperfusion. For sham surgery the rabbits were treated as above but cardiac ischemia was not induced by tightening the ligatures around the coronary vessel. After 120 minutes of reperfusion, all animals were sacrificed with an intravenous bolus of concentrated pentobarbital and the myocardial tissue was isolated and processed for either calculation of infarct size or histological analysis.
To assess the effect of functional inhibition of TF on I/R injury, 2 mg/kg of an inhibitory anti-rabbit TF monoclonal antibody (11F) in normal saline (0.9%) was administered intravenously to rabbits either 15 minutes before (n = 6) or 30 minutes after (n = 5) the onset of ischemia. Control rabbits (n = 5) for each experiment received saline. To determine the effect of inhibition of thrombin on myocardial I/R injury, rabbits (n = 5) were treated with recombinant hirudin (lepirudin; Hoechst Marion Roussel, Kansas City, MO). Hirudin specifically blocks thrombin activity through competitive inhibition of its catalytic site. 25 Hirudin treatment began with the intravenous administration of a 1 mg/kg bolus 30 minutes before ischemia and, 1 hour later, continued with an intravenous infusion of 1 mg/kg/hour for 1 hour and an infusion of 0.5 mg/kg/hour for 1 hour. 25 This dosing protocol prolonged the activated partial thromboplastin time to greater than twice baseline throughout the period of ischemia and reperfusion. Control rabbits (n = 4) received saline. To determine the contribution of fibrin deposition to myocardial I/R injury, rabbits (n = 5) were treated with ancrod, a defibrinogenating agent. 26 Ancrod cleaves only the A-chains of fibrinogen producing soluble, uncrosslinked fibrin-fibrinogen degradation products that are cleared by the reticuloendothelial system. Ancrod (Sigma Chemical Co., St. Louis, MO) was administered as previously reported. 26 Rabbits received ancrod intravenously beginning with a bolus dose of 1.0 IU/kg, followed by a second bolus dose (1.0 IU/kg) 1 hour later, and a third bolus dose (2.0 IU/kg) 3 hours later. The I/R protocol was initiated 6 hours after the first ancrod dose. This dosing schedule decreased circulating fibrinogen from 2.60 ± 0.09 mg/ml (n = 14) to undetectable levels (<0.20 mg/ml) after the first dose and throughout ischemia and reperfusion as determined by the von Clauss method. 27 This represents a >92% decrease in fibrinogen levels. Briefly, the von Clauss assay determines the clotting time of dilute plasma with exogenous thrombin. The formation of insoluble fibrin polymers is the end point of the reaction, and the concentration of fibrinogen in the test sample is obtained by comparing the clotting time of the sample with a standard curve. Control rabbits (n = 4) received saline. No serious bleeding complications were noted during any of the above treatments.
Determination of Infarct Size
At the completion of the 120-minute reperfusion period, the coronary artery was re-occluded and 6 ml of 20% Evans blue dye (Sigma Chemical Co.) was injected into the right atrium and allowed to circulate to identify all perfused tissue (blue). The area of myocardium receiving its blood supply from the ligated vessel remained pink, thus demarcating the myocardium of the LV at-risk (AR) for injury. After arrest with pentobarbital, the heart was rapidly excised, weighed, and cut into 2-mm-thick cross-sections in parallel with the atrioventricular groove. The LV was isolated from the remainder of the heart and weighed. The normal left ventricular myocardium (blue) was separated from the LV myocardial area AR for injury (pink). The AR area was then placed in a 37°C solution of 1% triphenyltetrazolium chloride (Sigma Chemical Co.) for 30 minutes. Triphenyltetrazolium chloride stains the viable tissue brick red, leaving the necrotic zone pale white. Red-stained (noninfarcted) tissue was separated from white-stained (infarcted; necrotic) tissue under a dissecting microscope and each area was weighed. The percentage of LV AR for infarction was calculated by dividing the weight of the LV AR area by the weight of the total LV. The percentage infarct size within the area of LV placed AR to injury was calculated by dividing the weight of necrotic tissue by the weight of the LV AR area. AR areas and infarct size for the anti-TF antibody-treated, hirudin-treated, ancrod-treated, and control rabbits were assessed by an investigator blinded to the treatment.
Anti-Rabbit TF Antibodies
The production of a sheep anti-rabbit TF polyclonal antibody and a mouse anti-rabbit TF monoclonal antibody (11F) has been described. 17 Briefly, BALB/c mice were immunized with 10 μg of purified rabbit TF in Freund’s complete adjuvant and their spleen cells were fused with NS1 cells to produce hybridomas by standard techniques. Supernatants from clones were screened for TF reactivity with immunoaffinity-purified antigen coated on microtiter plates. One clone designated 11F produced an IgG1 antibody with potent functional inhibitory activity in a one-stage coagulant assay and stained a 45-kd protein in acetone-extracted rabbit brain on Western blots. Both the anti-rabbit TF monoclonal (11F) (no. 4511) and polyclonal (no. 4513) antibodies are commercially available (American Diagnostica Inc., Greenwich, CT). We determined the inhibitory activity of 11F by performing a dose-titration experiment against rabbit brain TF. 11F (0.3 μg/ml) inhibits 50% of rabbit TF activity in a one-stage clotting assay (Figure 1) ▶ . We chose to use an intravenous dose of 11F of 2 mg/kg because we and others have used this dose with other antibodies for studies using the rabbit myocardial I/R model. 28,29 A 2 mg/kg dose of 11F would give a plasma concentration of ∼33 μg/ml, which is more than 10-fold above the IC50 observed in our in vitro inhibition studies.
Figure 1.

Inhibition of rabbit TF by 11F. Rabbit brain TF (3.3 mg/ml) resuspended in PBS was incubated with different concentrations of 11F at 37°C for 15 minutes and assayed for TF activity in a one-stage clotting assay.
Determination of Rabbit TF mRNA and Functional Activity
LV tissue from sham-operated control rabbits (n = 6) or from normal and AR areas of LV from I/R-injured rabbits (n = 8) was snap-frozen and stored in liquid nitrogen. TF mRNA levels were assessed by Northern blot as previously described using a rabbit TF cDNA probe. 30 To assess TF activity, LV tissue (50 mg) was homogenized in 15 mmol/L octyl-β-d-glucopyranoside and incubated at 37°C for 15 minutes. Samples were centrifuged at 12,000 × g for 1 minute and TF activity in the supernatant assayed in a one-stage clotting assay using human pooled plasma. 17 TF activity was calculated in arbitrary units by reference to a standard curve established with human brain TF, in which a clotting time of 50 seconds corresponds to 1,000 mU of TF activity, and normalized to the total protein concentration. Protein concentrations were determined using a Bio-Rad DC protein assay (Bio-Rad, Hercules, CA).
