

Abstract
Human amylin, a major constituent of pancreatic amyloid deposits, may be a pathogenetic factor for noninsulin-dependent diabetes mellitus (NIDDM). We demonstrated that the human amylin S20G gene mutation (S20G) was associated with a history of early onset, more severe type of NIDDM, linking the amylin gene to this disease. Also, we demonstrated that expression of human wild-type (WT) amylin in COS-1 cells leads to intracellular amyloidogenesis and induction of apoptosis, suggesting a possible mechanism for disease induction. Therefore we compared the abilities of S20G and WT amylin to induce apoptosis in transfected COS-1 cells and form amyloid in vitro. We transfected the rat (RAT), mutated human (MUT), WT, and S20G amylin genes into COS-1 cells and measured apoptosis using fluorescent-activated cell sorting analysis at 48, 72, and 96 hours. At 96 hours apoptosis increased significantly (P < 0.01) in cells transfected with WT and S20G over RAT or MUT (WT, 19%; S20G, 25%; RAT, 13%; and MUT, 12%) and the difference between WT and S20G was significant (P < 0.05). Synthetic WT and S20G monomeric peptides were used to generate amyloid fibrils in vitro as measured by the thioflavin T binding assay. The S20G amylin formed approximately twofold more amyloid at a rate approximately threefold higher than WT. Electron micrography indicated that the in vitro amyloid generated by WT and S20G amylins were morphologically indistinguishable. The results suggest that increased cytotoxicity by S20G is because of increased amyloidogenicity, which may be a causative factor in the early development of NIDDM, possibly through loss of β cell mass.
Noninsulin-dependent diabetes mellitus (NIDDM, or type II diabetes) is characterized by continuous hyperglycemia that results from insulin resistance in peripheral cells, particularly muscle, adipose tissue, and liver, or β cell dysfunction in pancreatic islets. 1,2 It is a complex, multifactorial disease, which results from an interaction between several hereditary and environmental factors. 1,2 Although susceptibility genes associated with certain types of diabetes have been reported, the majority of genetic factors which can be attributed to the development of the major form of NIDDM, represented by late-onset disease are still unknown. 1,2 For example, in the relatively rare maturity onset diabetes of the young, or MODY, mutations in five genes have been shown to play causative roles. 3 These genes include glucokinase (MODY2), hepatic nuclear factor-1α (HNF-1α; MODY3), hepatic nuclear factor-4α (HNF-4α; MODY1), insulin promoter factor 1 (IPF-1; MODY4) and hepatic nuclear factor-1β (HNF-1β; MODY5). Nevertheless, these five genes only account for a small percentage of MODY cases and the possibility that any of these genes is associated with late-onset NIDDM has been excluded. 2 Although very rare instances of NIDDM have been associated with mutations in the genes for insulin, insulin receptor, mitochondrial tRNA-Leu, and the GLUT2 glucose transporter, 2 the genetic factors that account for late-onset NIDDM are unknown. Given the apparent heterogeneity of the disease, it is likely our understanding of the genetics of late-onset NIDDM will require a very extensive genetic screening effort.
In previous studies of Japanese families, who exhibited a relatively early age onset of NIDDM (≤35 years), we reported the missense S20G mutation of the human amylin gene. 4 Human amylin (or islet amyloid polypeptide) is a 37 amino acid peptide derived from a larger precursor, that is co-secreted with insulin from β cells. 5,6 Amylin has 46% identity with calcitonin gene-related peptide and has been implicated in regulating insulin and glucose metabolism, 7,8 although its precise function is unknown. Of the 12 patients carrying the S20G amylin mutation, eight had severe, early-onset disease with a family history of late-onset NIDDM. The other four patients developed mild NIDDM after age 51 and did not have family histories of diabetes. These data clearly implicate the amylin gene itself as a causative factor in the genesis of NIDDM; however, the incomplete penetrance associated with the mutation suggests that other genes are also likely to be involved. That the amylin gene product was a causative factor in the disease phenotype was strengthened by a high pressure liquid chromatography analysis of the peptide, which confirmed the amylin-like immunoreactivity in the postprandial plasma extracted from affected patients and indicated that the bulk (84%) of the amylin was represented by the S20G mutant. 4 This indicates that the mutant amylin is expressed efficiently in vivo, and suggests the possibility that its rate of metabolic clearance is lower than wild type (WT); however, we cannot exclude the possibility that its overall rate of synthesis is higher than WT amylin.
From a pathogenetic viewpoint, one of the prominent features of amylin is its ability to form amyloid fibrils. The fibrils represent highly ordered rod-shaped structures that contain large amounts of cross β-pleated sheet structures. These have been shown to be the constituent peptide of islet amyloid deposits, which are characterized by birefringence under polarizing light with Congo red staining and which are detected in the pancreata of 70 to 90% of NIDDM patients at autopsy along with up to 50% loss of β cell mass. 8 It has been demonstrated that the region between amino acids 20 to 29 are responsible for the highly amyloidogenic character of human amylin, properties which are not shared by rodent amylins. 9 Interestingly, the S20G mutation, substituting a glycine for a serine makes the 20 to 29 region even more hydrophobic, suggesting that this mutation might have increased amyloidogenic characteristics, which may somehow be tied to the pathogenesis of NIDDM.
