

Abstract
Mycobacterium tuberculosis catalase-peroxidase (Mtb KatG) is a bifunctional enzyme that possesses both catalase and peroxidase activities and is responsible for the activation of the antituberculosis drug isoniazid. Mtb KatG contains an unusual adduct in its distal heme pocket that consists of the covalently linked Trp107, Tyr229, and Met255. The KatG(Y229F) mutant lacks this adduct and has decreased steady-state catalase activity and enhanced peroxidase activity. In order to test a potential structural role of the adduct that supports catalase activity, we have used resonance Raman spectroscopy to probe the local heme environment of KatG(Y229F). In comparison to wild-type KatG, resting KatG(Y229F) contains a significant amount of 6-coordinate, low-spin heme and a more planar heme. Resonance Raman spectroscopy of the ferrous-CO complex of KatG(Y229F) suggest a non-linear Fe-CO binding geometry that is less tilted than in wild-type KatG. These data provide evidence that the Met-Tyr-Trp adduct imparts structural stability to the active site of KatG that seems to be important for sustaining catalase activity.
1. Introduction
The catalase-peroxidase of Mycobacterium tuberculosis (Mtb. KatG) is a bifunctional enzyme that exists as a homodimer of 80-kDa subunits, both of which bind one heme [1, 2]. Mtb. KatG is of great interest in the field of tuberculosis research because of its activation of isoniazid (INH), a potent antituberculosis drug which has been the cornerstone in tuberculosis chemotherapy for more than half a century since its discovery in 1952 [3]. Resistance to INH was observed immediately after its introduction and, in the majority of the clinical isolates, mutations on the katG gene encoding the catalase-peroxidase have been linked to INH resistance [4–6].
Mtb. KatG is classified as a class I peroxidase on the basis of sequence homologies with cytochrome c peroxidase (CCP) and ascorbate peroxidases (APXs), and the same amino acid triads Arg/Trp/His in the distal heme pocket and His/Trp/Asp in the proximal heme pocket that are critical for peroxidase activity, are conserved in these enzymes [7]. In addition, KatGs have a C-terminus, non heme-binding domain that probably results from gene duplication and three loop insertions (LL1, LL2, and LL3) that are absent from the class I peroxidases [8–11]. In Mtb KatG, one important loop (LL2) runs from Pro280 to Thr314 and terminates at Ser315, a residue that plays an important role in Mtb resistance to INH [4], while a second loop (LL1) runs from Glu195 to Asn231 and includes Tyr229 [8]. In Mtb KatG, Tyr229 is the central amino acid in a unique, covalently-linked three amino acid adduct, Trp107, Tyr229, and Met255, in its distal heme pocket [11]. This adduct seems to be conserved in catalase-peroxidases and has also been observed in Haloarcula marismortui and Burkholderia pseudomallei KatG [9,10,12,13]. It has been proposed that resting KatG must turnover through high valent intermediates to form the Met-Tyr-Trp crosslink [14]. This unique adduct is not found in monofunctional peroxidases, and mutagenesis studies have shown that it is required for catalase but not for peroxidase activity of KatG [10, 15–20]. In E.coli KatG, deletion of the entire LL1 loop insertion that contains the central Tyr of the adduct results in increased peroxidase activity and no detectable catalase activity, while peroxide-dependent inactivation was observed (Doug Goodwin, private communication).
Although it has been established that modification of the adduct lowers or abolishes the catalase function in KatG, it is not yet clear how the adduct supports catalase function in the enzyme. Both structural and mechanistic roles have been proposed. It has been suggested that the adduct holds in place two long loops on the surface of the enzyme that cover the substrate access channel to the active site [9] and that it may prevent further modification of the oxidation-susceptible methionine residue or contribute to the rigidity of the enzyme [10]. It has also been proposed that it plays a role in electron transfer from a potential substrate binding region to the heme [10] and that the sulfonium ion may modify pKa's, redox potentials, or both of Tyr299 or Trp107 [14]. It was recently suggested that Trp107 may form the binding site for the second H2O2 in the catalase reaction and that the adduct stabilizes its proper binding geometry [16,19,21]. Finally, Mtb. KatG(Y229F) has a significantly reduced affinity for INH [20], and it has been proposed that the adduct may be required for establishing the proper geometry of residues that contribute to the formation of a small-molecule binding site [10]. Although many proposals involve an expected structural perturbation in the active site upon disruption of the adduct, recent resonance Raman and EPR studies on Synechocystis KatG Y249F and Mtb KatG Y229F, respectively, reported only changes in heme spin state distributions compared to WT KatG [20,22,23], but no other evidence for structural changes has been reported.
Besides structural differences, there are also significant differences in the catalytic activity and substrate specificity between KatGs and class I peroxidases. KatGs exhibit both catalase activity comparable with catalases and typical peroxidase activity with broad specificity [24]. In the general peroxidase cycle, the resting enzyme reacts with H2O2 to form Compound I (Cmpd I), a ferryl (FeIV=O) porphyrin π cation radical, though the radical can appear on a nearby amino acid residue [25]. Reduction of Compound I by a substrate yields Compound II (Cmpd II), a ferryl porphyrin, which can further react with a substrate molecule to regenerate the resting enzyme. Cmpd I is the common intermediate in peroxidase and catalase cycles. In catalases, reduction of Cmpd I is accomplished by a second H2O2 molecule, resulting in the regeneration of the resting enzyme and release of molecular oxygen [25]. Since catalase-peroxidases share common heme-pocket features with peroxidases [26], it is not well understood how a Cmpd I intermediate in a catalase-peroxidase can react with hydrogen peroxide, while avoiding the reaction of H2O2 with Cmpd II which would result in Compound III (Cmpd III), FeII-O2 or FeIII-O2•−, formation and reduced catalase activity [25]. The structure of Compound II is not yet clear. Our resonance Raman data have indicated that Cmpd II is a ferryl porphyrin in WT Mtb KatG [27], but recent absorption stopped-flow studies have proposed that Cmpd II in WT KatG is a ferric low-spin heme with hydroxide as the sixth ligand and an unknown amino acid radical [(KatG•)FeIII-OH] [8, 14, 28, 29, 23].
Replacement of the redox active tyrosine Y229 with phenylalanine prevents the formation of the distal side adduct in Mtb. KatG. In this work, we use resonance Raman spectroscopy to study the effect of the Y229F mutation in Mtb. KatG and the loss of the cross-linked adduct on the conformation of the heme cofactor and its protein environment. We use the small exogenous ligands carbon monoxide (CO) and nitric oxide (NO) to probe modifications of the distal heme pocket. The vibrations associated with these ligands are useful to monitor steric hindrance, electrostatic interactions, and substrate binding in the distal heme pocket, because they are sensitive to changes in their local environment [30, 31]. Our study of Mtb KatG(Y229F) shows that the heme cofactor is more planar than in WT KatG and that the distal heme-pocket has been modified, probably, due to disruption of a hydrogen-bonding network. Our results underscore the importance of the integrity of the covalent adduct both in the structure and catalytic mechanism of the enzyme. The implications of our findings for the role of the adduct in KatG are discussed.
2. Experimental
2.1 Materials
CO (99.5%) and NO (99%) gases were purchased from Matheson Tri-Gas. 13CO (99%) and 15NO (98%) were purchased from Cambridge Isotope Laboratories. All other reagents were from Sigma-Aldrich.
2.2 Isolation and purification of M. tuberculosis KatG
The plasmid pKATII was used for site-directed mutagenesis to produce KatG(Y229F) as described previously [20,32,33]. The mutant was overexpressed in E. coli strain UM262 (KatG minus) expressing the M. tuberculosis katG gene provided by Stewart Cole (Institute Pasteur, Paris). The bacteria were grown in the presence of the heme biosynthetic precursor, δ-aminolevulinic acid. KatG(Y229F) was isolated and purified according to published procedures [32–34]. Purified enzyme was stored in 20 mM potassium phosphate buffer at pH 7.2. For the pH studies, KatG(Y229F) samples were exchanged into 20 mM citrate buffer at pH 5 or 20 mM borate buffer at pH 10.
2.3 Sample preparation
The samples for the UV-vis and resonance Raman experiments were prepared and contained in a spinning cell that was sealed with a rubber septum. Typically, 60 μl sample of 40 μM KatG was used for each experiment. Ferrous KatG (Y229F) was prepared by injection of buffered, anaerobic sodium dithionite solution into a spinning cell containing ferric enzyme under N2 atmosphere. The CO adduct of ferrous KatG(Y229F) was made by purging the cell with CO gas. The NO samples were prepared by injecting 200 μl NO gas into a spinning cell containing ferric KatG(Y229F) under N2 atmosphere. The NO gas was first bubbled through a saturated KOH solution, and 15NO was taken from a vial containing nitrogen and KOH solution. The formation of the KatG(Y229F)-CO and KatG(Y229F)-NO complexes was verified by UV-vis spectroscopy.