Determination of Levels of TF, IL-8, and MCP-1 Protein
TF antigen was measured by enzyme-linked immunosorbent assay as previously described. 17 Briefly, 50 mg of tissue was homogenized in 1 ml of 0.5 mol/L ethylenediaminetetraacetic acid, 50 mmol/L Tris (pH7.5), 150 mmol/L NaCl, and 2% Triton X-100. Samples were incubated at 4°C for 4 hours and centrifuged at 12,000 × g for 5 minutes. The supernatant was removed and stored at −20°C overnight. TF antigen concentrations for each sample were calculated by reference to a standard curve generated with rabbit brain thromboplastin powder and normalized to the total protein concentration. Levels of IL-8 and MCP-1 in the heart were measured using enzyme-linked immunosorbent assays that are specific for rabbit IL-8 and MCP-1. 31,32
In Situ Hybridization
Cell type-specific TF mRNA expression was determined by in situ hybridization using an anti-sense TF riboprobe with a sense control. 33 Briefly, sections of heart tissue were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) and paraffin-embedded. Slides were hybridized with 600,000 cpm of 35S-labeled riboprobe for 18 hours at 55°C. Sections were counterstained with hematoxylin and eosin (H&E).
Immunohistochemistry
Regions of ischemic cardiac tissue were identified with acid-fuchsin, 34 which stains ischemic myocardium reddish-brown. However, acid-fuchsin staining is not a definitive indicator of ischemic injury. Immunohistochemical staining of TF was performed on frozen and fixed sections using either 11F or goat anti-human TF polyclonal antibody (American Diagnostica, Inc.), respectively. 11F administered in vivo to rabbits (n = 2) was detected with a biotinylated horse anti-mouse IgG (1:150; Vector Laboratories, Burlingame, CA). Staining used the Vectastain Elite ABC-HRP kit and the 3,3′-diaminobenzidine chromogen kit (Vector), which produces a brown reaction product. Sections were counterstained with hematoxylin. Dual staining of TF and endothelial cell-specific antigens was performed on frozen sections. TF protein was detected with either 11F (24 μg/ml) or sheep anti-rabbit TF antibody (20 μg/ml) and a fluorescein isothiocyanate-labeled donkey anti-mouse or donkey anti-sheep F(ab′)2 fragment (Jackson ImmunoResearch, West Grove, PA). Endothelial cells were detected with either a mouse anti-CD31 antibody (1:20) (DAKO, Carpinteria, CA) or a goat anti-rabbit von Willebrand Factor (vWF) antibody (1:1,000) (kindly provided by J. Ware, The Scripps Research Institute) and a Texas Red-labeled donkey anti-mouse or donkey anti-goat antibody (Jackson). Nonspecific staining was assessed using normal serum, normal immunoglobulin, and nonreactive monoclonal antibodies. Staining was visualized using Vectashield Mounting Media for fluorescence (Vector) and images captured with a scanning confocal microscope (MR 1000; Bio-Rad).
Fibrin Staining
Fixed sections of LV were chemically stained by the Carstairs’ method, which stains fibrin bright red. Liver and kidney sections from lipopolysaccharide-treated rabbits were used as positive controls. 35 Dual immunolocalization of fibrin and endothelial cells was performed on frozen sections. Cross-linked fibrin was detected with a mouse anti-human fibrin β-chain monoclonal antibody (59D8) (20 μg/ml) that binds to human fibrin but not fibrinogen. 36 59D8 was kindly provided by M. Runge, University of Texas Medical Branch, and binds to rabbit fibrin but not to rabbit fibrinogen. 59D8 was detected with a fluorescein isothiocyanate-labeled donkey anti-mouse antibody (Jackson). Endothelial cells were detected with a goat anti-rabbit vWF antibody (1:1,000) kindly provided by J. Ware and a Texas Red-labeled donkey anti-goat antibody (Jackson). Staining was visualized using a scanning confocal microscope (MR 1,000).
Ultrastructural Analysis of Tissue
Myocardial tissue was collected for electron microscopy from rabbits subjected to I/R injury (n = 2) or sham surgery (n = 2). Hearts were perfused with PBS followed by perfusion with 2% glutaraldehyde and 4% paraformaldehyde in PBS under a constant pressure of 100-mmHg to prevent damage to the vascular endothelium. After perfusion, tissue samples (1 mm3) were taken from normal LV of sham-operated rabbits and the grossly necrotic area of LV, the LV AR area, and the normal area of LV of I/R-treated rabbits. Tissue was fixed overnight in the perfusate, washed in 0.1 mol/L cacodylate buffer, fixed with osmium, and embedded in resin. Thick sections were cut and stained with toluidine blue for orientation and ultra-thin sections were stained with uranyl acetate and lead citrate for assessment by electron microscopy (Philips, GM 100 Eindhoven, Netherlands). For the sham animals, we examined one grid from one tissue sample. For the I/R-injured animals, we examined eight grids from one tissue sample of the LV AR area.
PMN Accumulation
Light microscopic examination was performed on zinc formalin (Anatec Ltd., Battle Creek, MI) fixed 3-μm-thick sections of heart stained with H&E to assess cellular infiltrate and to identify areas of tissue necrosis. PMN accumulation in the LV AR area of myocardium from I/R-injured rabbits treated with saline, 11F, or hirudin was quantified by an investigator blinded to the treatment. The number of PMNs in 10 high-powered fields (×400) selected from the PMN-dense regions of myocardium AR to I/R injury was counted in 4 to 6 different randomly selected tissue sections obtained from the hearts of different rabbits in each group (saline-treated, n = 6; 11F-treated, n = 2; hirudin-treated, n = 3).
Statistics
The data analysis was performed using Statview version 4.5 (SAS Institute, Cary, NC) for Apple Power Macintosh (Apple Computer, Cupertino CA). All quantitative data were presented as the mean ± SE (SE) and the statistical significance between each group was determined using a Mann-Whitney U test. P values <0.05 were considered statistically significant.
Results
Induction of TF Expression after Myocardial I/R Injury
We used a well-characterized rabbit model of acute myocardial I/R injury for these studies. 24 TF activity, TF antigen, and TF mRNA in the LV AR areas of rabbits subjected to myocardial I/R injury were compared with TF expression in the LV of sham-operated animals. TF activity and antigen levels in the LV of I/R-injured rabbits were 2.6 ± 0.5 (mean ± SE, n = 6) and 3.3 ± 0.8 (mean ± SE, n = 6) fold higher, respectively, than levels in LV of sham animals. TF mRNA levels were evaluated by Northern blotting in the AR and non-AR areas of the LV of three independent I/R-injured rabbits (Figure 2) ▶ . TF mRNA levels in the AR areas were 3.7 ± 0.3 (mean ± SE, n = 3) fold higher than the levels in the non-AR areas, whereas sham animals exhibited similar levels of TF mRNA in two separate samples of LV (Figure 2) ▶ . Thus, TF expression was increased in the AR region of the LV of I/R-injured rabbits.
Figure 2.

TF mRNA expression after myocardial I/R injury. TF mRNA levels were determined by Northern blotting and normalized to levels of glyceraldehyde-3-phosphate dehydrogenase (G3PDH). A: Determination of TF mRNA levels in two separate samples from non-AR areas (N) of LV of three independent sham-operated rabbits. B: Comparison of TF mRNA levels in the non-AR areas (N) of LV with levels in the AR areas of LV of three independent I/R-injured rabbits. TF mRNA levels were 3.7 ± 0.3 (mean ± SE, n = 3) fold higher in the AR areas compared with the levels in the non-AR areas of the three I/R-injured rabbits.