We previously demonstrated that COS-1 cells expressing human WT amylin exhibited intracellular amyloid deposits that were correlated with cell death by an apoptotic mechanism. Cells that expressed nonamyloidogenic rat amylin or mutated human amylin were shown to be present within the endoplasmic reticulum and Golgi apparatus, but did not induce apoptosis. 10,11 Immunohistochemical analyses demonstrated that the amyloid deposits accumulated in the endoplasmic reticulum and the Golgi apparatus. These results suggested that intracellular amyloid accumulation activated specific intracellular apoptotic signaling pathways, 11 and suggested that such a mechanism might be involved in the loss of β cell mass in NIDDM and thereby contributing to the genesis of NIDDM. We therefore undertook the current study to compare directly the cytotoxic and amyloidogenic properties of WT and S20G amylin. The data indicate that S20G is a more potent intracellular cytotoxin than is WT amylin. The enhanced cytotoxicity is correlated with the enhanced ability of the S20G mutant amylin to form amyloid fibrils in vitro. The data provide further support for the concept that intracellular amyloid formation may be a pathogenetic factor in β cell pathophysiology and the genesis of NIDDM.
Materials and Methods
Plasmids
Human WT amylin cDNA 5 was excised with HindIII/KpnI and subcloned into pKF19K. 10 The S20G amylin cDNA, containing the AGCSer to GGCGly mutation, was generated by site-directed mutagenesis using Mutan-Super Express Km Kit (Takara, Shiga, Japan) and mutagenic oligonucleotide: 5′-AGTTCATTCCGGCAACAACTTTG-3′. WT and S20G cDNA fragments were isolated by HindIII/SacI digestion, blunt-ended, and subcloned into the pMT2 expression vector. 10 The pMT2 plasmids containing rat amylin cDNA (pMT2-RAT) and a mutated, nonamyloidogenic human amylin (pMT2-MUT), converting amino acids AILSS (residues 25 to 29) to PVLPP have been previously described (Figure 1) ▶ . 10,11
Figure 1.

Amino acid sequence of human WT, S20G, and MUT, and RAT amylin. Only the amino acids that differ are shown. The underlined sequence between amino acids 20 to 29 represents the amyloidogenic domain. 9
Cell Culture and Transfection
COS-1 cells were purchased from American Type Cell Culture Collection (Rockville, MD) and grown in Dulbecco’s modified Eagle medium (Hyclone, Logan, UT) supplemented with 10% Fetal Clone II (Hyclone), 100 U/ml of penicillin (Life Technologies, Inc., Rockville, MD), 100 μg/ml of streptomycin-sulfate (Life Technologies, Inc.) and 2 mmol/L of l-glutamine at 37°C in 100% humidified air containing 5% CO2. All cells in the present study were used within passage 20. Cells were rinsed with 5 ml of phosphate-buffered saline (PBS; Life Technologies, Inc.) and harvested by trypsinization with 0.5× trypsin-ethylenediaminetetraacetic acid solution (Sigma, St. Louis, MO) in PBS. Cells were collected by centrifugation (1,500 × g for 5 minutes), washed with PBS, and resuspended at 1.5 × 10 7 cells/ml with Cytomix 12 (120 mmol/L KCl, 0.15 mmol/L CaCl2, 10 mmol/L K2HPO4/KH2PO4, 25 mmol/L HEPES, pH 7.6, 2 mmol/L EGTA, 5 mmol/L MgCl2). Cells (3.0 × 106) were mixed with 200 μl of Cytomix and 7 to 10 μg of plasmid DNA and incubated on ice for 20 minutes. The cells were electroporated at 960 μFarad and 250 volts (Gene Pulser; Bio-Rad Laboratories, Hercules, CA) in standard 4-mm cuvettes (Bio-Rad). Transfected cells were cultured in 12 ml of medium in 10-cm tissue culture dishes and media was changed 24 hours after electroporation. Positive apoptosis controls were generated by treating cells with tunicamycin (10 μg/ml).
Apoptosis Assay
Apoptosis was assessed by Annexin-V binding to phosphatidylserine residues exposed on the cell surface and detection by fluorescent-activated cell sorting (FACS). 13 Intercalation of 7-amino actinomycin-D (7-AAD; Molecular Probes, Eugene, OR) into DNA was used to identify cells whose membrane integrity was lost, distinguishing cells that are dying by necrosis or damaged while harvesting. Cells were harvested by trypsinization and pooled with their culture medium containing floating cells that had lost their adherent properties during apoptosis. Cells were pelleted by centrifugation (1,500 × g for 5 minutes) at room temperature and washed with 2 ml of ice-cold PBS and 2 ml of ice-cold binding buffer (10 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2). Cells were resuspended with 300 μl of binding buffer and 50 μl of cells (0.5 to 1.0 × 106) were aliquoted for labeling. Prepared samples were incubated in 100 μl of binding buffer containing 8 μg/ml of Annexin-V-biotin conjugate (Trevigen, Inc., Gaithersburg, MD) in the dark for 20 minutes and washed with 1 ml of binding buffer. Samples were incubated in 100 μl of binding buffer containing 8 μg/ml streptavidin-phycoerythrin (PE) (Molecular Probes) and 5 μg/ml 7-AAD and then washed under the same conditions. Finally, samples were resuspended with 400 μl of binding buffer and analyzed within 2 hours of labeling. FACS was performed on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). FACS gating based on the forward and side scatter was used to exclude cellular debris and larger aggregates. In each case 30,000 ± 2,000 cells/sample were analyzed with Cell Quest software (Becton Dickinson).