2.4 Electronic absorption and resonance Raman spectroscopy
Electronic absorption spectra were recorded at room temperature on a UV-vis spectrophotometer (Lambda 40 P, Perkin-Elmer). The resonance Raman (RR) spectra were obtained using a single spectrograph (TriAx 550, JY/Horiba) and a N2(l)-cooled CCD detector (Spectrum One, JY/Horiba) with a UV-enhanced 2048 x 512 pixels chip (EEV). The spectral resolution is 4 cm−1, and peak positions are determined with an accuracy of 1 cm−1. The samples were excited with 406.7 and 413.1 nm light from a Kr+ laser (Coherent, I-302) and kept at 6 ± 2 °C during the experiments. Rayleigh scattering was blocked by a 413.1 nm holographic notch filter (Kaiser Optical). The incident laser power was 10 and 1 mW for the KatG(Y229F)-NO and KatG(Y229F)-CO complexes, respectively. KatG(Y229F)-INH complexes were made by addition of 500 equivalents of INH. Background correction of RR spectra was done by subtraction of a smooth polynomial. Vibrations were labeled and assigned following the literature unless indicated differently [35–37], and toluene was used to calibrate the RR spectra.
3. Results
3.1 Ferric Mtb KatG(Y229F)
Figure 1 shows the high-frequency RR spectra of KatG(Y229F) excited at 406.7 nm at pH values of 5, 7.2, and 10, and in the presence of INH at pH 7.2. The heme vibrations above 1300 cm−1 are sensitive markers of the oxidation state, the coordination number, and the spin state of the heme iron [38]. The oxidation-state marker ν4 occurs at 1376 cm−1 for KatG(Y229F), which is typical of an Fe(III) heme and the same as for WT KatG [34]. The spin- and coordination-state marker ν3 is observed as a broad feature in KatG(Y229F). Four contributing bands at 1484, 1492, 1502 and 1509 cm−1 are revealed by curve fitting of the ν3 band as shown in Figure 2, and the curve fitting results of four independent experiments are listed in Table 1. The ν3 modes at 1484 and 1492 cm−1 are attributed to a 6-c HS and a 5-c HS ferric heme, respectively, and have slightly lower frequencies than the same species in WT KatG (1487 and 1495 cm−1) [34]. Although a 1492 cm−1 vibration has been associated with a 6-c QS heme in peroxidases at 12 K [39], we prefer the assignment of this vibration in KatG(Y229F) to a 5-c HS heme in our data that were obtained at 6 °C. This assignment is supported by that of a similar vibration to a 5-c HS heme in Coprinus cinereus peroxidase at room temperature [22] and by the absence of an identifiable 6-c QS heme signal in the EPR spectrum of Mtb KatG(Y229F) [20]. Furthermore, the EPR spectrum contains a signal due to 5-c, HS heme (g1 = 6.6 , g2 = 5.2) that is also present in WT Mtb KatG but with low intensity compared to the predominant 5-c, HS heme signal (g1 = 6.3, g2 = 5.14) [20]. The latter species is not found in EPR spectra of the Y229F mutant. Although heme spin state populations can be temperature dependent [40], low temperature EPR spectra of KatG(Y229F) contain the axial signal characteristic of 6-c heme [20] as the majority of species, which is also the major heme species (44%) at 280 K in our RR spectra (Table 1). The ν3 vibration at 1502 cm−1 is attributed to a 5-c, QS heme, which results from the admixture of high (S=5/2) and intermediate (S=3/2) heme spin states [41, 42], while the vibration at 1509 cm−1 is assigned to a 6-c LS ferric heme. Excitation at 413.1 nm results in an increase of the 1509 cm−1 band (Figure 1, inset; Figure 2) favoring its assignment to a 6-c LS heme, which is enhanced at this excitation wavelength. Low-temperature EPR spectroscopy of KatG(Y229F) did not allow a definitive assignment of the identity of the 6-c LS heme species but room temperature absorption spectra contain bands near 540 and 580 nm that can be attributed to a LS heme [20]. In WT KatG, we did not observe any evidence of a 6c-LS heme [34]. At pH 5, the heme spin-state distribution in the mutant changes little while at pH 10, the 6-c LS heme becomes more abundant at the expense of the other heme species. Addition of INH to KatG(Y229F) at pH 7.2 results in a small increase in the contribution of the 6-c LS and 5-c QS hemes at the expense of the other two heme species (Table 1).
Figure 1.
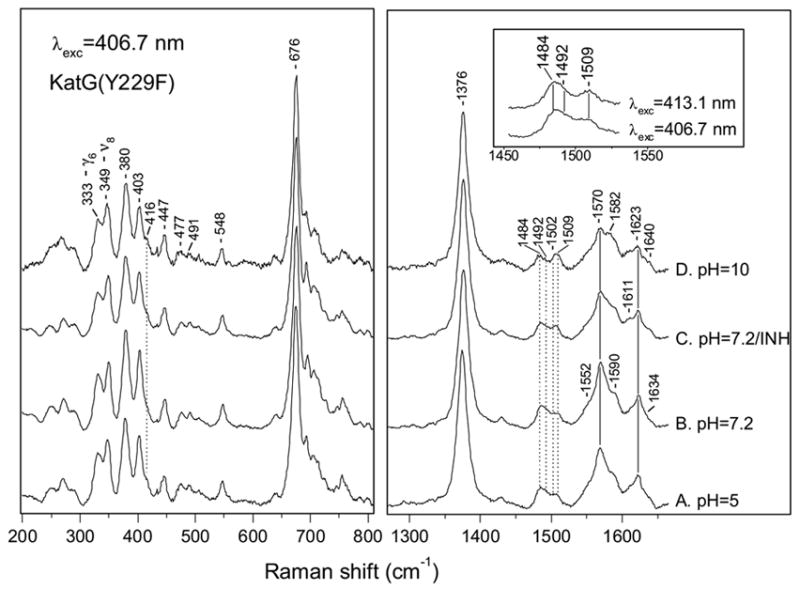
High- and low-frequency resonance Raman spectra of ferric KatG(Y229F) obtained with 406.7 nm excitation at pH 5 (A), pH 7.2 (B), pH 10 (D) and of its complex with INH at pH 7.2 (C). Inset: ν3 region of KatG(Y229F) at pH 7.2 with 406.7 and 413.1 nm excitation.
Figure 2.
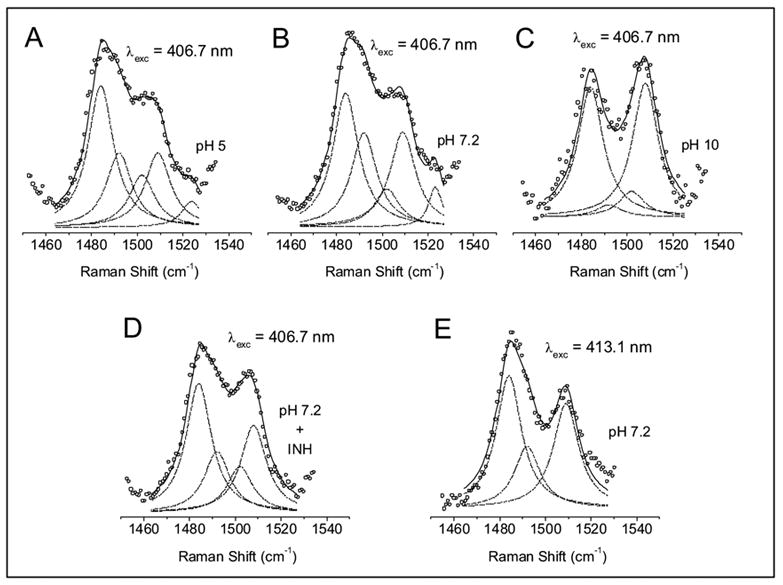
Deconvolution of the ν3 mode in KatG(Y229F). Lorentzian line shapes with a width of 13.5 cm−1 were used for the simulation of the experimental spectra (open circles) in the ν3 region following Ref. 82. The frequencies and the assignments of the fitted bands (dashed lines) are listed in Table 1. The total fit is indicated by the solid line. The spectra were collected with 406.7 nm excitation at pH 5 (A), pH 7.2 (B), pH 10 (C), pH 7.2 + INH (D) and with 413.1 nm excitation at pH 7.2 (E).
Table 1.
