

Abstract
Proteases and their inhibitors play key roles in physiological and pathological processes. Cerebral amyloid plaques are a pathological hallmark of Alzheimer’s disease (AD). They contain amyloid-β (Aβ) peptides in tight association with the serine protease inhibitor α1-antichymotrypsin. 1,2 However, it is unknown whether the increased expression of α1-antichymotrypsin found in AD brains counteracts or contributes to the disease. We used regulatory sequences of the glial fibrillary acidic protein gene 3 to express human α1-antichymotrypsin (hACT) in astrocytes of transgenic mice. These mice were crossed with transgenic mice that produce human amyloid protein precursors (hAPP) and Aβ in neurons. 4,5 No amyloid plaques were found in transgenic mice expressing hACT alone, whereas hAPP transgenic mice and hAPP/hACT doubly transgenic mice developed typical AD-like amyloid plaques in the hippocampus and neocortex around 6 to 8 months of age. Co-expression of hAPP and hACT significantly increased the plaque burden at 7 to 8, 14, and 20 months. Both hAPP and hAPP/hACT mice showed significant decreases in synaptophysin-immunoreactive presynaptic terminals in the dentate gyrus, compared with nontransgenic littermates. Our results demonstrate that hACT acts as an amyloidogenic co-factor in vivo and suggest that the role of hACT in AD is pathogenic.
The major protein component of plaques in Alzheimer’s disease (AD) brains is amyloid β (Aβ), which is derived proteolytically from the amyloid protein precursor. 6-8 The majority of Aβ produced in the normal brain terminates at amino acid 40 (Aβ40). 9 Mutations linked to autosomal dominant forms of familial AD (FAD) and formation of plaques in all forms of AD are associated with an abnormal accumulation of Aβ ending at amino acid 42 (Aβ42), which aggregates more readily than Aβ40. 10,11
High-level neuronal expression of FAD-mutant forms of human amyloid precursor protein (hAPP)/Aβ, directed by the platelet-derived growth factor (PDGF) β chain promoter, elicits age-related synaptic transmission deficits and AD-like neuropathological alterations in transgenic mice, including typical amyloid plaques. 4,5,12-15 AD-like pathology has also been observed in a variety of other transgenic models in which neuronal expression of FAD-mutant hAPP is directed by different promoters. 16
Diverse proteins bind to amyloid plaques including apolipoprotein E, extracellular matrix proteins, amyloid P component, complement, and cytokines. 17 Most of these proteins also bind to other forms of amyloid (eg, in primary AL amyloidosis, secondary AA amyloidosis, FAP amyloidosis, or prion-associated spongiform encephalopathies). In contrast, human α1-antichymotrypsin (hACT) seems to be associated primarily with Aβ amyloidosis, 18 suggesting a more specific role for hACT in AD pathogenesis. However, this role remains to be determined.
ACT is a serine protease inhibitor (serpin) and an acute phase protein. 19 In AD, hACT mRNA levels are increased in the cortex 1 where hACT is produced primarily by astrocytes. 20,21 In vitro, hACT increases, 22,23 decreases, 24,25 or does not alter 17,26 amyloid fibril formation. Similarly controversial results have been reported for the effects of hACT on Aβ-induced neurotoxicity. 27-30 Here we demonstrate that astroglial expression of hACT increases amyloid deposition in vivo and that this effect does not augment Aβ-induced synaptotoxicity.
Materials and Methods
Generation of Transgenic Mice
An ∼1.7-kb fragment containing a full-length hACT cDNA 1 was isolated from pGEM4 by EcoRI digest, subcloned in pGEMEX-2 after NotI linker ligation, and ligated via NotI with a glial fibrillary acidic protein (GFAP) expression construct (C-445) described previously. 31 The resulting GFAP-hACT transgene was freed of vector sequences by SfiI digestion, purified, and microinjected into C57Bl/6xSJL F2 one-cell embryos. The PDGF-hAPP transgene 12,13 and the generation of PDGF-hAPP line J9 on the C57Bl/6xDBA/2 background 4 have been described.
Mice were crossed as outlined in the Results section and genomic tail DNA was analyzed with a touchdown polymerase chain reaction protocol essentially as described. 32 hACT primers: forward (5′-CTCGAGCTCGAGAGTTAGTCCTGAAGGCCC-3′), reverse (5′-AGATCTAGATCTCGGAGGTGCTGGAAGCTC-3′). hAPP primers: forward (5′-GGTGAGTTTGTAAGTGATGCC-3′), reverse (5′-TCTTCTTCTTCCACCTCAGC-3′). Genotypes were confirmed by slot-blot analysis with 32 P-labeled cDNA probes specific for hAPP or hACT coding sequences. All transgenic mice were heterozygous with respect to individual transgenes. Nontransgenic littermates served as controls.