Amylin Radioimmunoassay (RIA)
Cells were pelleted by centrifugation (13,000 × g for 10 seconds) and resuspended in the cell lysis buffer (10 mmol/L NaH2PO4, pH 7.6, 80 mmol/L NaCl, 25 mmol/L ethylenediaminetetraacetic acid, 0.01% NaN3, 0.05% Triton X-100, 0.002 mg/ml antipain hydrochloride (Sigma), 0.05 mg/ml elastatinal (Sigma), and 0.0008 mg/ml leupeptin (Sigma) on ice. Pelleted cells were homogenized by sonication in the cell lysis buffer on ice and cell debris was removed by centrifugation (13,000 × g for 15 minutes). Protein concentration of the supernatant was determined by colorimetric assay using the Detergent Compatible Protein Assay Kit (Bio-Rad). Amylin concentrations in cell lysates were performed by RIA according to a modification of the method of Permert et al. 14 Briefly, cell lysates were extracted with Sep-Pak C18 (Waters, Franklin, MA). The extracts were evaporated to dryness and the residues were resuspended with RIA cell lysis buffer as described above with 0.25% of bovine serum albumin (BSA). Amylin levels in the extracts were determined by RIA using [125I]-radiolabeled synthetic human amylin (1–37) prepared by the Iodogen method and rabbit anti-human amylin antibody (Peninsula Laboratories, Belmont, CA). The antibody reacted equally well with synthetic human and rat amylin amides, and nonamidated forms of human WT and S20G amylin. The recovery of S20G and rat amylin extracts through Sep-Pak C18 were in the range of 70 to 80% (data not shown). The sensitivity of this assay system for amylin determination in cell lysates was 2.5 fmol/μg protein.
In Vitro Amyloidogenesis Assay
WT and S20G amylin peptides were synthesized using solid phase methods by the Mayo Protein Core Facility (Rochester, MN) on an ABI 433A Peptide Synthesizer (PE Biosystems, Foster City, CA) using Fmoc/HBTU/HOBt/DIPEA chemistry. The amylin peptides were deprotected and removed from the polystyrene resin by treatment with a mixture of 90% trifluoroacetic acid (TFA), 5.0% water, 2.5% ethanedithiol, 2.5% thioanisole for 90 minutes in the dark at room temperature. Each peptide was washed by precipitation in 3 × 50 volumes of cold methyl t-butyl ether, and then purified by high-temperature reversed phase high pressure liquid chromatography at 60°C on a Vydac C18 column (2.1 × 25 cm) in 0.1% TFA-water with a 60-minute gradient of 10 to 80% acetonitrile in 0.1% TFA. The disulfide bond at Cys-2 and Cys-7 was formed by iodine oxidation in 80% acetic acid for 6 hours at 20°C. The iodine and excess reagents were extracted from the oxidized peptide with CCl4 and the peptides were purified by reverse phase high pressure liquid chromatography at 60°C as described above. WT and S20G peptides were analyzed for in vitro amyloid formation by a modified thioflavin T (ThT) binding assay. 15 Briefly, each peptide was dissolved in dimethyl sulfoxide at a final concentration of 1 mmol/L and then diluted with 10 mmol/L Tris-HCl, pH 7.5, 100 mmol/L NaCl, in a final volume of 400 μl so that the final concentration of each peptide was 10 or 25 μmol/L and the dimethyl sulfoxide concentration was 5% (v/v). Reaction mixtures were stirred constantly at room temperature in 1.5-ml cryogenic screw cap tubes, whose inside wall was treated with Sigmacote (Sigma). Fluorescence was measured on a SLM 8000 spectrofluorometer (Spectronic Instruments, Inc.). Excitation and emission wavelengths were 450 and 485 nm, respectively. For each time point duplicate 5-μl aliquots of the reaction mixture were mixed with 1.5 ml of 1 μmol/L ThT (Sigma) and 50 mmol/L of glycine/NaOH, pH 9.0, in quartz cuvettes. The emitted fluorescence of each sample was measured at 30-minute intervals for 2 hours, and hourly thereafter for 8 hours.
Electron Microscopy
Synthetic WT and S20G peptide suspensions from the ThT assays were applied to the surface of Formvar-coated carbon grids and negatively stained with 1% phosphotungstic acid, pH 8.0. The specimens were examined with a JEOL-1200 EXII transmission electron microscope operated at 40 to 80 kV. Photomicrographs of each peptide were taken for morphological comparison. Measurements of amyloid fibrils were taken from enlarged prints using standard calibration bars.
Statistical Analysis
Data were subjected to repeated measures or one-way analysis of variance analysis using post hoc Bonferroni/Dunn. Error bars represent standard errors.
Results
WT and S20G Amylin Expression Differentially Induce Apoptosis in COS-1 Cells
In separate studies we have established that cultured βTC-3 cells, a mouse pancreatic β cell line, display virtually the same sensitivity toward intracellular amyloidogenesis and subsequent cell death as COS-1 cells (Hiddinga HJ, Shakagashira S, and Eberhardt NL, unpublished observations), indicating that the differences in secretory capacity between these two cell lines does not abrogate amyloid-induced cell death. Because COS-1 cells are much easier to maintain in cell culture, we elected to compare the behavior of the S20G and WT amylins in this cell line. We first examined the relative apoptosis in COS-1 cells induced by the expression of WT and S20G amylin using FACS analysis. We used PE-conjugated Annexin-V as a probe to detect exposed phosphatidylserine on the outer layer of cell membranes during early apoptosis. 13 Simultaneously 7-AAD that intercalates into DNA was used to identify cells dying by necrosis or by physical damage. We previously demonstrated that amyloid accumulated in transfected COS-1 cells after 72 to 96 hours and was correlated with the occurrence of pycnotic nuclei 10 and apoptosis 11 within this same time period. Accordingly, we examined apoptosis by FACS analysis at 48, 72, and 96 hours after transfection with pMT2 control vector, pMT2.WT, pMT2.S20G, pMT2.RAT, and pMT2.MUT expression cassettes in COS-1 cells. COS-1 cells transfected with pMT2 vector (Figure 2A) ▶ showed little change in the number of apoptotic cells at all time points. Similar results were obtained after transection of the nonamyloidogenic pMT2.RAT and pMT2.MUT expression cassettes (Figure 2, D and E) ▶ . In contrast, when COS-1 cells were transfected with pMT2.WT and pMT2.S20G expression vectors, there was a twofold to threefold increase in the number of apoptotic cells at 72 and 96 hours compared to control or pMT2.RAT and pMT2.MUT transfected cells (Figure 2, B and C) ▶ .