Contribution (%) of the four heme spin states in KatG(Y229F) with excitation at 406.7 nm. The numbers for the WT enzyme are shown in parentheses for comparison [34]. In the WT enzyme, ν3 occurs at 1487, 1492, and 1503 cm−1 for 6-c HS, 5-c HS, and QS heme, respectively.
| ν3 = 1484 cm−1 6-c HS heme | ν3 = 1492 cm−1 5-c HS heme | ν3 = 1502 cm−1 QS heme | ν3 = 1509 cm−1 6-c LS heme | |
|---|---|---|---|---|
| pH=5 | 39 ± 2 (34 ± 1) | 29 ± 5 (37 ± 2) | 13 ± 3 (29 ± 1) | 19 ± 5 |
| pH=7.2 | 44 ± 5 (33 ± 1) | 26 ± 2 (35 ± 4) | 9 ± 2 (32 ± 3) | 21 ± 4 |
| pH=7.2/INH | 39 ± 2 (27) | 22 ± 5 (36) | 12 ± 4 (37) | 27 ± 1 |
| pH=10 | 37 ± 5 (22 ± 3) | 22 ± 12 (37 ± 1) | 7 ± 2 (40 ± 3) | 34 ± 9 |
| λ = 413.1 nm pH=7.2 | 45 | 20 | - | 35 |
The large overlap of bands in the ν2 region and the presence of four heme species complicate a straightforward assignment of the observed vibrations. At pH 10, two shoulders at 1570 and 1582 cm−1 can be attributed to the ν2 mode of a 6-c HS and a 6-c LS ferric heme, respectively, in agreement with the relative abundance of those heme species. We assign the band at 1623 cm−1 and the shoulder at 1634 cm−1 to the in-plane and the out-of-plane vinyl stretching vibrations, νCC, respectively [43, 44]. The ν10 modes of the 5-c HS heme are hidden by the νCC mode, and the ν10 vibrations of the 6-c HS and 6-c LS hemes are observed at 1611 and 1640 cm−1, respectively.
The low-frequency RR spectra of KatG(Y229F) at pH values of 5, 7.2, and 10 and in the presence of INH at pH 7.2 are also shown in Figure 1. On the basis of the assignment for metmyoglobin [45], which is widely used for vibrational modes in heme proteins, we assign the bands at 403 and 447 cm−1 to the vinyl bending modes, δ(CβCaCb). The modes at 380, 349, and 333 cm−1 are attributed to the propionate bending vibration (δ (CβCcCd)), ν8, and γ6, respectively. The frequency of δ(CβCcCd) is relatively high, indicating hydrogen-bonding interactions between the propionate groups and the protein-matrix [46–48]. The presence of a shoulder around 416 cm−1 is additional evidence for the presence of a 6-c LS heme [49], and the small increase in its intensity at pH 10 correlates well with the increase in the contribution of the 6-c LS heme at this pH (Table 1). The vibrations at 430 and 521 cm−1 that are observed in WT KatG and KatG(S315T) and arise from a ruffling deformation of the heme [34], are not present in KatG(Y229F) which suggests much less or no ruffling of the heme cofactor. The ruffling deformation is described by opposite twisting of the porphyrin pyrrole rings about the metal-nitrogen bonds [50].
3.2 Ferrous KatG(Y229F)
The high-frequency RR spectrum of ferrous KatG(Y229F) (Figure 3) obtained with excitation at 413.1 nm is similar to that of ferrous WT KatG [51]. The ν4 oxidation state marker at 1360 cm−1 is characteristic of a ferrous heme. The spin state marker ν3 has one main contribution at 1475 cm−1, indicating a 5-c, HS ferrous heme [38], consistent with the absorption spectrum with a Soret band at 439 nm and Q-bands at 554 and 593 nm (data not shown) of a 5-c, HS ferrous heme [52]. A small, residual contribution of ferric 5-c, QS heme cannot be excluded as indicated by a weak ν3 mode at 1500 cm−1, which is similar to our observations after reduction of WT KatG [51]. The ν10 and νCC vibrations at 1608 and 1620 cm−1 also indicate a 5-c, HS ferrous heme. In the low-frequency region (Figure 3, inset), the prominent band at 244 cm−1 is assigned to the stretching vibration of the FeII and the proximal histidine, ν (Fe-His). This is a typical frequency for a proximal histidine with imidazolate character in peroxidases and is due to strong hydrogen-bonding to its Nδ hydrogen by a proximal residue [53]. This vibration occurs at the same frequency as in WT KatG [51], indicating that the Y229F mutation does not significantly alter the hydrogen-bonding environment of the proximal histidine.
Figure 3.
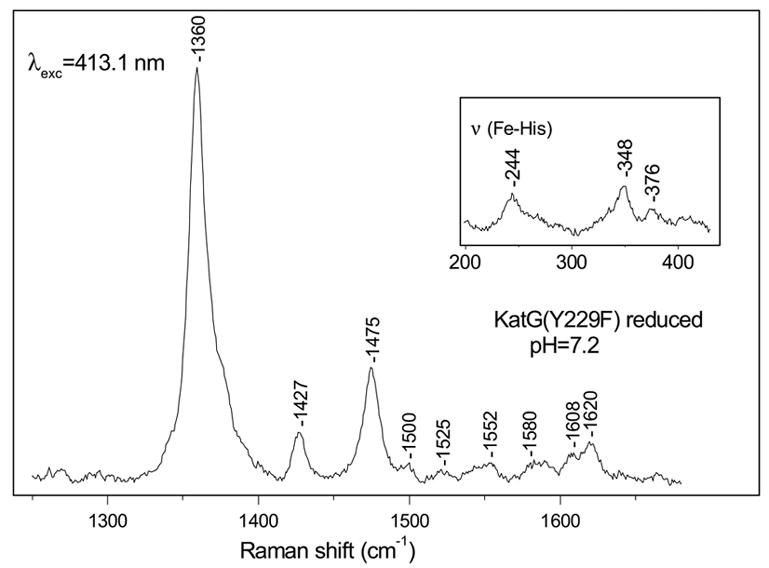
High-frequency resonance Raman spectrum of ferrous KatG(Y229F) at pH 7.2 with excitation at 413.1 nm. Inset: the 200–430 cm−1 region of ferrous KatG(Y229F) at pH 7.2.
3.3 KatG(Y229F)FeII-CO complex
Carbon monoxide (CO) binds to ferrous heme proteins, preferably, in a nearly linear Fe-C-O geometry. It is an excellent probe of the distal heme pocket because the Fe-CO and C-O stretching frequencies are sensitive to electrostatic and steric interactions with nearby amino acid residues [30, 54–56]. In peroxidases, the relatively high ν (Fe-CO) frequency is attributed to an increase in Fe-CO bond order due to imidazolate character of the proximal histidine ligand and hydrogen bonding interactions of a distal pocket residue with CO [55]. In our previous work, we observed an unusually intense Fe-C-O bending mode in WT KatG and in its S315T mutant, which was interpreted to result from a strongly bent Fe-C-O binding geometry [51].
Figure 4 shows the low-frequency RR spectra of KatG(Y229F)-CO at pH 5 (trace A), pH 7.2 (trace B), pH 7.2 with INH (trace C), pH 7.2 with 13CO (trace D), pH 10 (trace E), and pH 10 with 13CO (trace F). At pH 5 and pH 7.2, the broad band centered at 515 cm−1 can be deconvoluted with three Gaussians (see Supplementary data); two bands at 523 and 506 cm−1 with a 16 cm−1 width, and one band at 486 cm−1 with a 22 cm−1 width. At pH 7.2, only the bands at 523 and 587 cm−1 are isotope sensitive and shift to 519 and 572 cm−1, respectively, in the 13CO-bound adduct (see inset I, trace a for the isotope difference spectrum). We assign the 523 and 587 cm−1 modes to ν (Fe-CO) and δ (Fe-C-O), respectively, of KatG(Y229F)-CO. The C-O stretching mode, ν (CO), is at 1930 cm−1 and shifts to 1885 cm−1 for 13CO (Figure 4, inset II). The ν (CO) frequency does not shift in D2O buffer (data not shown), which argues against hydrogen-bonding to CO in contrast with results for WT KatG [51]. The frequencies of the vibrations of the Fe-CO moiety are slightly different from those in WT KatG-CO [51]. In WT KatG-CO, δ(Fe-C-O) is anomalously intense at pH 7.2, but it is significantly less intense in KatG(Y229F)-CO with an intensity comparable to that in WT KatG-CO at pH 10 [51]. In KatG(Y229F)-CO at pH 10, ν (Fe-CO) is observed at 523 cm−1 but with decreased intensity compared to pH 7.2. The difference spectrum of KatG(Y229F)-CO at pH 7 and pH 10 indicates that a small amount of the 523 cm−1 form is converted to a conformation with ν(Fe-CO) at 503 cm−1 (Inset I, trace B) that shifts to 497 cm−1 in the 13CO-complex, which is supported by deconvolution of the Raman bands (see Supplementary data). Addition of INH to KatG(Y229F)-CO at pH=7.2 (trace C) does not change the frequencies of the Fe-CO unit, which can be clearly noted in the difference spectrum (Inset I, trace c).
Figure 4.