Preparation of Brain Tissues
For analysis, mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline. Brains were removed and divided sagittally. One hemibrain was postfixed in phosphate-buffered 4% paraformaldehyde (pH 7.4) at 4°C for 48 hours for vibratome sectioning at 40 μm; the other hemibrain was snap-frozen, either in its entirety or after rapid dissection into subregions, and stored at −70°C for RNA and protein analyses. Postmortem brain tissues from humans without neurological disease were obtained from the tissue bank of the Alzheimer’s Disease Research Center at the University of California at San Diego.
RNase Protection Assays
RNA extraction and mRNA quantitation by solution hybridization RNase protection assay were performed as described, 13 using 10 μg of total RNA per sample in combination with the following 32P-labeled antisense riboprobes (protected nucleotides are indicated with reference to GenBank accession numbers): hAPPSV40 containing nucleotides 2468 to 2657 (X06989) of hAPP fused by NotI linker with nucleotides 2532 to 2656 (M24914) of SV40, hACT containing nucleotides 200 to 380 (K01500) of hACT, and actin containing nucleotides 480 to 559 (X03672) of mouse β-actin sequence.
Primary Astrocyte Cultures
Cultures of primary astrocytes were established from whole brains of neonatal mice as described. 33 Cells were grown to confluence, washed in serum- and methionine-free Dulbecco’s modified Eagle’s medium, placed in t-80 flasks, and labeled with 4 ml of 35S-methionine (25 μCi/ml) for 18 hours. Conditioned media were then collected and spun at 100,000 × g for 1 hour. Supernatants were first precleared with preimmune serum followed by protein A Sepharose and spun at 3,000 rpm in an Eppendorf centrifuge for 5 minutes. The supernatants were collected and hACT was immunoprecipitated for 18 hours at 4°C with polyclonal goat (Atlantic, Stillwater, MN) or rabbit (MBL, Japan) anti-hACT antibodies (diluted 1:500). Complexes were brought down with protein A Sepharose for 1 hour at 4°C. The pellets were rinsed in STEN buffer, boiled in Laemmli sample buffer, and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The gels were then fixed, enhanced with Enlightening (New England Nuclear, Boston, MA), and autoradiographed. Similar results were obtained with either of the primary antibodies.
Quantitations of Aβ
Snap-frozen hippocampus was homogenized in guanidine buffer, and human Aβ (Aβ1-x versus Aβ1-42) was quantitated by enzyme-linked immunosorbent assay as described. 15 Sagittal vibratome sections (40 μm) of postfixed hemibrains were incubated overnight at 4°C with biotinylated mouse monoclonal antibody 3D6 (diluted to 5 μg/ml; Elan Pharmaceuticals, South San Francisco, CA), which specifically recognizes Aβ1-5. 15,34 Binding of primary antibody was detected with an Elite kit (Vector Laboratories, Burlingame, CA) using diaminobenzidine and H2O2 for development. Sections were counterstained with 1% hematoxylin and examined with a Vanox light microscope (Olympus, Tokyo, Japan) using a ×2.5 objective. The percent area of the hippocampus covered by 3D6-immunoreactive deposits (plaque load) was determined morphometrically with a Quantimet 570C (Leica, Deerfield, IL) in four immunolabeled sections per mouse. One section per mouse was immunostained and analyzed in each of four independent experiments and average values were calculated from the pooled data. Additional sections were pretreated with formic acid (99%) for 45 seconds and labeled with rabbit polyclonal antibodies (diluted 1:1,000; a gift from Dr. F. Checler, IPMC du CNRS, Valbonne, France) that specifically recognize the C-terminus of Aβ40 or Aβ42. 35 Specific binding of these antibodies was detected essentially as described. 34 For detection of mouse Aβ, sections were incubated with the rabbit polyclonal antibody RAT 1-28 (Elan Pharmaceuticals) and then with biotinylated goat anti-rabbit IgG. Immunoperoxidase activity was revealed with an Elite kit (Vector Laboratories) using diaminobenzidine and H2O2 for development.
Evaluation of Presynaptic Terminals
To ensure objective assessments and reliability of results, brain sections from mice to be compared in any given experiment were blind-coded and processed in parallel. Codes were broken after the analysis was complete. Vibratome sections were labeled with monoclonal antibodies against synaptophysin (1 μg/ml; Boehringer-Mannheim, Indianapolis, IN) or GAP-43 (1:100; Sigma) as described. 36 Synaptophysin-immunofluorescence-labeled sections were imaged with a laser-scanning confocal microscope (MRC1024; Bio-Rad Laboratories, Hercules, CA) as described. 12,14 Three sections were analyzed per mouse and four confocal images (each covering 7,282 μm2) of the molecular layer of the dentate gyrus were obtained per section. One section per mouse was immunostained and analyzed in each of three independent experiments and average values were calculated from the pooled data. Digitized images were transferred to a Macintosh computer and analyzed with NIH Image. For each experiment, we first determined the linear range of the intensity of immunoreactive terminals in nontransgenic control sections. This setting was then used, as described, 37 to collect all images analyzed in the same experiment. The area of the outer molecular layer occupied by synaptophysin-immunoreactive (SYN-IR) presynaptic terminals was quantified and expressed as a percentage of the total image area. 12,36 As reviewed recently, 5,37 this method has been used to assess neurodegenerative alterations in diverse experimental models and in diseased human brains and has been validated by comparisons with quantitative immunoblots, quantitations of synaptic proteins by enzyme-linked immunosorbent assay, and the optical disector approach. GAP-43-immunoperoxidase-labeled sections were analyzed microdensitometrically with the Quantimet 570C as described 36 to determine the level of GAP-43 immunoreactivity in the molecular layer of the dentate gyrus.