Figure 2.

Induction of apoptosis in COS-1 cells expressing intracellular WT and S20G. FACS analysis was performed at 48, 72, and 96 hours using COS-1 cells labeled with AnnexinV-PE after transfection with pMT2 vector (A), pMT2.WT (B), pMT2.S20G (C), pMT2.RAT (D), and pMT2.MUT (E). AnnexinV-PE-positive cells, representing early stage apoptotic cells, are delineated by the bar designated M1.
Composite histogram profiles of the percentage of apoptotic cells from several experiments are shown in Figure 3 ▶ . The percentage of apoptotic cells was calculated by the gated number of AnnexinV-PE-positive cells divided by the total number of cells. The number of Annexin-PE-positive and 7-AAD-positive cells representing the late apoptotic/necrotic cells was <5% in all samples (data not shown) and are included in the histogram plots shown in Figure 3 ▶ . No significant difference between any of the groups was observed at 48 hours. At 72 hours there was a significantly larger number of cells in the early apoptotic stage when transfected with pMT2.S20G than with pMT2 or pMT2.MUT (P < 0.05) but not significant when compared with pMT2.RAT or pMT2.WT transfections. At 96 hours after transection with pMT2.WT and pMT2.S20G there were significantly higher levels of apoptosis than with pMT2, pMT2.RAT, and pMT2.MUT (P < 0.01). In addition, pMT2.S20G-transfected cells showed significantly (P < 0.05) higher levels of apoptosis than cells transfected with the pMT2.WT at both 72 and 96 hours. These results support the concept that expression of the different amylin forms might have a direct link to cell apoptosis and that the S20G mutant is a more potent intracellular cytotoxin than WT amylin. This concept is supported by our previous results demonstrating that inhibition of amylin expression by co-transection of the antisense pMT2.WT DNA blocked apoptosis. 11 Accordingly, it was important to compare the levels of expression of WT and S20G amylin peptide expression, to be able to compare their relative cytotoxicity. Thus we next measured the levels of exogenously expressed WT and S20G amylin in COS-1 cells by RIA.
Figure 3.

Histogram profiles of the induction of apoptosis in COS-1 cells transfected with pMT2 vector (open bars), pMT2.MUT (cross-hatched bars), pMT2.RAT (shaded bars), pMT2.WT (diagonal hatched bars), and pMT2.S20G (solid bars) after 48, 72, and 96 hours. The Annexin-positive and 7-AAD-positive cells representing the late apoptotic or necrotic cells accounted for <5% of the cells in all samples (data not shown). Statistically significant differences are indicated by * (P < 0.05) and ** (P < 0.01). Error bars are standard errors. The data represent the average of seven independent experiments.
Content of WT and S20G Amylin in COS-1 Cells
COS-1 cell lysates were measured by RIA to determine the expression levels of WT, S20G, MUT, and RAT amylin peptides (Figure 4) ▶ . In cell lysates transfected with pMT2 plasmid the levels of amylin were below the sensitivity of the assay (5 fmol/μg) at all time points. Importantly, there were no significant differences between the level of expression of WT and S20G amylin (Figure 4) ▶ , indicating that the differences observed in the induction of apoptosis in transfected COS-1 cells (Figure 3) ▶ were attributable to differences in cytotoxicity between WT and S20G amylin. Nevertheless, significant differences between the expression of either MUT or RAT and S20G and WT were observed at later time points when significant apoptosis occurred. At 48 hours there were no significant differences among MUT, RAT, WT, and S20G amylin levels. At 72 hours, the levels of WT and S20G were both significantly lower than that of MUT or RAT (P < 0.01). At 96 hours, the levels of WT and S20G amylin were significantly lower than that of MUT (P < 0.05) but not that of RAT. The general trend for decreased expression of all peptides from 48 to 96 hours may be attributed in part to the degradation of the plasmid DNA, 16 effectively reducing the levels of DNA template from which to generate mRNA. However, the larger decline in amylin levels in cells transfected with either the S20G or WT expression cassettes are more likely attributed to increased DNA degradation, decreased protein synthetic capacity, and increased protein degradation as a result of the apoptosis that is induced in these cells. 17 Attempts to measure the amounts of amylin secreted by the cells were prevented because of the presence of an interfering substance in the medium. However, as we have previously demonstrated that the secreted extracellular amylin is not cytotoxic, 11 the relative levels of intracellular amylin are more important in comparing the relative expression. It should be emphasized that because of the apoptosis occurring in the later time points, increased DNA and protein degradation as well as decreased protein synthetic capacity may skew the normalized amylin content values. Accordingly, determination of the amylin contents at 48 hours, before any significant apoptosis, provides the best measure of assessing individual peptide expression in the various transfected cells.