Identification of the Cellular Source of Increased TF Expression after Myocardial I/R Injury
To determine the cellular source of induced TF expression in myocardial I/R injury, we performed immunohistochemistry and in situ hybridization experiments. Low levels of TF mRNA and protein are constitutively expressed by cardiomyocytes. 10,37,38 Immunohistochemical analysis of tissue sections of LV from I/R-injured rabbits demonstrated a regional increase in TF antigen (Figure 3A) ▶ that was identified as ischemic myocardium by acid-fuchsin staining of a serial section (Figure 3B) ▶ . TF antigen was increased in the sarcolemma of ischemic cardiomyocytes (Figure 3, C and E) ▶ compared with levels of TF antigen in nonischemic cardiomyocytes in the same section (Figure 3D) ▶ .
Figure 3.

TF antigen is up-regulated in ischemic cardiomyocytes. A: Regional increase in TF staining of ischemic cardiomyocytes in fixed heart tissue from a rabbit after I/R injury. Rabbit TF was detected using a goat anti-human TF polyclonal antibody. Original magnification, ×100. B: Serial section stained with acid-fuchsin to identify ischemic cardiomyocytes (reddish brown) Original magnification, ×100. TF expression in the AR area of LV (C) (original magnification, ×400) was compared with expression in normal LV (D) of the same I/R-injured rabbit. Minimal TF expression was observed in the vascular endothelium (E) (original magnification, ×1,000). Cardiomyocytes (arrow) and endothelium (arrowhead) are shown. F: Serial section stained with control antibody (original magnification, ×1,000). TF-positive cells were detected with 3,3′-diaminobenzidine and stain brown. G: Analysis of TF and CD31 expression. The sheep anti-rabbit TF polyclonal antibody was detected with a fluorescein isothiocyanate-labeled antibody and stains green. The mouse anti-CD31 monoclonal antibody was detected with a Texas Red-labeled antibody and stains red. H: Analysis of TF and vWF expression. The anti-TF monoclonal antibody (11F) was stained green and the anti-rabbit vWF antibody was stained red.
We detected minimal TF staining on the vascular endothelium in tissue sections of hearts from rabbits subjected to I/R injury (Figure 3E) ▶ . No staining was observed using a control antibody (Figure 3F) ▶ . Dual immunofluorescence studies were performed to further evaluate if TF was expressed by microvascular endothelial cells in the ischemic regions of hearts from I/R-injured rabbits. Figure 2, G and H ▶ , shows that TF (green) was expressed by cardiomyocytes but was not expressed at detectable levels by CD31- and vWF-positive endothelial cells (red). Thus, we conclude that endothelial cells express only minimal levels of TF after myocardial I/R injury. Myocardial I/R injury is associated with an inflammatory response characterized by infiltration of leukocytes into the myocardium. 4,6 We observed TF antigen on some leukocytes, including a small number of PMNs (not shown).
In situ hybridization studies demonstrated a high level of TF mRNA expression in cells within the AR area (Figure 4, A and B) ▶ . In contrast, we observed low levels of TF mRNA expression in cardiomyocytes within normal LV of I/R injured rabbits (Figure 4E) ▶ and sham-operated rabbits (data not shown). The TF mRNA-positive cells in the AR area possessed large fusiform nuclei and were aggregated in trabecular networks of muscle fibers (Figure 4F) ▶ . Analysis of cross sections through muscle fibers suggested that TF mRNA was expressed by the centrally placed cardiomyocytes (Figure 4G) ▶ . All these morphological features indicated that TF mRNA expression was up-regulated in ischemic cardiomyocytes. Epicardial cells (Figure 4B) ▶ , endocardial cells (not shown), and vascular endothelial cells of larger vessels (Figure 4C) ▶ and capillaries (Figure 4G) ▶ did not express detectable levels of TF mRNA. Hybridization with a sense TF riboprobe yielded no specific signal (Figure 4D) ▶ .
Figure 4.
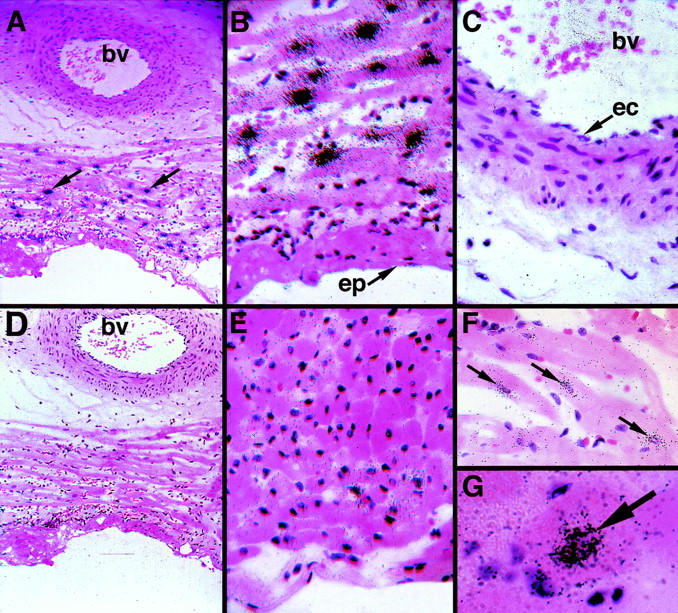
TF mRNA expression in rabbit hearts after I/R injury. In situ hybridization experiments were performed on heart sections from I/R-injured rabbits. Tissue sections were hybridized with anti-sense TF riboprobes (A–C, E–G) or sense control (D). A: Low-power view (original magnification, ×100) of a heart section with a blood vessel (bv) from an I/R-injured rabbit (arrows indicate TF mRNA-positive cells). Higher magnifications are shown in B and C. B: TF mRNA-positive cells were observed in the LV, whereas epicardium (ep) is negative (original magnification, ×400). C: No TF mRNA signal is observed in the vascular endothelium (ec). bv, blood vessel. D: Serial section of D demonstrating no positive signal using a TF sense probe (original magnification, ×100). E: TF mRNA expression was very low in normal myocardium from I/R-injured rabbits (original magnification, ×400). F: View of the longitudinal axis of cardiac muscle bundles shows that the TF mRNA signal is localized to cardiomyocyte (arrows) (original magnification, ×400). G: Cross sections of TF mRNA-positive (arrow) cardiomyocytes located within a cardiac muscle bundle (original magnification, ×1,000).
Localization of Anti-TF Antibody Administered in Vivo
To identify the possible sites of action of an inhibitory anti-rabbit TF monoclonal antibody (11F), we localized in vivo administered 11F antibody using a horse anti-mouse IgG antibody. We determined the AR area of LV by staining sections with acid-fuchsin, which identified AR (reddish-brown) versus non-AR areas (yellow) of LV of I/R-injured rabbits (Figure 5A) ▶ . 11F antibody bound to the sarcolemma of cardiomyocytes in AR areas but not to cardiomyocytes in non-AR areas (Figure 5, B–E) ▶ . Minimal levels of 11F localized to vascular endothelial cells and to the majority of leukocytes (Figure 5E) ▶ . Dual immunofluorescence studies did not reveal significant co-localization between in vivo administered 11F and vWF-positive microvascular endothelial cells (data not shown). These studies demonstrated that 11F predominately bound to TF expressed by extravascular cardiomyocytes in the AR area.