Statistical Analysis
Statistical analyses were performed with the StatView 5.0 program (SAS Institute Inc., Cary, NC). Differences among normally distributed sets of data were evaluated by one-way analysis of variance and Tukey-Kramer post hoc test. Differences in plaque load were assessed by the Mann-Whitney U test. Correlation studies were performed by simple regression analysis. The null hypothesis was rejected at the 0.05 level.
Results
Expression of hACT in Astrocytes of Transgenic Mice
We used regulatory sequences of the murine GFAP gene to target expression of hACT to astrocytes (Figure 1) ▶ . Six GFAP-hACT transgenic founders were identified and their offspring analyzed for cerebral transgene expression by RNase protection assay. Mice from the highest expresser line (528-13) had robust hACT mRNA levels in the brain (Figure 1b) ▶ , and primary astrocytes from these mice released hACT into the extracellular milieu (Figure 1c) ▶ . hACT mice displayed no overt behavioral phenotype, and inspection of their hematoxylin and eosin-stained brain sections revealed a normal cytoarchitecture (data not shown). Line 528-13 was selected for further analysis in this study.
Figure 1.
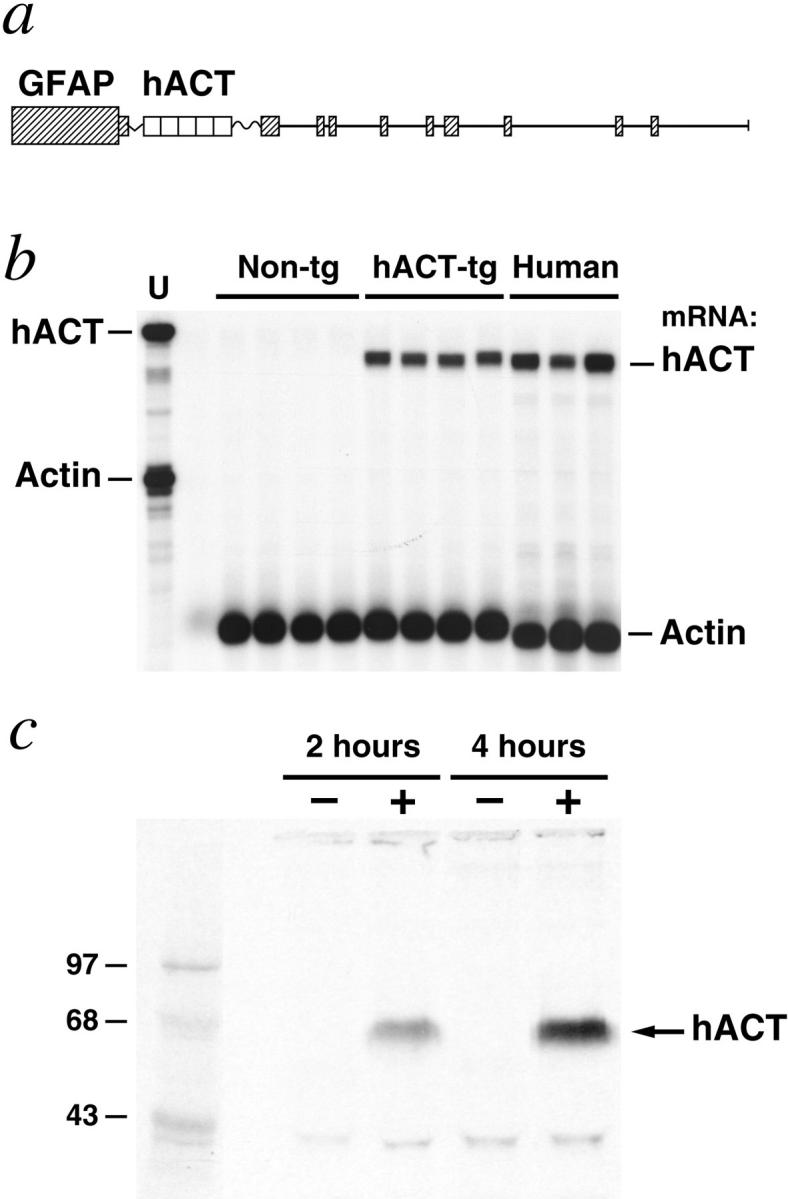
Expression of hACT in astrocytes of transgenic mice. a: Astroglial expression of an hACT cDNA 1 was directed by regulatory sequences of a modified murine GFAP gene. 3 SV40 polyadenylation signals at the 3′ end of the hACT cDNA prevent expression of downstream GFAP-coding sequences. Elements are not drawn to scale. b: hACT mRNA levels in brains of humans and transgenic mice. A representative autoradiograph is shown. Total RNA extracted from mouse hemibrains or from the midfrontal gyrus of humans without neurological disease was analyzed by RNase protection assay. The leftmost lane shows signals of undigested radiolabeled riboprobes. The other lanes contained the same riboprobes plus brain RNA (10 μg/lane) from different mice or humans, digested with RNases. Protected mRNA segments are indicated on the right. Non-tg = nontransgenic. Signals were quantitated by phosphorimager analysis: hACT/actin mRNA ratios in hACT mice (0.18, 0.15, 0.17, 0.18) were less variable than those in postmortem human brain tissues (0.18, 0.39, 0.48). c: Production of hACT by transgenic astrocytes. Primary astrocytes were established from transgenic (+) and nontransgenic (−) neonatal mice and metabolically labeled for 2 or 4 hours. hACT was immunoprecipitated with anti-hACT antibodies from conditioned culture medium, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and detected by autoradiography. Results similar to those shown were obtained with a different hACT antibody. The left lane contains 14C-labeled molecular weight standards.