Figure 4.

RIA analysis of intracellular WT (solid square), S20G (solid circle), MUT (solid triangle), and RAT (solid diamond) amylin peptide expression in COS-1 cell lysates after transection with the appropriate expression cassette for 48, 72, and 96 hours. Statistically significant differences are denoted by single (P < 0.05) and double (P < 0.01) asterisks. The data represent the average of seven independent experiments. Error bars represent standard errors.
In Vitro Characterization of Amyloid Formation by WT and SG20 Amylin
To assess the amyloidogenic properties of WT and S20G directly, we measured ThT binding to in vitro-generated amyloid fibrils from the two synthetic peptides. This assay has been well characterized for a variety of amyloidogenic substrates 18-20 and we have used it previously to monitor the amyloid formation by WT amylin 21 and the Alzheimer Aβ1-42 peptide. 15 We used newly synthesized WT and S20G amylin that were prepared under identical reaction and purification conditions, as well as oxidation of the C2-C7 disulfide bond. Unlike our previous WT peptide that required BSA and Triton X-100 in the reaction mixture to enable observation of the efficient generation of amyloid fibrils that bound ThT, the newly synthesized peptides formed amyloid efficiently in the absence of BSA/Triton X-100. In the absence of BSA/Triton X-100, there was a well-behaved time- and concentration-dependent (25, 50, and 100 μmol/L) increase in ThT binding up to 8 hours after initiating the reaction (Figure 5) ▶ . Although the data in Figure 5 ▶ suggest that the reaction had reached a plateau by 8 hours, there were much smaller increases in the fluorescence up to 48 hours (data not shown). The exact reason for the difference in peptide behavior in the different buffer conditions is not known, but we suspect that more efficient C2-C7 disulfide bond formation in the latest preparations may have reduced amorphous aggregation, that can lead to a loss of substrate for the generation of amyloid fibrils. All of the studies reported here were performed in the absence of BSA/Triton X-100.
Figure 5.

In vitro generation of amyloid by WT and S20G synthetic peptides as assessed by a ThT binding assay. Open and closed squares represent 10 μmol/L and 25 μmol/L of WT, respectively Open and closed circles represent 10 and 25 μmol/L of S20G, respectively. The data represent the average results from three independent experiments. Error bars represent standard errors.
After an initial lag of ∼1 hour ThT fluorescence of WT amylin showed nearly linear increases up to 8 hours (Figure 5) ▶ . In contrast, there was a much greater increase in the fluorescence signal with S20G amylin. After a 30-minute lag the S20G fluorescence signal increased more rapidly than that of WT and nearly attained the maximal levels after 2 hours (Figure 5) ▶ . The maximum fluorescence signal attained by S20G was approximately twofold higher that of WT. T1/2max (time to achieve half-maximal fluorescence signal) of S20G and WT were 1 and 3 hours, respectively. With both peptides, the kinetics showed dose-dependent behavior and both exhibited lag-times before the main amyloid assembly reaction leading to a plateau. The lag time probably reflects the time necessary for significant seed formation, after which the formation of amyloid proceeds relatively rapidly. 22 These results confirm our previous preliminary report that S20G had significantly higher affinity than WT in a Congo red binding absorption assay (unpublished results). These results indicate that S20G is more highly amyloidogenic than WT amylin.
Electron micrographs of the in vitro-generated amyloid from WT and S20G amylin are shown in Figure 6, A and B ▶ . In both cases filamentous amyloid fibers were detected with diameters between 5 to 10 nm and fibril windings and branches could be observed. No significant morphological differences were observed between WT or S20G amyloid, confirming previous preliminary data (unpublished results).
Figure 6.
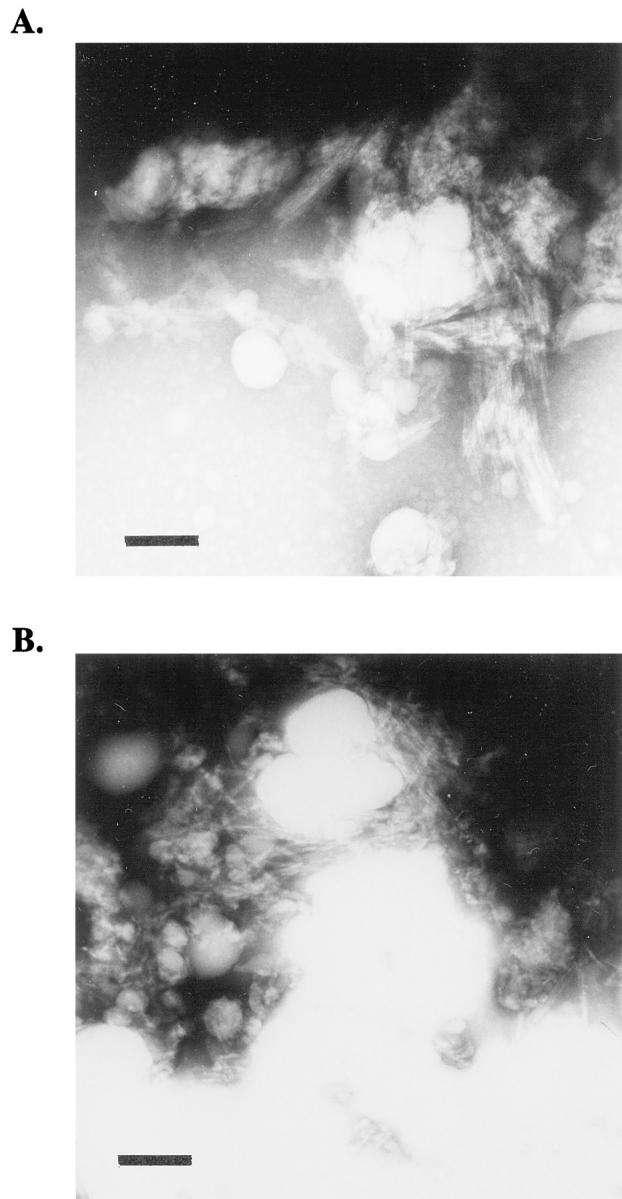
Electron microscopy of in vitro-generated amyloid fibers from synthetic WT (A) and S20G (B) amylin. Scale bars, 100 nm.