Figure 5.

Localization of the anti-TF antibody (11F) in the myocardium of rabbits treated intravenously with 11F before I/R injury. Immunohistochemical analysis of mouse IgG was performed on zinc-formalin-fixed heart sections using a horse anti-mouse IgG polyclonal antibody. A: Acid-fuchsin staining was used to identify areas of ischemic myocardium (original magnification, ×100). Staining of horse anti-mouse IgG demonstrates localization of 11F to the sarcolemma (arrow) and intercalated disks (arrowhead) of cardiomyocytes. Original magnifications, ×250 (B) and ×1,000 (C). Cardiomyocyte is indicated by the arrow whereas minimal staining was detected on endothelium (arrowhead). D: Section from a saline control rabbit reveals no staining (original magnification, ×1,000). Horse anti-mouse IgG was detected with 3,3′-diaminobenzidine and stains brown.
Analysis of Fibrin Deposition in Ischemic Myocardium
Fibrin deposition in ischemic myocardium was assessed by histochemical staining. Using this technique, intravascular fibrin deposition and thrombi could be observed in the macro- and microvasculature of livers from lipopolysaccharide-treated rabbits (Figure 6A) ▶ . In contrast, we could not observe any intravascular fibrin deposition or thrombi in the microvasculature of normal and AR areas of LV from sham-operated and I/R animals, respectively (Figure 6, B and C) ▶ . Next, we analyzed fibrin deposition by immunohistochemical staining using a monoclonal antibody that specifically detects cross-linked fibrin. 36 Despite detecting abundant intravascular fibrin deposition that co-localized with endothelial cell staining in a kidney section from a lipopolysaccharide-treated rabbit, we did not detect intravascular fibrin deposition in normal LV of sham-operated animals or AR areas of LV from I/R rabbits (Figure 6, D and E) ▶ . However, we did observe a low level of extravascular fibrin in AR areas of LV from I/R rabbits (Figure 6E) ▶ , which are the same regions that exhibit up-regulated TF expression by cardiomyocytes.
Figure 6.

Analysis of fibrin deposition in myocardium from I/R-injured rabbits. Fibrin deposition was assessed by Carstairs’ staining (A–C), which stains fibrin bright red, and dual immunohistochemistry using an anti-fibrin and an anti-rabbit vWF antibody (D–F). A: A liver section from a lipopolysaccharide-treated rabbit was used as a positive control and shows a large fibrin-rich thrombus (arrow) as well as fibrin deposition in the microvasculature. Minimal fibrin staining was observed in myocardium from sham (B) or I/R-injured (C) rabbits. Immunohistochemical analysis of fibrin deposition was performed by dual immunolocalization of fibrin (green) and endothelial cells (vWF) (red). D: A kidney section from a lipopolysaccharide-treated rabbit was used as a positive control and shows co-localization (yellow) of abundant fibrin deposition (green) and endothelial cells (red), indicating intravascular fibrin deposition. Minimal fibrin deposition was observed in myocardium from sham rabbits (E) but higher levels of fibrin were observed extravascularly in I/R-injured rabbits (F). All panels were photographed at an original magnification of ×400.
Ultrastructural Analysis of the Heart after Myocardial I/R Injury
We analyzed the microvasculature of hearts of I/R-injured rabbits to determine whether there was disruption of the endothelium after injury. Ultrastructural analysis of endothelial cells of capillaries in areas of nonischemic myocardium from I/R-injured rabbits were completely intact (data not shown) and indistinguishable from the capillaries in LV from sham-operated animals (Figure 7A) ▶ . In contrast, analysis of the AR area of LV of I/R-injured rabbits revealed endothelial cell damage that ranged from vacuolization (not shown) to disruption of the endothelial barrier (Figure 7B) ▶ . In addition, ultrastructural analysis of AR tissue did not reveal significant levels of intravascular fibrin deposition (not shown). These studies indicated that our model of I/R injury induced disruption of the endothelium in AR regions of myocardium, which would allow plasma-clotting factors to contact TF expressed by extravascular cardiomyocytes.
Figure 7.

Ultrastructural analysis of myocardium from I/R-injured rabbits. A: Normal capillary from the left ventricular myocardium of a sham rabbit (original magnification, ×8,900). B: Injured capillary in an AR area of myocardium of an I/R-injured rabbit (original magnification, ×5,200). Arrow indicates disruption of the endothelium.
Inhibition of TF Activity Reduces Infarct Size after Myocardial I/R Injury
We have recently shown that inhibition of IL-8 with an anti-IL-8 antibody (2 mg/kg) significantly reduces the degree of necrosis in a rabbit model of myocardial I/R injury. 28 In this study, we found that an isotype control antibody had no effect. Another study examining the role of ICAM-1 in myocardial I/R injury showed that an anti-ICAM-1 antibody (2 mg/kg) was cardioprotective. 29 This study showed no difference between the control antibody group and a saline vehicle group. Therefore, we chose to use saline-treated rabbits as a control for our current studies. Saline-treated rabbits were also used as controls for the hirudin and ancrod studies (see below).
We determined the functional role of TF in myocardial I/R injury by administration of the inhibitory anti-rabbit TF monoclonal antibody 11F to rabbits. We generated a consistent area AR for ischemic damage (Figure 8) ▶ . Administration of 11F to rabbits (n = 6) 15 minutes before coronary ligation significantly reduced infarct size compared with saline-treated rabbits (n = 5; 16 ± 1% versus 41 ± 2%, P = 0.004). This represented a 61% reduction in infarct size (Figure 8A) ▶ . Moreover, administration of anti-TF antibody to rabbits (n = 5) 30 minutes after the onset of ischemia resulted in a 44% reduction in infarct size compared with saline-treated rabbits (n = 5; P = 0.014) (Figure 8B) ▶ . These data indicated that TF contributed to myocardial I/R injury in our model.
Figure 8.

Effect of anti-TF antibody treatment on infarct size. A: Rabbits were treated intravenously with either saline (n = 5) (hatched bars) or an anti-rabbit TF monoclonal antibody (11F) (n = 6) (black bars) 15 minutes before ischemia. Similar AR areas of LV were observed in both groups (42 ± 2% for 11F-treated rabbits and 39 ± 5% for saline-treated rabbits). The mean infarct sizes were 16 ± 1% for the 11F-treated rabbits and 41 ± 1% for the saline-treated rabbits, indicating that 11F reduced infarct size by 61% (P = 0.004). B: Rabbits were treated intravenously with either saline (n = 5) or 11F (n = 5) 30 minutes after the onset of ischemia. Similar areas of LV were placed AR to I/R injury (47 ± 4% for 11F-treated and 44 ± 1% for saline-treated rabbits). The mean infarct sizes were 24 ± 4% for the 11F-treated rabbits and 43 ± 1% for the saline-treated rabbits, indicating that 11F reduced infarct size by 44% (P = 0.014). Data are expressed as the mean ± SE.