Astroglial Expression of hACT Increases Amyloid Deposition in hAPP Transgenic Mice
Heterozygous GFAP-hACT transgenic mice (line 528-13) were crossed with heterozygous PDGF-hAPP transgenic mice (line J94,5) expressing an FAD-mutant hAPP minigene (Figure 2a) ▶ in neurons. These crosses yielded four groups of littermates (n = 20 to 21 per genotype): hACT mice, hAPP mice, hAPP/hACT mice, and nontransgenic controls.
Figure 2.

Cerebral transgene expression in hAPP/hACT mice and singly transgenic controls. a: Neuronal expression of an alternatively spliced hAPP minigene was directed by the human PDGF β-chain promoter as described previously. 12,13 The hAPP minigene expressed in line J9 carries FAD-linked mutations (670/671KM → NL and 717V → F, hAPP770 numbering) that increase the production of Aβ42. 4,5,10 Elements are not drawn to scale. b: hACT expression does not alter cerebral hAPP mRNA levels. A representative autoradiograph is shown. Total RNA extracted from mouse hemibrains was analyzed by RNase protection assay. Conventions are as in Figure 1b ▶ . The chimeric hAPPSV40 probe protects human but not mouse amyloid precursor protein; it also protects SV40 sequences in transgene-derived mRNAs. These SV40 sequences provide polyadenylation signals and are of slightly different length in the two transgenes allowing for differentiation of GFAP-hACT-derived (hACT(SV40)) and PDGF-hAPP-derived (hAPP(SV40)) mRNA segments. Signals were quantitated by phosphorimager analysis: singly and doubly transgenic mice (n = 4 per genotype) did not differ significantly in hAPP/actin (0.298 ± 0.053 versus 0.307 ± 0.096) or hACT(SV40)/actin (0.153 ± 0.021 versus 0.160 ± 0.012) mRNA ratios (mean ± SD).
Hippocampal deposition of Aβ was assessed in all four groups of mice at 7 to 8, 14, and 20 months of age. No amyloid deposits were detected in hACT mice or nontransgenic controls using antibodies that recognize human or mouse Aβ (data not shown). In contrast, hAPP and hAPP/hACT mice developed AD-like amyloid plaques around 7 months of age, and progressive accumulation of plaques was observed at 14 and 20 months (Figures 3 and 4) ▶ ▶ .
Figure 3.

Astroglial expression of hACT increases amyloid deposition in hAPP/hACT mice. Brain sections from hAPP mice (left) and hAPP/hACT mice (right) were labeled with the anti-Aβ antibody 3D6 at 7 or 14 months (m) of age. Aβ-immunoreactive deposits in the hippocampus were visualized by immunoperoxidase reaction and light microscopy.
Figure 4.

Quantitation of plaque load in hAPP and hAPP/hACT mice at different ages. Brain sections of hAPP and hAPP/hACT mice were labeled with the 3D6 antibody at 7 to 8 months (n = 9 per genotype), 14 months (n = 7 per genotype), or 20 months (n = 4 to 6 per genotype) of age. The hippocampal area occupied by Aβ deposits was greater in hAPP/hACT than in hAPP mice at all ages examined. Note the lower scale of the y axis in the youngest age group. Values represent group means ± SEM. *, P < 0.05 versus age-matched hAPP mice (Mann-Whitney U test).