Discussion
In the present study, we have compared the cytotoxic and amyloidogenic properties of human WT and S20G mutant amylin. The studies address the hypothesis that altered S20G amyloidogenic properties might represent the basis for its association with early onset, more severe NIDDM. Examination of 294 unrelated patients with NIDDM in the Japanese population revealed that the S20G mutation was present with a relative frequency of 4.1%. 4 However, when limited to relatively early-onset diabetes with age of onset (<35 years), the relative frequency increased to ∼10%. From clinical data and pedigree analysis of the affected patients, it seems that the S20G mutation may create mild glucose intolerance on its own. However, when this mutation is combined with other presently unknown susceptibility gene(s) for late-onset NIDDM, it may contribute to accelerate the age of onset and severity of disease. Recent studies from our laboratory have raised the question whether WT amylin itself might contribute to the pathogenesis of NIDDM through its inherent tendency to form amyloid fibrils which may disturb β cell function, possibly leading to the apoptosis of affected cells. 10,11 Although the mechanism(s) by which S20G amylin contributes to the development of NIDDM is unclear, we reasoned that comparative studies of the amyloidogenicity and cellular cytotoxicity of WT and S20G amylin would be a first step in examining its potential role in the development of NIDDM. That the amyloidogenic properties of the mutant amylin might be a potential pathogenic factor, is supported by the fact that the relatively hydrophobic amino acid 20 to 29 region has been shown to be responsible for amyloid formation. 9 Thus the S20G mutation would result in an even more hydrophobic amyloidogenic domain, suggesting that this peptide might be more highly fibrillogenic. Therefore, in this study we compared the cytotoxicity of intracellular S20G with WT amylin expression in COS-1 cells and assessed their ability to form amyloid fibrils in vitro.
As in our previous studies apoptosis was induced in COS-1 cells at 72 to 96 hours after transfection and increased apoptosis was only observed with amylin peptides that were amyloidogenic. 10,11 Although S20G-induced apoptosis was greater than WT at 72 hours, this difference did not reach statistical significance until 96 hours (Figure 3) ▶ . That the increased apoptosis was because of enhanced intracellular cytotoxicity of S20G was confirmed by analysis of intracellular peptide expression levels by RIA, which established that equivalent WT and S20G expression occurred over the course of the study (Figure 4) ▶ . At 48 hours the levels of S20G, WT, RAT, and MUT peptides were indistinguishable. However, by 72 and 96 hours WT and S20G peptide expression decreased sharply and was coincident with the induction of apoptosis. These findings suggested that expression of the amyloidogenic WT and S20G might retard normal cellular metabolism. Whether such metabolic perturbation precedes or follows the induction of apoptosis is not known; however, the data suggest that reduction of β-cell protein synthetic capacity, including that of insulin, before the onset of apoptosis could represent another mechanism by which intracellular amyloid formation could contribute to the pathogenesis of NIDDM. Like our previous studies, 10,11 the levels of secreted WT and S20G amylin detected in cell culture medium by RIA were several thousand-fold lower than the levels of WT amylin required to induce cell death by adding amylin directly to cultured cells. 23,24 Thus, like WT the cytotoxicity of S20G amylin seems to be because of its intracellular expression and possible accumulation of intracellular amyloid.
In considering mechanisms by which amylin may contribute to the genesis of NIDDM, the amyloidogenic properties of the peptide are a prominent feature. NIDDM patients demonstrate the characteristic islet amyloidosis in high prevalence. 8 On the other hand in rodents that express a nonamyloidogenic amylin, islet amyloidosis is not observed. Although it has long been considered that the amyloidosis observed in post mortem pancreatic islets is a bystander effect unrelated to the pathogenesis of NIDDM, recent advances in other diseases associated with amyloidoses are causing reexamination of this issue. For example, rare mutations in the APP gene which result in the increased proteolytic processing and generation of the highly amyloidogenic Aβ1-42 peptides have been clearly associated with premature onset Alzheimer’s diseases 25 and evidence for increased levels of these amyloidogenic peptides in Alzheimer’s disease has been found. 26,27 Accordingly, our finding that S20G peptides form amyloid in vitro rapidly and more efficiently than WT amylin (Figure 5) ▶ , provides a strong association between the cytotoxicity after intracellular expression (Figure 3) ▶ and the association of the S20G mutation with early-onset and severe NIDDM in individuals with a family history of late-onset NIDDM.