Inhibition of Thrombin Reduces Infarct Size after Myocardial I/R Injury
We determined the contribution of thrombin to myocardial I/R injury by administration of hirudin, a direct thrombin inhibitor. Administration of hirudin to rabbits significantly reduced infarct size compared with saline-treated rabbits (18 ± 1% versus 44 ± 1%, P = 0.014, n = 5). This represented a 59% reduction in infarct size (Figure 9A) ▶ . This result suggests that thrombin, in part, mediates myocardial I/R injury.
Figure 9.

The effect of hirudin and ancrod treatment on infarct size. A: Rabbits were treated intravenously with either saline (n = 4) (hatched bars) or hirudin (n = 5) (black bars) before and during myocardial I/R injury. Similar areas of the LV were placed AR to I/R injury in both groups (43 ± 5% for hirudin-treated rabbits and 46 ± 5% for saline-treated rabbits). The mean infarct sizes were 18 ± 1% for the hirudin-treated rabbits and 44 ± 1% for the saline-treated rabbits, indicating that treatment with hirudin reduced infarct size by 59% (P = 0.014). B: Rabbits were treated intravenously with either saline (n = 4) (hatched bars) or ancrod (n = 5) (black bars) before I/R injury. Similar areas of the LV were placed AR to I/R injury (50 ± 3% for ancrod-treated rabbits and 43 ± 4% for saline-treated rabbits). The mean infarct sizes were not significantly different (42 ± 2% for ancrod-treated rabbits and 41 ± 4% for saline-treated rabbits). All data are expressed as mean ± SE.
Fibrinogen Depletion Does Not Affect Infarct Size after Myocardial I/R Injury
We investigated the role of fibrin deposition in myocardial I/R injury by administration of ancrod, a defibrinogenating enzyme (Figure 9B) ▶ . The ancrod treatment resulted in >92% decrease in fibrinogen levels from 2.60 ± 0.09 mg/ml (n = 14) to functionally undetectable levels. Importantly, administration of ancrod to rabbits did not affect the infarct size compared with saline-treated animals (42 ± 2% versus 41 ± 4%, P = 0.62, n = 5). Although these studies do not exclude a role of low levels of fibrin deposition in I/R injury, these results together with our analysis of fibrin deposition suggest that TF-dependent thrombin generation may contribute to myocardial I/R injury by mechanisms beyond simply initiating intravascular fibrin deposition.
Hirudin Reduces Chemokine Expression after Myocardial I/R Injury
We investigated the hypothesis that the TF-thrombin pathway contributed to I/R injury by enhancing chemokine expression and inflammation. We measured the levels IL-8 and MCP-1 in non-AR and AR areas of LV of I/R-injured rabbits with or without hirudin. The induction of both IL-8 and MCP-1 was reduced by hirudin (Figure 10) ▶ . Although these results did not achieve statistical significance, they suggest that functional inhibition of thrombin has an impact on chemokine expression that may affect the recruitment of leukocytes into the AR areas of myocardium.
Figure 10.

Functional inhibition of thrombin reduces chemokine expression. Levels of IL-8 (A) and MCP-1 (B) were assessed by enzyme-linked immunosorbent assay in non-AR (N) and AR areas of LV of I/R-injured rabbits with (n = 3) or without (n = 3) hirudin. Data are shown as mean ± SD.
Functional Inhibition of TF or Thrombin Reduces PMN Infiltration
We further investigated the mechanism by which TF contributes to infarct size by quantitating the recruitment of PMNs in anti-TF antibody-treated and saline-treated I/R-injured rabbits. Histological assessment of tissue from saline-treated rabbits revealed a large infiltrate of marginating leukocytes (predominantly PMNs) within capillaries and postcapillary venules in the AR area of the LV (Figure 11A) ▶ . Most of the PMNs were associated with the endothelium and subendothelium in the parenchymal vessels. Animals treated with 11F antibody 15 minutes before the onset of ischemia showed a profound reduction in PMN margination and transendothelial migration (Figure 11B) ▶ . To more accurately assess the infiltration of PMNs, the number of PMNs in the AR areas of LV in saline- and 11F-treated I/R-injured rabbits was scored. Functional inhibition of TF significantly reduced the infiltration of PMNs (Figure 11C) ▶ . Importantly, there was no difference in the number of circulating leukocytes between 11F-treated and control groups after I/R injury (data not shown). We observed a similar reduction in PMN infiltration in hirudin-treated rabbits compared with a separate group of saline-treated rabbits (Figure 11C) ▶ . This data indicated that functional inhibition of TF or thrombin reduced the recruitment of PMNs during I/R injury.
Figure 11.

Functional inhibition of TF or thrombin reduces PMN infiltration. H&E representative photomicrographs (original magnification, ×400) of the viable area of myocardium in the AR area of LV from rabbits treated with saline (A) or anti-TF antibody (B) 15 minutes before ischemia. Note the large infiltration of PMNs packing the venule of the saline-treated animal whereas the venule from the 11F-treated animal is almost devoid of PMNs. C: Quantification of PMNs within rabbit heart sections. An investigator blinded to the treatment counted the number of PMNs per high-powered field in the LV of I/R-injured rabbits treated with either saline (n = 30 sections) (six different rabbits) or anti-TF antibody 11F (n = 12 sections) 15 minutes before to the onset of ischemia (two different rabbits). In addition, we analyzed I/R-injured rabbits treated with hirudin (n = 14 sections) (three different rabbits). Data were presented as the mean ± SE.
Discussion
A recent study demonstrated that administration of a different inhibitory anti-rabbit TF antibody (AP-1) partially restored blood flow to ischemic myocardium during reperfusion. 23 It was proposed that TF was expressed by vascular endothelial cells, 23 which would suggest that the AP-1 antibody may be acting by reducing TF-initiated intravascular thrombosis. However, TF may contribute to myocardial I/R injury by other mechanisms, such as by increasing the extravasation of monocytes or by enhancing extravascular thrombin generation and inflammation. 15-17,39 We demonstrated that administration of an inhibitory anti-TF antibody either before or after ischemia significantly reduced infarct size after I/R injury, indicating that TF contributes to myocardial I/R injury. We used an acute model of I/R injury, however, further studies are required to examine the role of TF in models of more chronic reperfusion.
TF activity may be increased after myocardial I/R injury by de novo protein synthesis as well as by de-encryption of pre-existing TF. There are a variety of cell types that may contribute to the pathological expression of TF during myocardial I/R-injury that include cardiomyocytes, which constitutively express low levels of TF, and vascular cells, such as endothelial cells and circulating leukocytes, which can be induced to express TF. Our study was not designed to identify all possible sites of TF expression during myocardial I/R-injury but simply to document the cell types that exhibit increased or induced expression. We analyzed TF protein and mRNA expression in the hearts of I/R-injured rabbits by immunohistochemistry and in situ hybridization, respectively. We observed a small number of TF-positive leukocytes, which included monocytes and PMNs (not shown). We were unable to detect significant TF expression by endothelial cells using in situ hybridization and immunohistochemistry, despite the proposal by Golino and colleagues 23 that myocardial I/R injury induces TF expression in the vascular endothelium. Our study and that of Golino and colleagues used different experimental models (45 minutes of ischemia and 2 hours of reperfusion versus 5 minutes of ischemia and 2 hours of reperfusion), which may, in part, explain the different conclusions. Nevertheless, we propose that TF generated by intravascular cell types, such as the TF-positive leukocytes observed in our model, does not seem to play a major role in myocardial I/R injury because we failed to observe significant intravascular thrombosis or fibrin deposition.