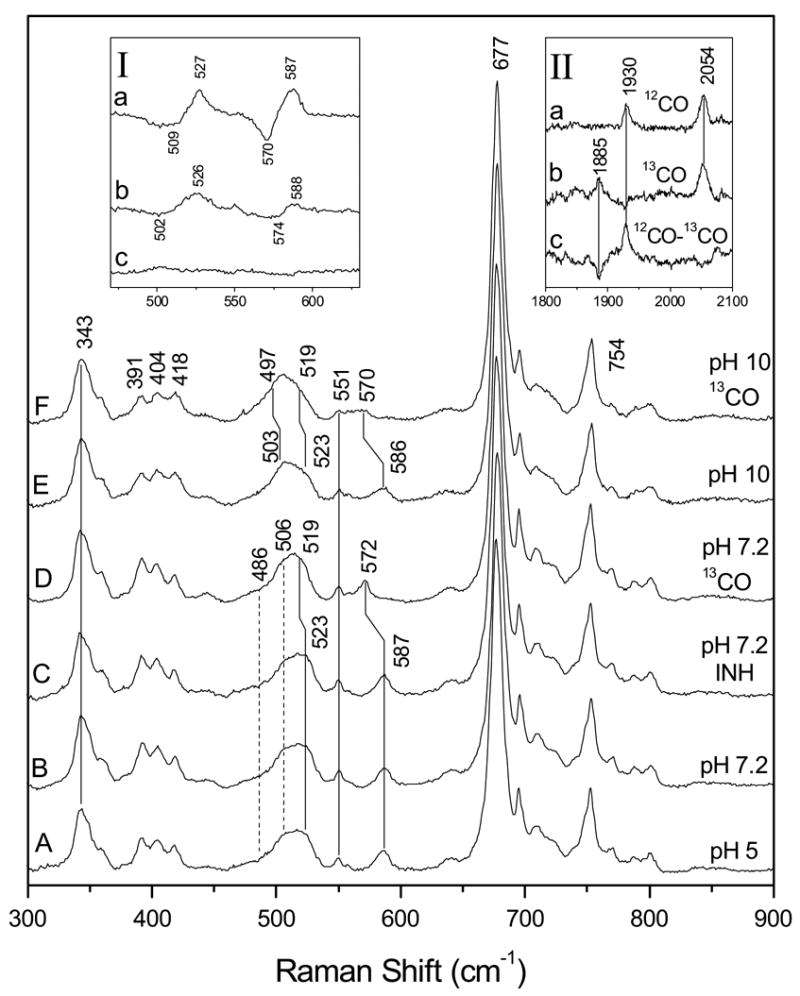
Low-frequency resonance Raman spectra of ferrous KatG(Y229F)-CO at pH 5 (A), at pH 7.2 (B), with INH at pH 7.2 (C), at pH 7.2 with 13CO (D), at pH 10 (E) and of ferrous KatG(Y229F)-13CO at pH 10 (F). Inset I shows the difference between spectra B and D, 12CO - 13CO at pH 7.2 (a), the difference between spectra B and E, pH 7.2 minus pH 10 (b), and the difference between spectra B and C, without INH minus with INH (c). Inset II: resonance Raman spectra in the 1800-2030 cm−1 region; ferrous KatG(Y229F) with 12CO (a), with 13CO (b), and their difference spectrum, 12CO - 13CO (c) at pH 7.2.
The ν(Fe-C) and ν(C-O) of the CO complexes follow an inverse linear correlation due to π-electron back-donation from the dπ(dxz,dyz) orbitals of FeII to the empty π * orbitals of CO (30). The Fe-CO π-backbonding is modulated by polar interactions with distal residues and by variation in donor strength of the proximal ligand. Figure 5 shows the ν(Fe-CO) versus ν(C-O) correlation that compares KatG(Y229F) to WT KatG and to peroxidases. In general, CO-complexes with ν(Fe-CO) > 520 cm−1 and ν(C-O) < 1935 cm−1 reflect increasing distal hydrogen-bonding interactions to CO and a positively charged distal heme pocket [57]. Although the H/D exchange experiment suggests a lack of hydrogen-bonding to CO in KatG(Y229F), the frequency of ν(CO) and the small displacement of KatG(Y229F) in the correlation plot suggest that at best, weakening of the distal hydrogen-bond to CO occurs. Changes in hydrogen-bonding to the proximal histidine could also be responsible for the increase in ν(CO), but that would require also an increase or, at best, no change in ν(Fe-CO) (58), which is not observed, nor were changes in ν(Fe-His) observed that might indicate a change in proximal H-bonding in KatG(Y229F). Therefore, our data suggest that CO is the hydrogen-bonded in KatG(Y229F) and weakened with respect to the WT enzyme.
Figure 5.
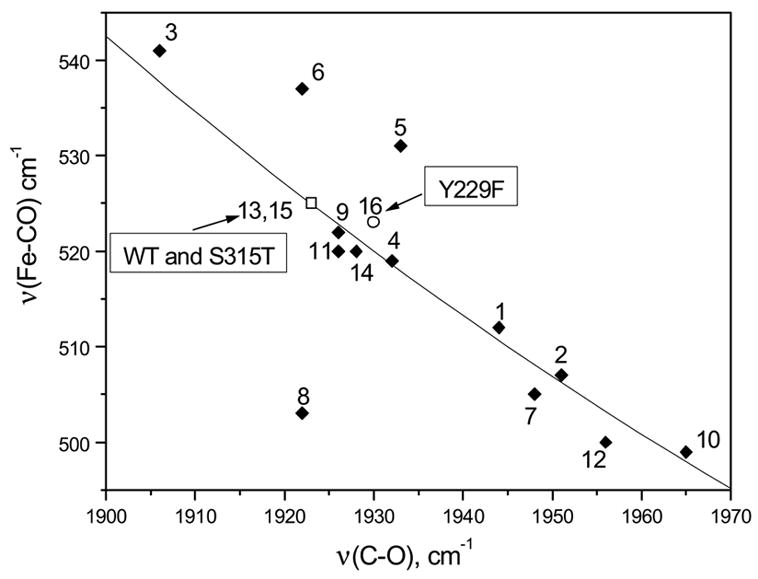
Correlation line of ν (Fe-CO) vs. ν (C-O) for various peroxidases, Mtb. catalase-peroxidase KatG and KatG(Y229F): KatG at pH 7.2 [13], KatG(S315T) at pH 7.2 [15] (□) and KatG(Y229F) at pH 7.2 [16] (○). All entries are tabulated in the Supporting Information.
3.4 KatG(Y229F)FeIII-NO complex
The NO complex of ferric KatG(Y229F) has an absorption spectrum with a Soret band at 421 nm and Q-bands at 533 and 570 nm (data not shown), similar to WT KatGIII-NO and HRPIII-NO [51, 59]. Figure 6 shows the high- and low-frequency RR spectra of the ferric-NO complex of KatG(Y229F) at pH 5 (A), pH 7.2 (B) and pH 10 (E), and in the presence of excess INH at pH 7.2 (D). The ν4, ν3, and ν10 modes appear at 1378, 1513, and 1644 cm−1, respectively, and are similar to those in WT KatGIII-NO and indicative of a 6-c-ferric-nitrosyl complex [51, 60]. These vibrations are not sensitive to pH, nor did the addition of INH at pH 7.2 cause any changes in the vibrational frequencies of ferric KatG(Y229F)-NO in the high- or low-frequency region. Only minor changes in the relative intensities of the 393 and 402 cm−1 bands are observed. Isotope-sensitive bands are observed at 598 and 574 cm−1 for KatG(Y229F)-NO that shift to 593 and 563 cm−1 in KatG(Y229F)-15NO (trace C). The difference spectrum (14NO-15NO) is shown in the inset of Figure 6. On the basis of the assignments for the Mb-NO complex [60] and WT KatG-NO (51), we assign the 598 and 574 cm−1 bands of KatG(Y229F)-NO to ν (Fe-NO) and δ(Fe-N-O), respectively. These values are slightly higher than those observed in the WT enzyme [51].
Figure 6.
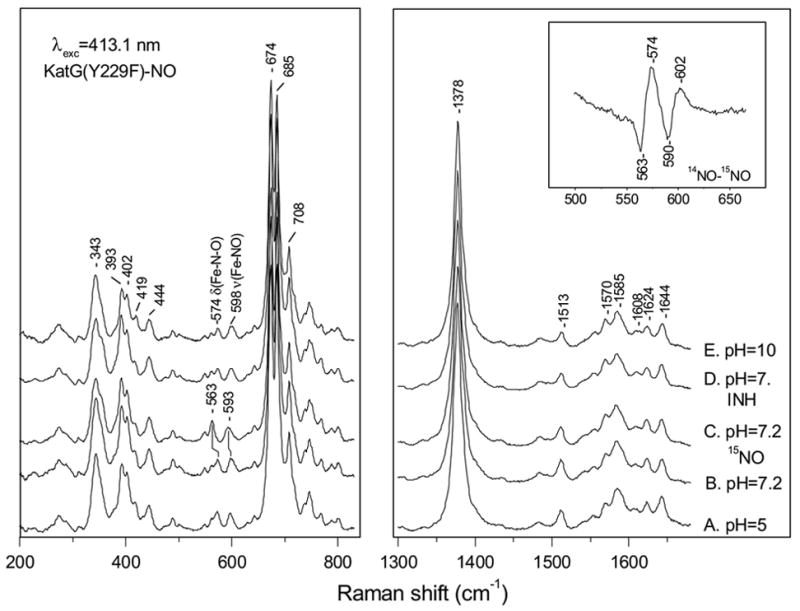
High- and low-frequency resonance Raman spectra of ferric KatG(Y229F)-NO at pH 5 (A), at pH 7.2 (B), at pH 7.2 with 15NO (C) with INH at pH 7.2 (D) and at pH 10 (E). Inset: difference spectrum of ferric KatG(Y229F)-NO minus KatG(Y229F)-15NO at pH 7.2.