It should be emphasized that although our data indicate that increased amyloidogenicity is associated with increased cytotoxicity, the data do not allow the conclusion that amyloid fibrils per se are the pathogenic factor. Several lines of evidence gathered from other diseases that are associated with the accumulation of amyloid fibrils have given rise to the suggestion that a protofibrillar intermediate might be the pathogenic factor. 28 According to this model the amyloid produced from the protofibrillar intermediate might even serve as a protective mechanism, because it would serve to remove or neutralize the protofibrils. In support of this concept, studies of β cells of transgenic mice homozygous for the human amylin gene, which spontaneously develop diabetes mellitus, seem to lack amyloid deposits, but do contain amorphous intra- and extracellular aggregates. 29 In addition, smaller islet amyloid polypeptide aggregates derived from freshly prepared amylin solutions, but not larger, mature amyloid deposits, have been shown to be cytotoxic when added to dispersed mouse and human islet cells, provoking the formation of abnormal vesicle-like membrane structures in association with vacuolization and cell death. 28
Several groups have reported that there exists a racial difference in prevalence and clinical character of the patients who possess the S20G mutation. 30-32 It is natural that the onset of NIDDM is affected by racial background and circumstances because it is a multifactorial and genetically heterogeneous disorder. Needless to say, it is not a common mutation, nor is it likely to represent a single gene that can cause diabetes, because there is incomplete phenotypic penetration in individuals that possess the genotype. 4 Thus the S20G mutation seems to mimic the situation observed with MODY, because each of the genes that have been shown to be involved in the pathogenesis of this diseases accounts for only a few percent of all MODY cases and the clinical features of MODY also vary among different races. 33-36
In summary we have compared the intracellular cytotoxicity and in vitro amyloidogenicity of human WT and S20G amylin. In comparison to WT, S20G amylin possessed elevated amyloidogenicity and increased intracellular cytotoxicity in transfected COS-1 cells. These studies suggest that the increased amyloidogenicity and intracellular cytotoxicity may be linked to the pathogenesis of early onset, relatively severe NIDDM found in patients with this genotype, who also have a family history of late-onset NIDDM. Further studies will be required to understand this relationship in more detail.
Acknowledgments
We thank James Tarara, Holly Lamb, Teresa Halsey, Colleen Moe, and Chris Anderson for assistance with the flow cytometry; Dr. Daniel McCormick and Robert Oda for synthesis and purification of WT and S20G peptides; Jon Charlesworth for the technical assistance with electron microscopy; and Ruth Kiefer for editorial and secretarial assistance.
Footnotes
Address reprint requests to Norman L. Eberhardt, Division of Endocrinology, Department of Medicine, 5-194 Joseph, Mayo Clinic, 200 First St., SW, Rochester, MN 55905. E-mail: eberhardt@mayo.edu.
This study was supported in part by a National Institutes of Health Grant DK56890 (to N. L. E.), a Grant-in Aid for Creative Basic Research (10NPO201) from the Ministry of Education, Science, Sports, and Culture, Japan (to K. N.), and Health Science Grants from the Ministry of Health and Welfare in Japan (to T.S.).
References
- 1.Polonsky KS, Sturis J, Bell GI: Seminars in: medicine of the Beth Israel Hospital, Boston. Non-insulin-dependent diabetes mellitus—a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med 1996, 334:777-783 [DOI] [PubMed] [Google Scholar]
- 2.Kahn CR, Vicent D, Doria A: Genetics of non-insulin-dependent (type-II) diabetes mellitus. Annu Rev Med 1996, 47:509-531 [DOI] [PubMed] [Google Scholar]
- 3.Guazzarotti L, Bartolotta E, Chiarelli F: Maturity-onset diabetes of the young (MODY): a new challenge for pediatric diabetologists. J Pediatr Endocrinol Metab 1999, 12:487-497 [DOI] [PubMed] [Google Scholar]
- 4.Sakagashira S, Sanke T, Hanabusa T, Shimomura H, Ohagi S, Kumagaye KY, Nakajima K, Nanjo K: Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 1996, 45:1279-1281 [DOI] [PubMed] [Google Scholar]
- 5.Sanke T, Bell GI, Sample C, Rubenstein AH, Steiner DF: An islet amyloid peptide is derived from an 89-amino acid proteolytic processing. J Biol Chem 1988, 263:17243-17246 [PubMed] [Google Scholar]
- 6.Nishi M, Sanke T, Nagamatsu S, Bell GI, Steiner DF: Islet amyloid polypeptide. A new beta cell secretory product related to islet amyloid deposits. J Biol Chem 1990, 265:4173-4176 [PubMed] [Google Scholar]
- 7.Clark A, Charge SB, Badman MK, MacArthur DA, de Koning EJ: Islet amyloid polypeptide: actions and role in the pathogenesis of diabetes. Biochem Soc Trans 1996, 24:594-599 [DOI] [PubMed] [Google Scholar]
- 8.O’Brien TD, Butler PC, Westermark P, Johnson KH: Islet amyloid polypeptide: a review of its biology and potential roles in the pathogenesis of diabetes mellitus. Vet Pathol 1993, 30:317-332 [DOI] [PubMed] [Google Scholar]
- 9.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C: Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA 1990, 87:5036-5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Brien TD, Butler PC, Kreutter DK, Kane LA, Eberhardt NL: Human islet amyloid polypeptide expression in COS-1 cells. A model of intracellular amyloidogenesis. Am J Pathol 1995, 147:609-616 [PMC free article] [PubMed] [Google Scholar]