The percent area of the hippocampus covered by Aβ-immunoreactive deposits (plaque load) was determined morphometrically as described in Materials and Methods. At all ages analyzed, hAPP/hACT mice had a higher plaque load than hAPP mice (Figure 4) ▶ . The distribution of the plaques was similar in both groups (Figure 3 ▶ and data not shown). Diffuse amyloid deposits first developed in a laminar distribution in the molecular layer of the dentate gyrus and the hippocampal alveus, followed by the formation of more mature compact plaques (3 to 15 μm in diameter) in the stratum radiatum of the hippocampus, the subiculum, and the neocortex. Areas that are relatively spared in AD, such as the thalamus, basal ganglia, and cerebellum, displayed only occasional small amyloid deposits (≤5 μm in diameter) in these mice. Plaques in both hAPP and hAPP/hACT mice showed stronger immunoreactivity for Aβ42 than for Aβ40 (data not shown), consistent with results obtained in humans. 11 In contrast to hACT mice and nontransgenic controls, hAPP and hAPP/hACT mice developed a reactive astrocytosis that was most prominent at 20 months of age and of comparable magnitude (data not shown).
Similar Transgene Expression and Aβ Production in hAPP/hACT and hAPP Mice before Plaque Formation
The increased plaque load in hAPP/hACT mice could result from effects of hACT on the production, removal, or aggregation of Aβ. Expression of hAPP and hACT in hAPP/hACT mice did not alter the expression levels of either transgene compared with singly transgenic controls (Figure 2b) ▶ . To determine whether hACT affects the production or clearance of soluble Aβ, hippocampal steady-state levels of human Aβ1-x (approximates total Aβ) and Aβ1-42 were measured by enzyme-linked immunosorbent assay. 15 To avoid confounding contributions of Aβ released from plaques during the homogenization of tissues, these measurements were performed at 5 weeks of age, which is before plaques are detected in hAPP and hAPP/hACT mice. No significant differences were identified between hAPP and hAPP/hACT mice (n = 6 per genotype) in Aβ1-x (42.9 ± 3.7 versus 45.4 ± 3.0) or Aβ1-42 (6.7 ± 0.4 versus 7.1 ± 0.4) levels (means ± SD in nmol/L). Aβ1-42/Aβ1-x ratios in hAPP and hAPP/hACT mice were also similar (0.157 ± 0.008 versus 0.158 ± 0.008). Thus, hACT does not increase the overall tissue levels of Aβ before plaque formation.
Synaptotoxicity Depends on hAPP/Aβ but Not on hACT or Plaque Load
One of the best neuropathological correlates of cognitive decline in AD is the loss of SYN-IR presynaptic terminals in specific brain regions. 38-42 Increased expression of Aβ in PDGF-hAPP mice is associated with a significant decrease in SYN-IR presynaptic terminals in the outer molecular layer of the dentate gyrus. 4,12
Nontransgenic controls and hACT mice without hAPP/Aβ expression had comparable levels of SYN-IR presynaptic terminals (Figure 5a) ▶ , suggesting that expression of hACT does not by itself affect the integrity of these structures. In contrast, hAPP mice with or without hACT expression had decreased levels of SYN-IR presynaptic terminals, but the presence or absence of hACT in these mice did not significantly affect the extent of neurodegeneration (Figure 5a) ▶ . Similar results were obtained for growth-associated protein 43 (GAP-43) (Figure 5b) ▶ , another marker of synaptic integrity. 43 No correlation was identified between the density of SYN-IR presynaptic terminals and the hippocampal plaque load in hAPP or hAPP/hACT mice (Figure 5c) ▶ .
Figure 5.

Comparable levels of synaptic damage in hAPP and hAPP/hACT mice. The density of SYN-IR presynaptic terminals (a) and the level of GAP-43 immunoreactivity (b) in the molecular layer of the dentate gyrus were determined in nontransgenic controls, singly transgenic mice (hAPP or hACT) and hAPP/hACT doubly transgenic mice (n = 11 to 13 mice per genotype) at 14 to 20 months of age. Data represent group means ± SD. *, P < 0.05 versus nontransgenic controls (Tukey-Kramer test). c: The density of SYN-IR presynaptic terminals did not correlate with the plaque load in hAPP (P = 0.074) or hAPP/hACT (P = 0.88) mice. At 7 to 8 months of age (n = 9 mice per genotype), the density of SYN-IR presynaptic terminals was also similar in hAPP (24.7 ± 1.4) and hAPP/hACT (24.2 ± 1.9) mice and it did not correlate with plaque load (P = 0.76).
Discussion
We have demonstrated in an hAPP transgenic mouse model that the serpin hACT increases the age-dependent accumulation of cerebral amyloid plaques. This finding may be relevant to AD where hACT is overexpressed in the brain 1 and present in most, if not all, amyloid plaques. 44 Previous studies that examined the effect of hACT on Aβ fibrillization in vitro yielded conflicting results. 17,22-26 Our study demonstrates that astroglial overproduction of hACT is clearly amyloidogenic in vivo, which suggests that it promotes rather than inhibits the development of AD.
Before plaque formation, similar steady-state levels of Aβ1-x and Aβ1-42 were found in the hippocampus of hAPP and hAPP/hACT mice. This indicates that hACT does not increase the production or decrease the degradation of soluble Aβ, at least not in young mice. It is possible that hACT promotes the aggregation of Aβ and its deposition into the brain parenchyma or that it interferes with the degradation and removal of Aβ aggregates. These mechanisms are not mutually exclusive and could involve direct interactions between hACT and Aβ, 22,23,45 as well as indirect effects of hACT on the levels or activities of extracellular matrix proteins, 46 pathological chaperones, 47 or enzymes that may be involved in the deposition or clearance of amyloid. 48 Additional studies are required to differentiate among these possibilities.