4. Discussion
Substitution of Tyr229 with Phe prevents posttranslational formation of the Met-Tyr-Trp covalent adduct in Mtb. KatG [14, 20]. Loss of this adduct results in a significant reduction in catalase activity and a small enhancement in peroxidase activity in KatG(Y229F) with respect to WT KatG [20]. Our data indicate that loss of the adduct results in significant structural perturbation of the heme cofactor and its distal pocket. These perturbations and their implications for enzyme activity are discussed next.
4.1 Structural changes due to loss of Met-Tyr-Trp adduct in Mtb. KatG Y229F
The RR spectra in Figure 1 and the analysis of the heme spin and coordination states listed in Table 1 show that loss of the covalent adduct significantly affects the distribution of heme spin and coordination states. In KatG(Y229F), a significant amount of LS heme is present that appears to come largely at the expense of the QS heme contribution when compared to WT KatG, which does not have detectable levels of LS heme [33, 34]. The contributions of the four different heme species in KatG(Y229F) are rather insensitive to pH, though a small increase in LS heme is observed with increasing pH at the expense of the other heme species. The addition of INH also seems to result in a small increase in LS heme population.
The observation of a 6-c LS heme in KatG(Y229F) indicates the presence of an unknown strong field ligand at the sixth coordination position, which does not interfere with peroxidase activity in KatG(Y229F) compared to WT KatG [20]. In distal Trp mutants of Synechocystis KatG, the distal His was ruled out and hydroxide was proposed as the sixth ligand of a 6-c LS heme [16]. Although the abundance of LS heme increases with pH, we could not detect the characteristic Fe-OH stretching vibration in the low-frequency region to confirm this by using blue excitation [61–64], consistent with EPR results [20]. Previously, we had proposed that the LS heme was Cmpd III [20], which we now rule out on the basis of our present RR data.
The low abundance of QS heme in resting KatG(Y229F) and the absence of low-frequency vibrations at 473 and 520 cm−1 suggests that the heme cofactor is considerably less saddled and less ruffled, respectively, and more planar compared to WT KatG [34,65,66]. In the saddled deformation, the meso carbons remain in the mean porphyrin plane and the pyrrole rings are alternately displaced above and below the porphyrin plane, and the ruffled deformation is due to opposite twisting of the pyrrole rings about the metal-nitrogen bonds [50]. In WT KatG, we observed an increase in the population of the QS heme when the pH was raised from 7.2 to 10, and we proposed that this was due to perturbation of the hydrogen bonding of His276 to the heme propionates and their extended hydrogen-bonding network involving distal water molecules and distal Arg104. In KatG(Y229F), the amount of QS heme decreases at high pH, which suggests that the hydrogen-bonding network involving the heme propionates is disturbed in the absence of the Met-Tyr-Trp adduct.
Although ferric KatG(Y229F) is quite different from WT KatG, their ferrous forms are basically the same. In ferrous KatG(Y229F), the important ν (Fe-His) occurs at 244 cm−1, which is the same as in WT KatG and in the corresponding Y249F mutant of Synechocystis KatG [51, 65]. This suggests that the mutation and loss of the adduct do not affect hydrogen-bonding to the imidazole Nδ hydrogen of the proximal His ligand in KatG(Y229F), nor its imidazolate character. Therefore, the proximal imidazolate still provides increased electron density on the heme iron and helps stabilize higher oxidation states, which facilitates peroxidase activity by weakening the FeIV=O bond and increasing the chemical reactivity of Cmpd I. Therefore, changes in the chemical reactivity of Cmpd I in KatG(Y229F) are most likely due to modifications in the distal heme pocket.
We have used CO and NO as exogenous heme ligands to probe the distal pocket of KatG(Y229F). Similar to WT KatG and KatG(S315T), we observe only a single heme-CO conformer for KatG(Y229F)-CO with ν (Fe-CO) = 523 cm−1, δ(Fe-C-O) = 586 cm−1, and ν (CO) = 1930 cm−1 [51]. The position of ν (Fe-CO) and ν (CO) in the correlation plot (Figure 5) suggests that the distal pocket in KatG(Y229F) may be less positively charged than in WT KatG. We think that this indicates that the hydrogen bonding to CO by distal residues, presumably His108 and Arg104, has somewhat weakened with respect to WT KatG-CO [51].
The FeII-CO and FeIII-NO complexes of iron-porphyrins favor a linear binding geometry [30], and, without distortion of the heme-CO geometry or asymmetry in the electrostatic potential of the surrounding protein, the bending mode is not present or is very weak in the RR spectrum [55]. The ratio of δ(Fe-C-O) and ν (Fe-CO) intensities (Ibend/Istretch) is a measure of sterically induced tilting of CO away from the normal to the heme plane [66]. The Ibend/Istretch of 0.47 in KatG(Y229F)-CO at pH 7.2 is similar to that in lactoperoxidase, which has been interpreted in terms of a tilted Fe-CO geometry [67], but it is significantly lower than the ratio of 0.92 in WT KatG-CO [51]. At pH 10, the Ibend/Istretch of 0.30 is also lower than that in WT KatG-CO of 0.38 at pH 10, which is low due to weakening of hydrogen bonding [51]. The fact that the ratio in KatG(Y229F)-CO at pH 7.2 is closer to that of WT KatG-CO at pH 10 than to that of WT KatG at pH 7.2 supports the proposal that there is weakened hydrogen bonding between CO and a distal pocket residues in KatG(Y299F). At pH 7.2, addition of INH does not change the vibrational frequencies of KatG(Y229F)-CO, but it increases Ibend/Istretch from 0.47 to 0.56. This suggests that INH binding does not affect the distal heme pocket polarity or hydrogen-bonding but does result in a small increase in CO tilting, probably due to an increase in steric constraints, indicating that INH binds in or near the distal heme pocket.
The δ(Fe-N-O) intensity depends weakly on tilting [68], and its high intensity in KatG(Y229F)-NO supports the analysis of the CO results. The frequency difference between ν (Fe-NO) and δ(Fe-N-O) in KatG(Y229F)-NO and in WT KatG-NO indicates a similarly distorted FeIII-NO linkage, but δ(Fe-N-O) is less intense in KatG(Y229F)-NO than in WT KatG-NO [51], suggesting a less polar environment in KatG(Y229F).
For WT KatG, we have proposed that the strong activation of δ(Fe-C-O) and δ(Fe-NO) is due to a combination of steric and electrostatic effects in the distal heme pocket [51, 69, 70]. Although δ(Fe-C-O) and δ(Fe-N-O) are significantly weaker in KatG(Y229F) than in WT KatG, they are still strongly activated. This suggests that steric constraints and electrostatic interactions between the CO and NO ligands and distal residues in KatG(Y229F) are weaker than in WT KatG but still significant. For both WT KatG and KatG(S315T), we have concluded that the heme pocket seems optimized for binding H2O2 and O2 in their favored bent configuration [51]. Disruption of the cross-linked adduct releases stress in the distal pocket to make it more open, which is consistent with observed changes in the heme-CO vibrations. We do not expect that this will affect the binding mode of H2O2 and O2, since a more open heme pocket will still allow their bent-binding conformation. However, a more open distal heme pocket is likely to facilitate access to the heme iron, which can explain the observed 300-fold increase in the rate of Cmpd I formation and the increased peroxidase activity in KatG(Y229F) [20].
4.2 Role of the cross-link Met255-Tyr229-Trp107 in Mtb. KatG
The importance of the covalent adduct in the bifunctional activity of KatGs has been demonstrated by significant or complete loss of catalase activity in variants in which at least one of its components has been mutated [12,14,15,17,20]. Previous results have shown that WT KatG readily reacts with peroxides to rapidly generate a relatively stable Cmpd I, while Cmpd II was not observed [32], suggesting that it has a very short lifetime. Furthermore, during turnover in WT KatG a Tyr radical is formed including one on Tyr353 [71]. In KatG(Y229F), formation of Cmpd I is over 300-times more rapid than in WT KatG, and it decays much faster to a long lived Cmpd II and a Tyr radical, whose formation is kinetically coupled to Cmpd I decay [20]. Interestingly, WT KatG forms Cmpd III only in the presence of very large excess of H2O2, while KatG(Y229F) forms Cmpd III with micromolar concentrations of H2O2 [20,32]. Similar observations have been made with Synechocystis KatG [15,19,28]. In order for KatG to sustain catalatic activity, it is important to rapidly reduce Cmpd II to ferric heme and prevent it from reacting with H2O2 to produce Cmpd III, which does not participate in catalatic turnover. Therefore, the observation that KatG(Y229F) loses its catalatic activity concomitant with a decrease in the lifetime of Cmpd I and an increase in that of Cmpd II suggests that the covalent adduct plays an important role in WT KatG by stabilizing Cmpd I and destabilizing Cmpd II to ensure high catalatic activity, while maintaining peroxidatic activity. The cross-linked adduct may also be important for optimization of the heme pocket geometry to enhance the binding and reactivity of substrates.