- 11.Hiddinga HJ, Eberhardt NL: Intracellular amyloidogenesis by human islet amyloid polypeptide induces apoptosis in COS-1 cells. Am J Pathol 1999, 154:1077-1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Hoff MJ, Moorman AF, Lamers WH: Electroporation in ’intracellular’ buffer increases cell survival. Nucleic Acids Res 1992, 20:2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanengeland M, Ramaekers FCS, Schutte B, Reutelingsperger CPM: A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24:131-139 [DOI] [PubMed] [Google Scholar]
- 14.Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Westermark P, Adrian TE: Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med 1994, 330:313-318 [DOI] [PubMed] [Google Scholar]
- 15.Kudva YC, Hiddinga HJ, Butler PC, Mueske CS, Eberhardt NL: Small heat shock proteins inhibit in vitro A-beta(1–42) amyloidogenesis. FEBS Lett 1997, 416:117-121 [DOI] [PubMed] [Google Scholar]
- 16.Alwine JC: Transient gene expression control: effects of transfected DNA stability and trans-activation by viral early proteins. Mol Cell Biol 1985, 5:1034-1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori K: Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101:451-454 [DOI] [PubMed] [Google Scholar]
- 18.Naiki H, Higuchi K, Hosokawa M, Takeda T: Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem 1989, 177:244-249 [DOI] [PubMed] [Google Scholar]
- 19.Naiki H, Higuchi K, Matsushima K, Shimada A, Chen WH, Hosokawa M: Fluorometric examination of tissue amyloid fibrils in murine senile amyloidosis: use of the fluorescent indicator, thioflavine T. Lab Invest 1990, 62:768-773 [PubMed] [Google Scholar]
- 20.Naiki H, Higuchi K, Nakakuki K, Takeda T: Kinetic analysis of amyloid fibril polymerization in vitro. Lab Invest 1991, 65:104-110 [PubMed] [Google Scholar]
- 21.Kudva YC, Mueske C, Butler PC, Eberhardt NL: A novel in vitro assay of human islet amyloid polypeptide amyloidogenesis and effects of insulin secretory vesicle peptides on amyloid formation. Biochem J 1998, 331:809-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harper JD, Lansbury PT, Jr: Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 1997, 66:385-407 [DOI] [PubMed] [Google Scholar]
- 23.May PC, Boggs LN, Fuson KS: Neurotoxicity of human amylin in rat primary hippocampal cultures: similarity to Alzheimer’s disease amyloid-beta neurotoxicity. J Neurochem 1993, 61:2330-2333 [DOI] [PubMed] [Google Scholar]
- 24.Lorenzo A, Razzaboni B, Weir GC, Yankner BA: Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368:756-760 [DOI] [PubMed] [Google Scholar]
- 25.Hardy J, Allsop D: Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 1991, 12:383-388 [DOI] [PubMed] [Google Scholar]
- 26.Eckman CB, Mehta ND, Crook R, Perez-tur J, Prihar G, Pfeiffer E, Hinder P, Yager D, Zenk B, Refolo LM, Prada CM, Hutton M, Hardy J: A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43). Hum Mol Genet 1997, 6:2087-2089 [DOI] [PubMed] [Google Scholar]
- 27.Golde TE, Cai XD, Shoji M, Younkin SG: Production of amyloid beta protein from normal amyloid beta-protein precursor (beta APP) and the mutated beta APPS linked to familial Alzheimer’s disease. Ann NY Acad Sci 1993, 695:103-108 [DOI] [PubMed] [Google Scholar]
- 28.Lansbury PT, Jr: Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci USA 1999, 96:3342-3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK: Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA 1996, 93:7283-7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birch CL, Fagan LJ, Armstrong MJ, Turnbull DM, Walker M: The S20G islet-associated polypeptide gene mutation in familial NIDDM. Diabetologia 1997, 40:1113. [PubMed] [Google Scholar]
- 31.Yamada K, Yuan X, Ishiyama S, Nonaka K: Glucose tolerance in Japanese subjects with S20G mutation of the amylin gene. Diabetologia 1998, 41:125. [DOI] [PubMed] [Google Scholar]
- 32.Chuang LM, Lee KC, Huang CN, Wu HP, Tai TY, Lin BJ: Role of S20G mutation of amylin gene in insulin secretion, insulin sensitivity, and type II diabetes mellitus in Taiwanese patients. Diabetologia 1998, 41:1250-1251 [DOI] [PubMed] [Google Scholar]
- 33.Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, Lesage S, Stoffel M, Takeda J, Passa P, Permutt A, Beckmann JS, Bell GI, Cohen D: Familial hyperglycemia due to mutations in glucokinase. Definition of a subtype of diabetes mellitus. N Engl J Med 1993, 328:697-702 [DOI] [PubMed] [Google Scholar]
- 34.Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, Fajans SS, Signorini S, Stoffel M, Bell GI: Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 1996, 384:458-460 [DOI] [PubMed] [Google Scholar]
- 35.Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Cox RD, Lathrop GM, Boriraj VV, Chen X, Cox NJ, Oda Y, Le Beau MM, Yamada S, Nishigori H, Takeda J, Fajans SS, Iwasaki N, Hansen T, Pedersen O, Polonsky KS, Bell GI: Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 1996, 384:455-458 [DOI] [PubMed] [Google Scholar]
- 36.Iwasaki N, Oda N, Ogata M, Hara M, Hinokio Y, Oda Y, Yamagata K, Ohgawara H, Omori Y, Bell GI: Mutations in the hepatocyte nuclear factor-1alpha/MODY3 gene in Japanese subjects with early- and late-onset NIDDM. Diabetes 1997, 46:1504-1508 [DOI] [PubMed] [Google Scholar]