We found that TF expression was up-regulated in cardiomyocytes in the AR areas of LV and anti-TF antibody administered in vivo bound to these cardiomyocytes. We observed structural and functional disruption of the endothelium, which is consistent with a previous report showing increased permeability of the coronary microvasculature after brief ischemia (15 minutes) and reperfusion. 40 Damage to the endothelial barrier would permit plasma-clotting factors to gain access to TF expressed by extravascular ischemic and nonischemic cardiomyocytes, suggesting that these cells may participate in local thrombin generation and fibrin deposition. Indeed, we observed extravascular but not intravascular fibrin deposition consistent with TF-positive cardiomyocytes initiating the clotting cascade at this extravascular site.
We investigated the role of fibrin deposition in I/R injury by defibrinogenating the rabbits with ancrod. Ancrod treatment reduced fibrinogen to undetectable levels, but did not affect the infarct size. Consistent with these results, we did not observe intravascular fibrin deposition or microvascular thrombosis in heart tissue of rabbits subjected to I/R injury by immunohistochemistry and ultrastructural analysis, although we did observe a low level of extravascular fibrin deposition. Although these results do not exclude that a low level of intravascular fibrin deposition occurs in this model and impairs blood flow, our results suggest that TF contributed to myocardial I/R injury by mechanisms other than initiating intravascular fibrin deposition. Fibrin-independent mechanisms for the no reflow phenomenon in I/R injury have been described, such as capillary plugging by leukocytes and erythrocytes. 7,8 Other investigators found frequent leukocyte and erythrocyte capillary plugging and only occasional fibrin-containing microthrombi in the microvasculature by electron microscopy in a similar pig I/R model. 41
We found that hirudin treatment reduced the infarct size by 59%, which is similar to the effect observed using anti-TF antibody treatment (61%). Thrombin stimulates endothelial cells to express chemoattractants, such as IL-8 21 and MCP-1, 5 and adhesion molecules, 21,22 such as ICAM-1 and P-selectin. These molecules are required for the recruitment and extravasation of PMNs and monocytes, which contribute to myocardial I/R injury. Additional studies have suggested that thrombin contributes to inflammation in septic shock 42 and glomerulonephritis through PAR-1 signaling. 43 Vascular smooth muscle cells and endothelial cells express PAR-1 and both cell types exhibit inducible expression of MCP-1 in response to thrombin. 5
Examination of the mechanism by which the TF-thrombin pathway contributes to infarct size revealed that functional inhibition of thrombin decreased chemokine expression and inhibition of TF or thrombin reduced the infiltration of PMNs after myocardial I/R injury. PMN infiltration was assessed by counting the number of PMNs infiltrating into AR tissue. PMNs are an important component of myocardial cell death in I/R injury. Inhibition of PMN accumulation by blocking CD18 and ICAM-1 or by use of CD18- and ICAM-1-deficient mice has shown that PMNs contribute to infarct size in models of myocardial I/R injury. 4,44,45 We propose that extravascular TF, through the action of thrombin, has a proinflammatory role in myocardial I/R injury by increasing chemokine expression and enhancing the recruitment of leukocytes.
The beneficial effects of anti-thrombin therapy in rabbit models of vascular injury and thrombosis have been extensively studied. 46-48 In addition, administration of hirudin to patients with acute coronary syndromes decreased cardiovascular death, new myocardial infarction, refractory angina, surgical cardiac procedures, 49 and re-occlusion rates after thrombolysis. 50 Hirudin also decreased troponin T levels, a marker for myocardial infarction, in patients undergoing angioplasty for unstable angina. 51 However, anti-thrombin therapy was associated with both minor and major bleeding complications. 49,50,52 Other studies have examined the effects of anti-TF therapy in animal models of thrombosis. Administration of anti-TF antibody prevents re-occlusion in femoral vessels, 53 decreases the incidence of restenosis after carotid thrombosis, 54 and reduces the required dose of tPA for effective thrombolysis in carotid thrombosis models. 55 We can now extend this list of beneficial effects of anti-TF therapy to myocardial I/R injury. To date, there have been no clinical trials using anti-TF therapy.
The reduction in infarct size (44%) observed even when the anti-TF antibody was administered after the onset of ischemia indicates that anti-TF therapy should be of significant clinical benefit in the treatment of acute coronary syndromes before thrombolysis. Importantly, unlike anti-thrombin therapy, anti-TF therapy is not associated with bleeding complications in rabbits, baboons, or chimpanzees. 15,17,56 Moreover, the beneficial effects of this anticoagulant therapy may be, in part, because of inhibition of thrombin generation and inflammation. We propose that anti-TF therapy should be effective in decreasing myocardial I/R injury in patients and may have a superior safety profile to anti-thrombin therapy with fewer bleeding complications.
Acknowledgments
We thank J. Robertson for assistance with preparing this manuscript, Drs. T. Edgington, L. Curtiss, and M. Riewald for critical reading of the manuscript; Robert Thomas and Christine Rothnie for expert animal surgery assistance; Kent Osborn for pathological review of tissues; and Malcolm Wood for assistance in tissue processing for ultrastructural analysis of heart tissues.
Footnotes
Address reprint requests to Nigel Mackman, Ph.D., The Scripps Research Institute, Departments of Immunology and Vascular Biology, IMM-17, 10550 North Torrey Pines Rd., La Jolla, CA 92037-9701. E-mail: nmackman@scripps.edu.
Supported by National Institutes of Health Grants HL16411 (to N. M.), and GM46662 and GM07037 (to T. H. P.). This work was performed during the tenure of an Established Investigator (N. M.), a postdoctoral fellowship (J. E.) from the American Heart Association, fellowships from the Thoracic Surgery Foundation for Research and Education (E. M. B. and E. N. M.) and by fellowships from the Stanley J. Sarnoff Endowment for Cardiovascular Science (J. C. K. and J. L.).