Our RR data provide structural evidence that disruption of the cross linked adduct in Mtb. KatG results in conformational changes in the heme pocket that directly impact catalytic activities. In KatG(Y229F), the heme is more planar than in WT KatG, and significant changes occur in the distal heme pocket compared to WT KatG; (i) KatG(Y229F) has a greater abundance of 6-c LS heme, (ii) KatG(Y229F) shows a different pH-dependence, and (iii) in KatG(Y229F) the properties of the ferrous CO- and ferric NO-complexes are different. These observations support the idea that loss of the cross-linked triad by the Y229F mutation causes rearrangement of distal heme pocket residues and perturbs their steric and electrostatic interactions with ligands bound to the heme cofactor, which is most likely due to changes in hydrogen-bonding interactions [34,51]. The primary residue whose conformation is likely altered in this mutant is Trp107, which has a free side chain in the absence of the adduct. As a consequence, the binding environment O2 and H2O2 and of reaction intermediates may be modified in KatG(Y229F) compared to WT KatG. In cytochrome c peroxidase, a covalent crosslink between Trp51 and Tyr52 in its H52Y mutant causes significant rearrangement of two segments of residues [72]. We propose that the adduct stabilizes a distal hydrogen-bonding network that is disrupted in KatG(Y229F) due to rearrangement of distal residues. This network starts at Ile228 which forms a hydrogen-bond with Asp137. In Synechocystis KatG, the analogous Asp plays an important role in stabilization of the LL1 loop that is anchored by Tyr229 [8–11,21]. Asp137 forms a hydrogen bond with Arg104 which interacts with a water molecule that is hydrogen-bonded to the heme propionates [11]. Perturbation of hydrogen-bonding to the heme propionates can explain the difference in heme planarity and pH sensitivity between KatG(Y229F) and WT KatG [34]. Differences in the steric and electrostatic environment of their distal heme pockets are most likely due to rearrangement of distal pocket residues and to a lesser extent to perturbation of the hydrogen-bonding network. Figure 7 shows the proposed hydrogen-bonding network. The integrity of the covalent adduct seems crucial for the stability of the LL1 loop, the maintenance of the proposed hydrogen-bonding network, and, in general, of the heme pocket architecture. This hypothesis will be further tested in the near future by RR spectroscopy of site-directed mutants of the distal heme pocket of KatG.
Figure 7.
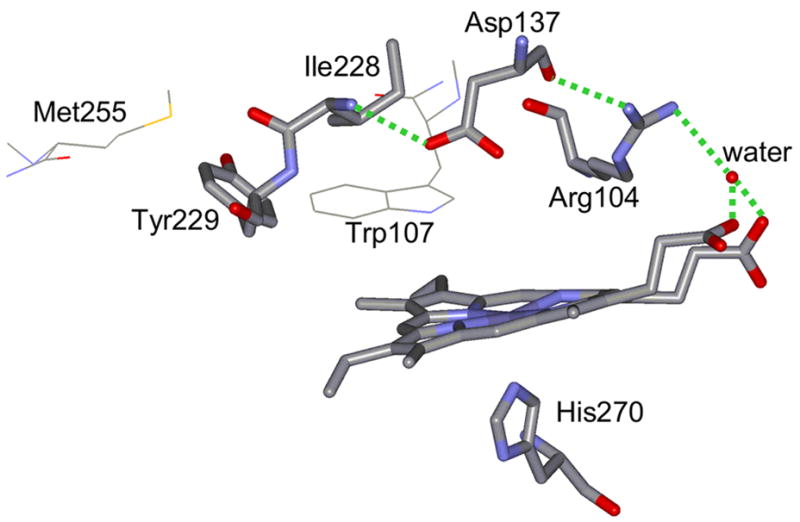
Hydrogen-bonding network between Tyr229 and heme propionate groups that may be perturbed in KatG(Y229F). PDB entry 2CCA was used to construct the figure [81]. The dotted lines indicate hydrogen bonds with lengths shorter than 3 Å.
Our observation of perturbation of the heme pocket in KatG(Y229F) supports the notion that the adduct may be important for stabilizing the proper geometry of Trp107 to provide (part of) the binding site for the second H2O2 in its reaction with Cmpd I and, therefore, for catalatic activity [16,19,21]. Conformational changes on the distal side may also be related to the effect of the sulfonium ion in the adduct on the pKa of nearby residues [14] or by the modulation of the pKa of Trp107 upon adduct formation.
Besides providing the correct heme pocket architecture and binding geometry for the second H2O2, the covalent adduct may also contribute to optimizing the reactivity of substrates (other than peroxides) and productive and non-productive intermediates to maintain KatG’s bifunctional activity. In WT KatG, reduction of Cmpd I by H2O2 must compete efficiently with electron transfer from aromatic residues and substrates, allowing high catalase activity. As previously suggested by stopped-flow experiments [32], efficient competition of H2O2 for reduction of Cmpd I is essential for catalase activity, because Cmpd II has been observed to form Cmpd III through reaction with H2O2 [20]. Formation of Cmpd III in the presence of H2O2 results in decreased catalase activity. Therefore, in the absence of the adduct, H2O2 may be less efficient in competing with amino acids and substrate for reduction of Cmpd I, resulting in formation of Cmpd II and decreased catalase and enhanced peroxidase activity. However, a different catalase mechanism that explicitly involves the reaction of Cmpd II to give Cmpd III has recently been proposed [23].
Perturbation of the distal side at Trp107 along with the observation of rapid formation of Cmpd II may not be the only reason for loss of catalase activity in KatG(Y229F). We propose that the covalent linkage between Met255-Tyr229-Trp107 modulates the redox potential of Trp107, Tyr229, or both. Such an effect has been proposed and observed for cross-linked Tyr residues in cytochrome c oxidase and galactose oxidase, respectively, [73–75], and it has been suggested that a covalent link between Cys356 and the proximal heme ligand Tyr379 may make Neurospora crassa Catalase-1 less prone to donate an electron to Cmpd I to form Cmpd II [76]. It has been suggested that the positively charged sulfonium group within the adduct in KatG may raise the redox potential of the covalently bound residues [14], hindering the oxidation of Tyr229, Trp107, or both by Cmpd I and the formation of Cmpd II (plus protein radical). However, other factors may also affect redox potentials in KatG(Y229F) compared to WT KatG [77–80]. We propose that the adduct formation is important for optimization of the redox potential of the Met-Tyr-Trp adduct as a whole compared to those of the non-covalently attached side chains in KatG(Y229F) to minimize Cmpd II formation and maximize Cmpd II decay to prevent inhibition of catalase activity, while its possible role in facilitating the required proton transfer step should also be investigated.
Finally, KatG(Y229F) has a decreased affinity for INH, and it has been proposed that the covalent adduct may be required for the integrity of a small molecule binding site [10, 20]. Our RR experiments show that addition of INH to KatG(Y229F) increases the amount of 6-c LS heme at the expense of the other heme spin states, while it changes the heme spin state distribution slightly in favor of the QS heme at the expense of the 6-c HS species in WT KatG (34). Addition of INH also seems to increase the distortion of the CO binding geometry in KatG(Y229F), while it decreases this distortion in WT KatG [51]. Therefore, our results support the idea that the covalent adduct may also be important for efficient drug binding [20], though the exact location of the INH binding site remains elusive.
5. Conclusion
We provide structural evidence that the Met255-Tyr229-Trp107 cross-link forms a unique protein environment in Mtb. KatG that is essential for the structural integrity of the active site and, therefore, plays an important functional role. Although disruption of the adduct in the Y229F mutant does not seem to affect hydrogen bonding in the proximal heme pocket, it does perturb the KatG active site as indicated by a more planar heme and a significantly higher abundance of 6-c LS heme in KatG(Y229F) than in WT KatG. Changes in the properties of the CO and NO vibrations indicate their binding geometry and their steric and electrostatic interactions with distal residues are modified in KatG(Y229F). This suggests that the binding environment of H2O2 and O2 is modified in the mutant compared to WT KatG. We propose that the adduct may stabilize the binding site for the second H2O2 and that the adduct as a whole has a modified redox potential as to minimize Cmpd II formation and maximize Cmpd II decay to prevent inhibition of catalase activity. We suggest that besides rearrangements of distal residues, disruption of a distal pocket hydrogen-bonding network is responsible for the observed structural and functional changes in KatG when the covalently linked adduct is not present. Our data also indicate that the INH binding site is perturbed in KatG(Y229F), though its exact location remains elusive.