Besides hACT, there are other factors that could influence the deposition of Aβ in AD without affecting Aβ production. Elimination of apolipoprotein E expression by crossing hAPP transgenic mice on the apoe knockout background prevents the formation of mature amyloid plaques. 49 Increased astroglial expression of the cytokine transforming growth factor-β1 results in a redistribution of amyloid deposits from the brain parenchyma into blood vessels. 34,50 In contrast, other molecules that have been implicated in the turnover of Aβ by cell culture studies have not had a significant impact on amyloid load when examined in transgenic models. For example, genetic ablation of the class A scavenger receptor did not affect the amyloid burden in the same line of hAPP mice that was analyzed in the current study. 32
Although there is increasing acceptance of the notion that Aβ plays a pivotal role in AD pathogenesis, the relationship between amyloid plaques and neurodegenerative alterations remains controversial. 51-57 It is interesting in this context that hAPP and hAPP/hACT mice had similar decreases in SYN-IR presynaptic terminals in the dentate gyrus and that the density of these structures did not correlate with the hippocampal plaque load in either group. These findings suggest that Aβ-induced synaptotoxicity is independent of plaque formation, a conclusion supported also by neuropathological studies in humans, 56,57 electrophysiological recordings from hippocampal slices of hAPP mice, 4 and correlations between SYN-IR presynaptic terminals and Aβ levels across multiple lines of hAPP mice. 4,5 In contrast, other AD-associated alterations are likely plaque-dependent. For example, plaques in AD and hAPP mice are tightly associated with dystrophic neurites and reactive glial cells 5,12,14 and it is possible that neuritic dystrophy 58,59 and glial inflammatory responses 60 contribute to the development of AD dementia. In view of the amyloidogenic effect of hACT observed in the current study, it is tempting to speculate that blocking the production or activity of hACT could inhibit the accumulation of amyloid plaques in the aging brain and, thereby, help prevent the development of plaque-associated alterations.
Acknowledgments
We thank Dr. F. Checler for Aβ antibodies, Drs. T. Wyss-Coray and K. Weisgraber for helpful comments on the manuscript, G. Howard and S. Ordway for editorial assistance, J. Carroll and S. Gonzales for preparation of graphics, and D. McPherson for administrative assistance.
Footnotes
Address reprint requests to Lennart Mucke, M.D., Gladstone Institute of Neurological Disease, P.O. Box 41900, San Francisco, CA 94141-9100. E-mail: Lmucke@gladstone.ucsf.edu.
Supported by National Institutes of Health Grants AG11385 (to L. M.), AG09905 (to C. A.), AG5131 and AG10869 (to E. M.).
References
- 1.Abraham CR, Selkoe DJ, Potter H: Immunochemical identification of the serine protease inhibitor α1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell 1988, 52:487-501 [DOI] [PubMed] [Google Scholar]
- 2.Shoji M, Hirai S, Yamaguchi H, Harigaya Y, Ishiguro K, Matsubara E: Alpha1-antichymotrypsin is present in diffuse senile plaques. A comparative study of beta-protein and alpha1-antichymotrypsin immunostaining in the Alzheimer brain. Am J Pathol 1991, 138:247-257 [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson WB, Ruppe MD, Rockenstein EM, Price J, Sarthy VP, Verderber LC, Mucke L: Indicator expression directed by regulatory sequences of the glial fibrillary acidic protein (GFAP) gene: in vivo comparison of distinct GFAP-lacZ transgenes. Glia 1995, 13:174-184 [DOI] [PubMed] [Google Scholar]
- 4.Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 1999, 96:3228-3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L: High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000, 20:4050-4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selkoe DJ: Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399:A23-A31 [DOI] [PubMed] [Google Scholar]
- 7.Storey E, Cappai R: The amyloid precursor protein of Alzheimer’s disease and the Aβ peptide. Neuropathol Appl Neurobiol 1999, 25:81-97 [DOI] [PubMed] [Google Scholar]
- 8.Wilson CA, Doms RW, Lee VM-Y: Intracellular APP processing and Aβ production in Alzheimer disease. J Neuropathol Exp Neurol 1999, 58:787-794 [DOI] [PubMed] [Google Scholar]
- 9.Gouras GK, Xu HX, Jovanovic JN, Buxbaum JD, Wang R, Greengard P, Relkin NR, Gandy S: Generation and regulation of beta-amyloid peptide variants by neurons. J Neurochem 1998, 71:1920-1925 [DOI] [PubMed] [Google Scholar]
- 10.Younkin SG: Evidence that Aβ42 is the real culprit in Alzheimer’s disease. Ann Neurol 1995, 37:287-288 [DOI] [PubMed] [Google Scholar]
- 11.Hosoda R, Saido TC, Otvas LJ, Arai T, Mann DMA, Lee VMY, Trojanowski JQ, Iwatsubo T: Quantification of modified amyloid β peptides in Alzheimer disease and Down-syndrome brains. J Neuropathol Exp Neurol 1998, 57:1089-1095 [DOI] [PubMed] [Google Scholar]