J. H. E. and E. M. B. contributed equally to this manuscript.
References
- 1.Matsumura K, Jeremy RW, Schaper J, Becker LC: Progression of myocardial necrosis during reperfusion of ischemic myocardium. Circulation 1998, 97:795-804 [DOI] [PubMed] [Google Scholar]
- 2.Entman ML, Michael L, Rossen RD, Dreyer WJ, Anderson DC, Taylor AA, Smith CW: Inflammation in the course of early myocardial ischemia. FASEB J 1991, 5:2529-2537 [DOI] [PubMed] [Google Scholar]
- 3.Herskowitz A, Choi S, Ansari AA, Wesselingh S: Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol 1995, 146:419-428 [PMC free article] [PubMed] [Google Scholar]
- 4.Ma XL, Lefer DJ, Lefer AM, Rothlein R: Coronary endothelial and cardiac protective effects of a monoclonal antibody to intercellular adhesion molecule-1 in myocardial ischemia and reperfusion. Circulation 1992, 86:937-946 [DOI] [PubMed] [Google Scholar]
- 5.Kranzhofer R, Clinton SK, Ishii K, Coughlin SR, Fenton JW, Libby P: Thrombin potently stimulates cytokine production in human vascular smooth muscle cells but not in mononuclear phagocytes. Circ Res 1996, 79:286-294 [DOI] [PubMed] [Google Scholar]
- 6.Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, LaRosa GJ, Hawkins HK, Smith CW, Michael LH, Entman ML, Rossen RD: Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation 1997, 95:684-692 [DOI] [PubMed] [Google Scholar]
- 7.Vanhaecke J, Flameng W, Borgers M, Jang IK, Van de Werf F, De Geest H: Evidence for decreased coronary flow reserve in viable postischemic myocardium. Circ Res 1990, 67:1201-1210 [DOI] [PubMed] [Google Scholar]
- 8.Engler RL, Schmid-Schonbein GW, Pavelec RS: Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol 1983, 111:98-111 [PMC free article] [PubMed] [Google Scholar]
- 9.Edgington TS, Mackman N, Brand K, Ruf W: The structural biology of expression and function of tissue factor. Thromb Haemost 1991, 66:67-79 [PubMed] [Google Scholar]
- 10.Drake TA, Morrissey JH, Edgington TS: Selective cellular expression of tissue factor in human tissues. Am J Pathol 1989, 134:1087-1097 [PMC free article] [PubMed] [Google Scholar]
- 11.Wilcox JN, Smith KM, Schwartz SM, Gordon D: Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci USA 1989, 86:2839-2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misumi K, Ogawa H, Yasue H, Soejima H, Suefuji H, Nishiyama K, Takazoe K, Kugiyama K, Tsuji I, Kumeda K, Nakamura S: Comparison of plasma tissue factor levels in unstable and stable angina pectoris. Am J Cardiol 1998, 81:22-26 [DOI] [PubMed] [Google Scholar]
- 13.Freeburn JC, Wallace JM, Strain JJ, Sinnamon DG, Craig BM, Johnson D, Gilmore WS: Monocyte tissue factor-like activity in post myocardial infarction patients. Br J Haematol 1998, 102:605-608 [DOI] [PubMed] [Google Scholar]
- 14.Ott I, Neumann FJ, Kenngott S, Gawaz M, Schomig A: Procoagulant inflammatory responses of monocytes after direct balloon angioplasty in acute myocardial infarction. Am J Cardiol 1998, 82:938-942 [DOI] [PubMed] [Google Scholar]
- 15.Taylor FB, Jr, Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R, Blick K, Edgington TS: Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock 1991, 33:127-134 [PubMed] [Google Scholar]
- 16.Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y: Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med 1997, 25:1820-1826 [DOI] [PubMed] [Google Scholar]
- 17.Tipping PG, Erlich JH, Apostolopoulos J, Mackman N, Loskutoff DJ, Holdsworth SR: Glomerular tissue factor expression in crescentic glomerulonephritis. Correlations between antigen, activity, and mRNA. Am J Pathol 1995, 147:1736-1748 [PMC free article] [PubMed] [Google Scholar]
- 18.Déry O, Corvera CU, Steinhoff M, Bunnett NW: Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol 1998, 274:C1429-C1452 [DOI] [PubMed] [Google Scholar]
- 19.Vu TK, Hung DT, Wheaton VI, Coughlin SR: Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64:1057-1068 [DOI] [PubMed] [Google Scholar]
- 20.Johnson K, Choi Y, DeGroot E, Samuels I, Creasey A, Aarden L: Potential mechanisms for a proinflammatory vascular cytokine response to coagulation activation. J Immunol 1998, 160:5130-5135 [PubMed] [Google Scholar]
- 21.Kaplanski G, Fabrigoule M, Boulay V, Dinarello CA, Bongrand P, Kaplanski S, Farnarier C: Thrombin induces endothelial type II activation in vitro: IL-1 and TNF-alpha-independent IL-8 secretion and E-selectin expression. J Immunol 1997, 158:5435-5441 [PubMed] [Google Scholar]
- 22.Sugama Y, Tiruppathi C, Offakidevi K, Andersen TT, Fenton JW, II, Malik AB: Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: a mechanism for stabilizing neutrophil adhesion. J Cell Biol 1992, 119:935-944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golino P, Ragni M, Cirillo P, Avvedimento VE, Feliciello A, Esposito N, Scognamiglio A, Trimarco B, Iaccarino G, Condorelli M, Chiariello M, Ambrosio G: Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat Med 1996, 2:35-40 [DOI] [PubMed] [Google Scholar]
- 24.Toombs CF, McGee S, Johnston WE, Vinten-Johansen J: Myocardial protective effects of adenosine. Infarct size reduction with pretreatment and continued receptor stimulation during ischemia. Circulation 1992, 86:986-994 [DOI] [PubMed] [Google Scholar]
- 25.Gertz SD, Fallon JT, Gallo R, Taubman MB, Banai S, Barry WL, Gimple LW, Nemerson Y, Thiruvikraman S, Naidu SS, Chesebro JH, Fuster V, Sarembock IJ, Badimon JJ: Hirudin reduces tissue factor expression in neointima after balloon injury in rabbit femoral and porcine coronary arteries. Circulation 1998, 98:580-587 [DOI] [PubMed] [Google Scholar]
- 26.Hatton MWC, Ross B, Soutward SMR, DeReske M, Richardson M: Pretreatment of rabbits with either hirudin, ancrod, or warfarin significantly reduces the immediate uptake of fibrinogen and platelets by the deendothelialized aorta wall after balloon-catheter injury in vivo. Arterioscler Thromb Vasc Biol 1998, 18:816-824 [DOI] [PubMed] [Google Scholar]
- 27.von Clauss A: Gerinnungsphysiologische schnellmethode zur bestimmung des fibrinogens. Acta Haematol 1957, 17:231-237 [DOI] [PubMed] [Google Scholar]
- 28.Boyle EM, Jr, Kovacich JC, Hébert CA, Canty TG, Jr, Chi E, Morgan EN, Pohlman TH, Verrier ED: Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg 1998, 116:114-121 [DOI] [PubMed] [Google Scholar]
- 29.Chen ZJ, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D: Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteosome pathway. Genes Dev 1995, 9:1586-1597 [DOI] [PubMed] [Google Scholar]