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by a New York University Whitehead Fellowship for Junior Faculty in Biomedical or Biological Sciences (J.P.M.S.), the Heiser Program of the New York Community Trust (S.M.K.), and the National Institutes of Health Grant Number AI-060014 (R.S.M.).
6. Table of Abbreviations
- M. tuberculosis
Mycobacterium tuberculosis
- WT
wild type
- 6-c
six coordinate
- 5-c
five coordinate
- LS
low-spin
- HS
high spin
- QS
quantum spin
- INH
isonicotinic acid hydrazide, isoniazid
- CCP
cytochrome c peroxidase
- HRP
horseradish peroxidase
- APXs
ascorbate peroxidase
- HmCP
Haloarcula marismortui catalase-peroxidase
- BpKatG
Burkholderia pseudomallei catalase-peroxidase
- Mtb KatG
Mycobacterium tuberculosis catalase-peroxidase KatG
- Cmpd I
II, or III, compound I, II, or III
Footnotes
Supplementary data
Supplementary data containing a table with the entries for Figure 5 and one figure with the deconvolution of the Fe-CO-vibrational modes can be found in the online version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johnsson K, Froland WA, Schultz PG. J Biol Chem. 1997;272:2834–2840. doi: 10.1074/jbc.272.5.2834. [DOI] [PubMed] [Google Scholar]
- 2.Magliozzo RS, Marcinkeviciene JA. J Am Chem Soc. 1996;118:11303–11304. [Google Scholar]
- 3.Robitzek EH, Selikoff IJ. Am Rev Tuberc. 1952;65:402–410. doi: 10.1164/art.1952.65.4.402. [DOI] [PubMed] [Google Scholar]
- 4.Marttila HJ, Soini H, Eerola E, Vyshnevskaya E, Vyshnevskiy BI, Otten TF, Vasilyef AV, Viljanen MK. Antimicrob Agents Chemother. 1998;42:2443–2445. doi: 10.1128/aac.42.9.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musser JM, Kapur V, Williams DL, Kreiswirth BN, van Soolingen D, van Embden JD. J Infect Dis. 1996;173:196–202. doi: 10.1093/infdis/173.1.196. [DOI] [PubMed] [Google Scholar]
- 6.Rouse DA, DeVito JA, Li Z, Byer H, Morris SL. Mol Microbiol. 1996;22:583–592. doi: 10.1046/j.1365-2958.1996.00133.x. [DOI] [PubMed] [Google Scholar]
- 7.Welinder KG. Curr Opin Struct Biol. 1992;2:388–393. [Google Scholar]
- 8.Zámocký M, Regelsberger G, Jakopitsch C, Obinger C. FEBS Letters. 2001;492:177–182. doi: 10.1016/s0014-5793(01)02237-2. [DOI] [PubMed] [Google Scholar]
- 9.Yamada Y, Fujiwara T, Sato T, Igarashi N, Tanaka N. Nat Struct Biol. 2002;9:691–695. doi: 10.1038/nsb834. [DOI] [PubMed] [Google Scholar]
- 10.Carpena X, Loprasert S, Mongkolsuk S, Switala J, Loewen PC, Fita I. J Mol Biol. 2003;327:475–489. doi: 10.1016/s0022-2836(03)00122-0. [DOI] [PubMed] [Google Scholar]
- 11.Bertrand T, Eady NAJ, Jones JN, Jesmin JM, Nagy B, Jamart-Gregoire EL, Raven KA, Brown J Biol Chem. 2004;279:38991–38999. doi: 10.1074/jbc.M402382200. [DOI] [PubMed] [Google Scholar]
- 12.Donald LJ, Krokhin OV, Duckworth HW, Wiseman B, Deemagarn T, Singh R, Switala J, Carpena X, Fita I, Loewen PC. J Biol Chem. 2003;278:35687–35692. doi: 10.1074/jbc.M304053200. [DOI] [PubMed] [Google Scholar]
- 13.Jakopitsch C, Kolarich D, Petutschnig G, Furtmuller PG, Obinger C. FEBS Lett. 2003;552:135–140. doi: 10.1016/s0014-5793(03)00901-3. [DOI] [PubMed] [Google Scholar]
- 14.Ghiladi RA, Knudsen GM, Medzihradszky KF, Ortiz de Montellano PR. J Biol Chem. 2005;280:22651–22663. doi: 10.1074/jbc.M502486200. [DOI] [PubMed] [Google Scholar]
- 15.Jakopitsch C, Auer M, Ivancich A, Rueker F, Furtmueller PG, Obinger C. J Biol Chem. 2003;278:20185–20191. doi: 10.1074/jbc.M211625200. [DOI] [PubMed] [Google Scholar]
- 16.Heering HA, Indiani C, Regelsberger G, Jakopitsch C, Obinger C, Smulevich G. Biochemistry. 2002;41:9237–9247. doi: 10.1021/bi025740u. [DOI] [PubMed] [Google Scholar]
- 17.Hillar A, Peters B, Pauls R, Loboda A, Zhang H, Mauk AG, Loewen PC. Biochemistry. 2000;39:5868–5875. doi: 10.1021/bi0000059. [DOI] [PubMed] [Google Scholar]
- 18.Regelsberger G, Jakopitsch C, Furtmuller PG, Rueker F, Switala J, Loewen P, Obinger C. Biochem Soc Trans. 2001;29:99–105. doi: 10.1042/0300-5127:0290099. [DOI] [PubMed] [Google Scholar]
- 19.Regelsberger G, Jakopitsch C, Rueker F, Krois D, Peschek GA, Obinger C. J Biol Chem. 2000;275:22854–22861. doi: 10.1074/jbc.M002371200. [DOI] [PubMed] [Google Scholar]
- 20.Yu S, Girotto S, Zhao X, Magliozzo RS. J Biol Chem. 2003;278:44121–44127. doi: 10.1074/jbc.M304757200. [DOI] [PubMed] [Google Scholar]
- 21.Jakopitsch C, Auer M, Regelsberger G, Jantschko W, Furtmueller PG, Rueker F, Obinger C. Biochemistry. 2003;42:5292–5300. doi: 10.1021/bi026944d. [DOI] [PubMed] [Google Scholar]
- 22.Smulevich G, Feis A, Howes BD. Acc Chem Res. 2005;38:433–440. doi: 10.1021/ar020112q. [DOI] [PubMed] [Google Scholar]
- 23.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. J Inorg Biochem. 2006;100:568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 24.Johnsson K, Schultz PG. J Am Chem Soc. 1994;116:7425–7426. [Google Scholar]
- 25.Dunford HB, editor. Heme Peroxidases. Wiley-VCH; New York: 1999. [Google Scholar]
- 26.Nicholls P, Fita I, Loewen PC. Adv Inorg Chem. 2001;51:51–106. [Google Scholar]
- 27.Kapetanaki SM, Chouchane S, Yu S, Magliozzo RS, Schelvis JPM. J Inorg Biochem. 2005;99:1401–1406. doi: 10.1016/j.jinorgbio.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 28.Regelsberger G, Jakopitsch C, Engleder M, Ruker F, Peschek GA, Obinger C. Biochemistry. 1999;38:10480–10488. doi: 10.1021/bi990886n. [DOI] [PubMed] [Google Scholar]
- 29.Engleder M, Regelsberger G, Jakopitsch C, Furtmüller PG, Ruker F, Peschek GA, Obinger C. Biochimie. 2000;82:211–219. doi: 10.1016/s0300-9084(00)00204-2. [DOI] [PubMed] [Google Scholar]
- 30.Yu N-T, Kerr EA. Biological Applications of Raman Spectroscopy. In: Spiro TG, editor. Resonance Raman Spectra of Heme and Metalloproteins. Vol. 3. Wiley; New York: 1988. pp. 39–95. [Google Scholar]
- 31.Wang J, Caughey WS, Rousseau DL. In: Methods in Nitric Oxide Research. Feelisch M, Stamler JS, editors. Wiley; Chichester, UK: 1996. pp. 427–454. [Google Scholar]
- 32.Chouchane S, Lippai I, Magliozzo RS. Biochemistry. 2000;39:9975–9983. doi: 10.1021/bi0005815. [DOI] [PubMed] [Google Scholar]
- 33.Chouchane S, Girotto S, Kapetanaki S, Schelvis JPM, Yu S, Magliozzo RS. J Biol Chem. 2003;278:8154–8162. doi: 10.1074/jbc.M208256200. [DOI] [PubMed] [Google Scholar]
- 34.Kapetanaki S, Chouchane S, Girotto S, Yu S, Magliozzo RS, Schelvis JPM. Biochemistry. 2003;42:3835–3845. doi: 10.1021/bi026992y. [DOI] [PubMed] [Google Scholar]