- 12.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J: Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373:523-527 [DOI] [PubMed] [Google Scholar]
- 13.Rockenstein EM, McConlogue L, Tan H, Gordon M, Power M, Masliah E, Mucke L: Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of transgenic mice and humans with Alzheimer’s disease. J Biol Chem 1995, 270:28257-28267 [DOI] [PubMed] [Google Scholar]
- 14.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D: Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer’s disease. J Neurosci 1996, 16:5795-5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L: Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA 1997, 94:1550-1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price DL, Sisodia SS: Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 1998, 21:479-505 [DOI] [PubMed] [Google Scholar]
- 17.Webster S, Rogers J: Relative efficacies of amyloid β peptide (Aβ) binding proteins in Aβ aggregation. J Neurosci Res 1996, 46:58-66 [DOI] [PubMed] [Google Scholar]
- 18.Abraham CR, Shirahama T, Potter H: α1-antichymotrypsin is associated solely with amyloid deposits containing the β-protein. Amyloid and cell localization of α1-antichymotrypsin. Neurobiol Aging 1990, 11:123-129 [DOI] [PubMed] [Google Scholar]
- 19.Potempa J, Korzus E, Travis J: The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem 1994, 269:15957-15960 [PubMed] [Google Scholar]
- 20.Pasternack JM, Abraham CR, Van Dyke BJ, Potter H, Younkin SG: Astrocytes in Alzheimer’s disease gray matter express α1-antichymotrypsin mRNA. Am J Pathol 1989, 135:827-834 [PMC free article] [PubMed] [Google Scholar]
- 21.Koo EH, Abraham CR, Potter H, Cork LC, Price DL: Developmental expression of alpha1-antichymotrypsin in brain may be related to astrogliosis. Neurobiol Aging 1991, 12:495-501 [DOI] [PubMed] [Google Scholar]
- 22.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H: Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 1994, 372:92-94 [DOI] [PubMed] [Google Scholar]
- 23.Janciauskiene S, Eriksson S, Wright HT: A specific structural interaction of Alzheimer’s peptide 65 β1-42 with α1-antichymotrypsin. Nat Struct Biol 1996, 3:668-671 [DOI] [PubMed] [Google Scholar]
- 24.Fraser PE, Nguyen JT, McLachlan DR, Abraham CR, Kirschner DA: α1-Antichymotrypsin binding to Alzheimer Aβ peptides is sequence-specific and induces fibril disaggregation in vitro. J Neurochem 1993, 61:298-305 [DOI] [PubMed] [Google Scholar]
- 25.Eriksson S, Janciauskiene S, Lannfelt L: α1-antichymotrypsin regulates Alzheimer β-amyloid peptide fibril formation. Proc Natl Acad Sci USA 1995, 92:2313-2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes SR, Khorkova O, Goyal S, Knaeblein J, Heroux J, Riedel NG, Sahasrabudhe S: α2-macroglobulin associates with β-amyloid peptide and prevents fibril formation. Proc Natl Acad Sci USA 1998, 95:3275-3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma J, Brewer HB, Jr, Potter H: Alzheimer Aβ neurotoxicity: promotion by antichymotrypsin, apoE4; inhibition by Aβ-related peptides. Neurobiol Aging 1996, 17:773-780 [DOI] [PubMed] [Google Scholar]
- 28.Aksenov MY, Aksenova MV, Carney JM, Butterfield DA: α1-antichymotrypsin interaction with Aβ(1-42) does not inhibit fibril formation but attenuates the peptide toxicity. Neurosci Lett 1996, 217:117-120 [PubMed] [Google Scholar]
- 29.Aksenova MV, Aksenov MY, Butterfield DA, Carney JM: α-1-Antichymotrypsin interaction with Aβ (1-40) inhibits fibril formation but does not affect the peptide toxicity. Neurosci Lett 1996, 211:45-48 [DOI] [PubMed] [Google Scholar]
- 30.Schubert D: Serpins inhibit the toxicity of amyloid peptides. Eur J Neurosci 1997, 9:770-777 [DOI] [PubMed] [Google Scholar]
- 31.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L: Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature 1994, 367:188-193 [DOI] [PubMed] [Google Scholar]
- 32.Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas RE, Mucke L: Elimination of the class A scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. Am J Pathol 1999, 155:1741-1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyss-Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L: Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-β1. Am J Pathol 1995, 147:53-67 [PMC free article] [PubMed] [Google Scholar]
- 34.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L: Amyloidogenic role of cytokine TGF-β1 in transgenic mice and Alzheimer’s disease. Nature 1997, 389:603-606 [DOI] [PubMed] [Google Scholar]
- 35.Barelli H, Lebeau A, Vizzavona J, Deleare P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Buxbaum JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter J-M, Mohr M, Checler F: Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid β peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer’s disease and cerebral amyloid angiopathy cases. Mol Med 1997, 3:695-707 [PMC free article] [PubMed] [Google Scholar]