- 30.Andrews BS, Rehemtulla A, Fowler BJ, Edgington TS, Mackman N: Conservation of tissue factor primary sequence among three mammalian species. Gene 1991, 98:265-269 [DOI] [PubMed] [Google Scholar]
- 31.Kajikawa O, Johnson MC, II, Goodman RB, Frevert CW, Martin TR: A sensitive immunoassay to detect the α-chemokine GRO in rabbit blood and lung fluids. J Immunol Methods 1997, 205:135-143 [DOI] [PubMed] [Google Scholar]
- 32.Kajikawa O, Goodman RB, Johnson MC, II, Konishi K, Martin TR: Sensitive and specific immunoassays to detect rabbit IL-8 and MCP-1: cytokines that mediate leukocyte recruitment to the lungs. J Immunol Methods 1996, 197:19-29 [DOI] [PubMed] [Google Scholar]
- 33.Mackman N, Sawdey MS, Keeton MR, Loskutoff DJ: Murine tissue factor gene expression in vivo: tissue and cell specificity and regulation by lipopolysaccharide. Am J Pathol 1993, 143:76-84 [PMC free article] [PubMed] [Google Scholar]
- 34.Lie JT: Detection of early myocardial infarction by the acid fuchsin staining technique. Am J Clin Pathol 1968, 50:317-319 [DOI] [PubMed] [Google Scholar]
- 35.Erlich JH, Fearns C, Mathison J, Ulevitch RJ, Mackman N: Lipopolysaccharide induction of tissue factor expression in rabbits. Infect Immun 1999, 67:2540-2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hui KY, Haber E, Matsueda GR: Monoclonal antibodies to a synthetic fibrin-like peptide bind to human fibrin but not fibrinogen. Science 1983, 222:1129-1132 [DOI] [PubMed] [Google Scholar]
- 37.Flössel C, Luther T, Müller M, Albrecht S, Kasper M: Immunohistochemical detection of tissue factor (TF) on paraffin sections of routinely fixed human tissue. Histochemistry 1994, 101:449-453 [DOI] [PubMed] [Google Scholar]
- 38.Luther T, Dittert D-D, Kotzsch M, Erlich JH, Albrecht S, Mackman N, Müller M: Functional implications of tissue factor localization to cell-cell contacts in myocardium. J Pathol 2000, 192:121-130 [DOI] [PubMed] [Google Scholar]
- 39.Randolph GJ, Luther T, Albrecht S, Magdolen V, Muller WA: Role of tissue factor in adhesion of mononuclear phagocytes to and trafficking through endothelium in vitro. Blood 1998, 92:4167-4177 [PubMed] [Google Scholar]
- 40.Dauber IM, VanBenthuysen KM, McMurtry IF, Wheeler GS, Lesnefsky EJ, Horwitz LD, Weil JV: Functional coronary microvascular injury evident as increased permeability due to brief ischemia and reperfusion. Circ Res 1990, 66:986-998 [DOI] [PubMed] [Google Scholar]
- 41.Mrak RE, Murphy ML, Peng CF, Straub KD: Microvasculature sparing with controlled reperfusion of ischemic myocardium. Am J Cardiovasc Pathol 1990, 3:253-258 [PubMed] [Google Scholar]
- 42.Minnema MC, Chang ACK, Jansen PM, Lubbers YTP, Pratt BM, Whittaker BG, Taylor FB, Hack CE, Friedman B: Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood 2000, 95:1117-1123 [PubMed] [Google Scholar]
- 43.Cunningham MA, Rondeau E, Chen X, Coughlin SR, Holdsworth SR, Tipping PG: Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. J Exp Med 2000, 191:455-461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma XL, Tsao PS, Lefer AM: Antibody to CD-18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J Clin Invest 1991, 88:1237-1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palazzo AJ, Jones SP, Girod WG, Anderson DC, Granger DN, Lefer DJ: Myocardial ischemia-reperfusion injury in CD18- and ICAM-1-deficient mice. Am J Physiol 1998, 275:H2300-H2307 [DOI] [PubMed] [Google Scholar]
- 46.Rubsamen K, Eschenfelder V: Reocclusion after thrombolysis: a problem solved by hirudin? Blood Coagul Fibrinolysis 1991, 2:97-100 [PubMed] [Google Scholar]
- 47.Gerdes C, Faber-Steinfeld V, Yalkinoglu O, Wohlfeil S: Comparison of the effects of the thrombin inhibitor r-hirudin in four animal models of neointima formation after arterial injury. Arterioscler Thromb Vasc Biol 1996, 16:1306-1311 [DOI] [PubMed] [Google Scholar]
- 48.Rudd MA, George D, Johnstone MT, Moore RT, Collins L, Rabbani LE, Loscalzo J: Effect of thrombin inhibition on the dynamics of thrombolysis and on platelet function during thrombolytic therapy. Circ Res 1992, 70:829-834 [DOI] [PubMed] [Google Scholar]
- 49.: Organization to Assess Strategies for Ischemic Syndromes (OASIS-2) Investigators: Effects of recombinant hirudin (lepirudin) compared with heparin on death, myocardial infarction, refractory angina, and revascularization procedures in patients with acute myocardial ischemia without ST elevation: a randomized trial. Lancet 1999, 353:429-438 [PubMed] [Google Scholar]
- 50.Molhoek GP, Laarman GJ, Lok DJ, Luz CM, Kingma JH, Van den Bos AA, Zijnen P, Bosma AH, Hertzberger DP, Takens LH: Angiographic dose-finding study with r-hirudin (HBW 023) for the improvement of thrombolytic therapy with streptokinase (HIT-SK). Interim results. Eur Heart J 1995, 16(Suppl D):33–37 [DOI] [PubMed]
- 51.Rupprecht HJ, Terres W, Ozbek C, Luz M, Jessel A, Hafner G, von Dahl J, Kromer EP, Pellwitz W, Meyer J: Recombinant hirudin (HBW 023) prevents troponin T release after coronary angioplasty in patients with unstable angina. J Am Coll Cardiol 1995, 26:1637-1642 [DOI] [PubMed] [Google Scholar]
- 52.Neuhaus KL, von Essen R, Tebbe U, Jessel A, Heinrichs H, Maurer W, Doring W, Harmjanz D, Kotter V, Kalhammer E, Simon H, Horacek T: Safety observations from the pilot phase of the randomized r-hirudin for improvement of thrombolysis (HIT-III) study. Circulation 1994, 90:1638-1642 [DOI] [PubMed] [Google Scholar]
- 53.Jang I-K, Gold HK, Leinbach RC, Fallon JT, Collen D, Wilcox JN: Antithrombotic effect of a monoclonal antibody against tissue factor in a rabbit model of platelet-mediated arterial thrombosis. Arterioscler Thromb 1992, 12:948-954 [DOI] [PubMed] [Google Scholar]
- 54.Pawashe AB, Golino P, Ambrosio G, Migliaccio F, Ragni M, Pascucci I, Chiariello M, Bach R, Garen A, Konigsberg WK, Ezekowitz MD: A monoclonal antibody against rabbit tissue factor inhibits thrombus formation in stenotic injured rabbit carotid arteries. Circ Res 1994, 74:56-63 [DOI] [PubMed] [Google Scholar]
- 55.Ragni M, Cirillo P, Pascucci I, Scognamiglio A, D’Andrea D, Eramo N, Ezekowitz MD, Pawashe AB, Chiariello M, Golino P: Monoclonal antibody against tissue factor shortens tissue plasminogen activator lysis time and prevents reocclusion in a rabbit model of carotid artery thrombosis. Circulation 1996, 93:1913-1918 [DOI] [PubMed] [Google Scholar]
- 56.Levi M, ten Cate H, Bauer KA, van der Poll T, Edgington TS, Büller HR, van Deventer SJH, Hack CE, Wouter ten Cate J, Rosenberg RD: Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J Clin Invest 1994, 93:114-120 [DOI] [PMC free article] [PubMed] [Google Scholar]