- 35.Abe M, Kitagawa T, Kyogoku Y. J Chem Phys. 1978;69:4526–4534. [Google Scholar]
- 36.Choi S, Spiro TG. J Am Chem Soc. 1983;105:3683–3692. [Google Scholar]
- 37.Choi S, Lee JJ, Wei YH, Spiro TG. J Am Chem Soc. 1983;105:3692–3707. [Google Scholar]
- 38.Spiro TG. Biological Applications of Raman Spectroscopy. In: Spiro TG, editor. Resonance Raman Spectra of Heme and Metalloproteins. Vol. 3. Wiley; New York: 1988. pp. 1–38. [Google Scholar]
- 39.Indiani C, Feis A, Howes BD, Marzocchi MP, Smulevich G. J Am Chem Soc. 2000;122:7368–7376. [Google Scholar]
- 40.Evangelista-Kirkup E, Crisanti M, Poulos TL, Spiro TG. FEBS Lett. 1985;190:221–226. doi: 10.1016/0014-5793(85)81288-6. [DOI] [PubMed] [Google Scholar]
- 41.Maltempo MM. J Chem Phys. 1974;61:2540–2547. [Google Scholar]
- 42.Fujii S, Yoshimura T, Kamada H, Yamaguchi S, Suzuki S, Shidara S, Takakuwa S. Biochim Biophys Acta. 1995;1251:161–169. doi: 10.1016/0167-4838(95)00092-9. [DOI] [PubMed] [Google Scholar]
- 43.Smulevich G. Biospectroscopy. 1998;4:S3–S17. doi: 10.1002/(SICI)1520-6343(1998)4:5+<S3::AID-BSPY2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 44.Kalsbeck WA, Robertson DE, Pandey RK, Smith KM, Dutton PL, Bocian DF. Biochemistry. 1996;35:3429–3438. doi: 10.1021/bi952662k. [DOI] [PubMed] [Google Scholar]
- 45.Hu S, Smith KM, Spiro TG. J Am Chem Soc. 1996;118:12638–12646. [Google Scholar]
- 46.Friedman JM, Peterson E. In: Asher SA, Stein PB, editors. Proc. XIVth Int. Conf. on Raman Spectrosc; New York: John Wiley & Sons; 1996. pp. 428–429. [Google Scholar]
- 47.Gottfried DS, Peterson ES, Sheikh AG, Wang J, Yang M, Friedman JM. J Phys Chem. 1996;100:12034–12042. [Google Scholar]
- 48.Cerda-Colón JF, Silfa E, López-Garriga J. J Am Chem Soc. 1998;120:9312–9317. [Google Scholar]
- 49.Smulevich G, Mauro JM, Fishel LA, English AM, Kraut J, Spiro TG. Biochemistry. 1988;27:5477–5485. doi: 10.1021/bi00415a014. [DOI] [PubMed] [Google Scholar]
- 50.Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 51.Kapetanaki SM, Chouchane S, Yu S, Zhao X, Magliozzo RS, Schelvis JPM. Biochemistry. 2005;44:243–252. doi: 10.1021/bi048097f. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Boldt NJ, Ondrias MR. Biochemistry. 1992;31:867–878. doi: 10.1021/bi00118a032. [DOI] [PubMed] [Google Scholar]
- 53.Kitagawa T. Biological Applications of Raman Spectroscopy. In: Spiro TG, editor. Resonance Raman Spectra of Heme and Metalloproteins. Vol. 3. Wiley; New York: pp. 97–131. [Google Scholar]
- 54.Kushkuley B, Stavrov SS. Biophys J. 1996;70:1214–1229. doi: 10.1016/S0006-3495(96)79680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spiro TG, Zgierski MZ, Kozlowski PM. Coord Chem Rev. 2001;219–221:923–936. [Google Scholar]
- 56.Phillips GN, Jr, Teodoro ML, Li T, Smith B, Olson JS. J Phys Chem B. 1999;103:8817–8829. [Google Scholar]
- 57.Smulevich G, Mauro MJ, Fishel LA, English AM, Kraut J, Spiro TG. Biochemistry. 1988;27:5486–5492. doi: 10.1021/bi00415a015. [DOI] [PubMed] [Google Scholar]
- 58.Franzen S. J Am Chem Soc. 2001;123:12578–12589. doi: 10.1021/ja0108988. [DOI] [PubMed] [Google Scholar]
- 59.Dawson JH, Kadkhodayan S, Zhuang C, Sono M. J Inorg Biochem. 1992;45:179–192. doi: 10.1016/0162-0134(92)80043-u. [DOI] [PubMed] [Google Scholar]
- 60.Benko B, Yu NT. Proc Natl Acad Sci USA. 1983;80:7042–7046. doi: 10.1073/pnas.80.22.7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sitter AJ, Shifflett JR, Terner J. J Biol Chem. 1988;263:13032–13038. [PubMed] [Google Scholar]
- 62.Sun J, Fitzgerald MM, Goodin DB, Loehr TM. J Am Chem Soc. 1997;119:2064–2065. [Google Scholar]
- 63.Howes BD, Feis A, Indiani C, Marzocchi MP, Smulevich G. J Biol Inorg Chem. 2000;5:227–235. doi: 10.1007/s007750050367. [DOI] [PubMed] [Google Scholar]
- 64.Nissum M, Feis A, Smulevich G. Biospectrosc. 1998;4:355–364. doi: 10.1002/(sici)1520-6343(1998)4:6<355::aid-bspy1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 65.Santoni E, Jakopitsch C, Obinger C, Smulevich G. Biopolymers. 2004;74:46–50. doi: 10.1002/bip.20041. [DOI] [PubMed] [Google Scholar]
- 66.Yu NT, Kerr EA, Ward B, Chang CK. Biochemistry. 1983;22:4534–4540. doi: 10.1021/bi00288a028. [DOI] [PubMed] [Google Scholar]
- 67.Hu S, Treat RW, Kincaid JR. Biochemistry. 1993;32:10125–10130. doi: 10.1021/bi00089a031. [DOI] [PubMed] [Google Scholar]
- 68.Hu S, Kincaid JR. J Am Chem Soc. 1991;113:2843–2850. [Google Scholar]
- 69.Li XY, Spiro TG. J Am Chem Soc. 1988;110:6024–6033. doi: 10.1021/ja00226a017. [DOI] [PubMed] [Google Scholar]
- 70.Ray GB, Li XY, Ibers JA, Sessler JL, Spiro TG. J Am Chem Soc. 1994;116:162–176. [Google Scholar]
- 71.Zhao X, Girotto S, Yu S, Magliozzo RS. J Biol Chem. 2004;279:7606–7612. doi: 10.1074/jbc.M311884200. [DOI] [PubMed] [Google Scholar]
- 72.Bhaskar B, Immoos CE, Shimizu H, Sulc F, Farmer P, Poulos TL. J Mol Biol. 2003;328:157–166. doi: 10.1016/s0022-2836(03)00179-7. [DOI] [PubMed] [Google Scholar]
- 73.Uchida T, Mogi T, Nakamura H, Kitagawa T. J Biol Chem. 2004;279:53613–53620. doi: 10.1074/jbc.M409719200. [DOI] [PubMed] [Google Scholar]
- 74.Hamilton GA, Adolf PK, de Jersey J, DuBois GC, Dyrkacz GR, Libby RD. J Am Chem Soc. 1978;100:1899–1912. [Google Scholar]
- 75.Johnson JM, Halsall HB, Heineman WR. Biochemistry. 1985;24:1579–1585. doi: 10.1021/bi00328a001. [DOI] [PubMed] [Google Scholar]
- 76.Díaz A, Horjales E, Rudiño-Piñera E, Arreola R, Hansberg W. J Mol Biol. 2004;342:971–985. doi: 10.1016/j.jmb.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 77.Das TP, Pecoraro C, Tomson FL, Gennis RB, Rousseau DL. Biochemistry. 1998;37:14471–14476. doi: 10.1021/bi981500w. [DOI] [PubMed] [Google Scholar]
- 78.Varadarajan R, Zewert TE, Gray HB, Boxer SG. Science. 1989;243:69–72. doi: 10.1126/science.2563171. [DOI] [PubMed] [Google Scholar]
- 79.Liu G, Shao W, Zhu S, Tang W. J Inorg Biochem. 1995;60:123–131. doi: 10.1016/0162-0134(95)00007-b. [DOI] [PubMed] [Google Scholar]
- 80.Sligar SG, Murray RI. In: Cytochrome P-450: Structure, Mechanism, and Biochemistry. Ortiz de Montellano PR, editor. Plenum; New York: 1986. pp. 429–503. [Google Scholar]
- 81.Zhao X, Yu H, Yu S, Wang F, Sacchettini JC, Magliozzo RS. Biochemistry. 2006;45:4131–4140. doi: 10.1021/bi051967o. [DOI] [PubMed] [Google Scholar]
- 82.Heering HA, Jansen MAK, Thorneley RNF, Smulevich G. Biochemistry. 2001;40:10360–10370. doi: 10.1021/bi0106033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.