- 36.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR: Synaptotrophic effects of human amyloid β protein precursor in the cortex of transgenic mice. Brain Res 1994, 666:151-167 [DOI] [PubMed] [Google Scholar]
- 37.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW: Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J Neurosci 1999, 19:4867-4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R: Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991, 30:572-580 [DOI] [PubMed] [Google Scholar]
- 39.Honer WG, Dickson DW, Glesson J, Davies P: Regional synaptic pathology in Alzheimer’s disease. Neurobiol Aging 1992, 13:375-382 [DOI] [PubMed] [Google Scholar]
- 40.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R: Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 1994, 174:67-72 [DOI] [PubMed] [Google Scholar]
- 41.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P: Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 1995, 16:285-298 [DOI] [PubMed] [Google Scholar]
- 42.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ: Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 1997, 56:933-944 [DOI] [PubMed] [Google Scholar]
- 43.Masliah E, Fagan A, Terry R, DeTeresa R, Mallory M, Gage R: Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP43 in the dentate gyrus of the adult rat. Exp Neurol 1991, 113:131-142 [DOI] [PubMed] [Google Scholar]
- 44.Rozemuller JM, Abbink JJ, Kamp AM, Stam FC, Hack CE, Eikelenboom P: Distribution pattern and functional state of α1-antichymotrypsin in plaques and vascular amyloid in Alzheimer’s disease. Acta Neuropathol 1991, 82:200-207 [DOI] [PubMed] [Google Scholar]
- 45.Janciauskiene S, Rubin H, Lukacs CM, Wright HT: Alzheimer’s peptide Aβ1-42 binds to two β-sheets of α1-antichymotrypsin and transforms it from inhibitor to substrate. J Biol Chem 1998, 273:28360-28364 [DOI] [PubMed] [Google Scholar]
- 46.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG: An important role of heparan sulfate proteoglycan (perlecan) in a model system for the deposition and persistence of fibrillar a β-amyloid in rat brain. Neuron 1994, 12:219-234 [DOI] [PubMed] [Google Scholar]
- 47.Wisniewski T, Frangione B: Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 1992, 135:235-238 [DOI] [PubMed] [Google Scholar]
- 48.Yamin R, Malgeri EG, Sloane JA, McGraw WT, Abraham CR: Metalloendopeptidase EC 3.4.24.15 is necessary for Alzheimer’s amyloid-β peptide degradation. J Biol Chem 1999, 274:18777-18784 [DOI] [PubMed] [Google Scholar]
- 49.Bales KR, Verina R, Dodel RC, Du R, Altstiel L, Bender B, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM: Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet 1997, 17:263-264 [DOI] [PubMed] [Google Scholar]
- 50.Wyss-Coray T, Lacombe P, Von Euw D, Lin C, McConlogue L, Masliah E, Mucke L: TGF-β1 modulates brain amyloid deposition and induces Alzheimer’s disease like microvascular abnormalities in transgenic mice. Soc Neurosci Abstr 1999, 25:22 [Google Scholar]
- 51.Cummings BJ, Pike CJ, Shankle R, Cotman CW: β-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 1996, 17:921-933 [DOI] [PubMed] [Google Scholar]
- 52.Terry RD: The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 1996, 55:1023-1025 [PubMed] [Google Scholar]
- 53.Bartoo GT, Nochlin D, Chang D, Kim Y, Sumi SM: The mean Aβ load in the hippocampus correlates with duration and severity of dementia in subgroups of Alzheimer disease. J Neuropathol Exp Neurol 1997, 56:531-540 [DOI] [PubMed] [Google Scholar]
- 54.Davis JN, Chisholm JC: The ‘amyloid cascade hypothesis’ of AD: decoy or real McCoy? Trends Neurosci 1997, 20:558-559 [DOI] [PubMed] [Google Scholar]
- 55.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT: Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 1997, 41:17-24 [DOI] [PubMed] [Google Scholar]
- 56.Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J: Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 1999, 155:853-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL: Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 1999, 46:860-866 [DOI] [PubMed] [Google Scholar]
- 58.Knowles RB, Gomez-Isla T, Hyman BT: Aβ associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol 1998, 57:1122-1130 [DOI] [PubMed] [Google Scholar]
- 59.Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT: Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc Natl Acad Sci USA 1999, 96:5274-5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akiyama H, Barher S, Barnum S, Bradt B, Bauer J, Cooper NR, Eikelenboom P, Emmerling M, Fiebich B, Finch CE, Frautschy S, Griffin WST, Hampel H, Landreth G, McGeer PL, Mrak R, McKenzie I, O’Banion K, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyama I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T: Inflammation and Alzheimer’s disease. Neuroinflammation working group. Neurobiol Aging 2000, 21:383-421 [DOI] [PMC free article] [PubMed] [Google Scholar]