

Abstract
Catalase-peroxidase (KatG) from Mycobacterium tuberculosis, a Class I peroxidase, exhibits high catalase activity and peroxidase activity with various substrates, and is responsible for activation of the commonly used antitubercular drug, isoniazid (INH). KatG readily forms amino acid based radicals during turnover with alkyl peroxides and this work focuses on extending the identification and characterization of radicals forming on the millisecond to seconds time scale. Rapid freeze- quench electron paramagnetic resonance spectroscopy (RFQ-EPR) reveals a change in the structure of the initially formed radical in the presence of INH. Heme-pocket binding of the drug, and knowledge that KatG[Y229F] lacks this signal provides evidence for radical formation on residue Y229. High-field RFQ-EPR spectroscopy confirmed a tryptophanyl radical signal and new analyses of X-band RFQ-EPR spectra also established its presence. High-field EPR spectroscopy also confirmed that the majority radical species is a tyrosyl radical. Site-directed mutagenesis, along with simulations of EPR spectra based on X-ray structural data for particular tyrosine and tryptophan residues enabled assignments based on predicted hyperfine coupling parameters. KatG mutants W107F, Y229F and the double mutant W107F/Y229F showed alteration in type and yield of radical species. Results are consistent with formation of a tyrosyl radical reasonably assigned to residue Y229 within the first few milliseconds of turnover. This is followed by a mixture of tyrosyl and tryptophanyl radical species, and finally to only a tyrosyl radical on residue Y353, which lies more distant from the heme. Radical processing of enzyme lacking the Trp107-Tyr229-Met255 adduct, found as a unique structural feature of catalase-peroxidases, is suggested to be a reasonable assignment of the phenomena.
M. tuberculosis catalase-peroxidase (KatG)1 is the enzyme responsible for activation of the anti-TB drug isoniazid (INH) in use for over fifty years, and mutations in this enzyme are the primary source of drug resistance in clinical strains of the TB pathogen throughout the world (1–3). KatG has been classified as a Class I peroxidase, but exhibits high catalase activity. Understanding the correspondence between specific structural features and catalytic function in KatG enzymes is in its early stages. In the course of work on KatG in this laboratory and others, it has become clear that amino acid based radicals formed during turnover of the enzyme with peroxide must be accounted for in order to understand mechanistic issues that distinguish the catalase-peroxidases from the corresponding mono-functional enzymes. Furthermore, a specific requirement for a radical in the catalase reaction path of KatG has been proposed and incorporated into a novel mechanism that diverges from the two-electron reactions defined for classical catalases (4,5). Additionally, a post-translational modification of amino acid side chains in the distal pocket of KatG enzymes is thought to result from radical formation catalyzed by the heme (6,7). A mechanistic role for KatG radicals may also be relevant to mammalian catalases in which a protein based radical is considered protective of dead-end catalytic paths such as Compound II formation (8,9). We suggested in a previous report that radicals could have a catalytic function because they were quenched in reactions with INH (10). In order to continue making progress toward understanding these issues, a survey of radical formation in Mtb KatG continues to be a worthwhile goal.
Recent reports have addressed the identification and characterization of radicals appearing within milliseconds and through 10 s of turnover of resting M. tuberculosis KatG with alkyl peroxides (10,11), and within 5 to 10 sec of reaction for the homologous enzyme from Synechocystis sp. (12,13). Critical questions then arise: are any of these radicals important for enzyme function in all KatG enzymes and which amino acids are involved? One approach to the analysis of radical production in peroxidases has historically involved examination of mutants, which may or may not yield confirmation of the location of radicals in the WT enzyme. Trapping and other techniques have also been applied to gain insights into radical formation and identity in a collection of metalloenzymes. Both these approaches have allowed identification of tyrosyl and tryptophanyl radicals in KatG enzymes and some assignment of the residues on which they reside (12,13). A combined approach, in which more than a single experimental method is applied, is expected to provide a more accurate picture of radical formation.
Evidence now points to a unique catalytic requirement in catalase-peroxidases of three amino acids on the distal side of the heme, which appear in a covalent adduct connecting the side chains of W107, Y229 and M255 in all KatG crystal structures (Fig. 1) (6,7,14). None of these amino acids can be mutated without damaging catalase function in a KatG, yet such mutants maintain or even exhibit improved peroxidase function (7,9,15,16). The detailed reasons for this are in some cases related to a change in the stability of intermediates in either mechanism in mutants; for example, in Mtb KatG[Y229F], a very rapid conversion of Compound I, the classical oxoferryl heme π-cation radical species, to a stable Compound II occurs, allowing peroxidase activity but eliminating catalase activity (8); and in Mtb KatG[W107F], facile conversion to and stable formation of Compound III also interferes with catalase activity (Yu, S. and Magliozzo, R. S., unpublished data).
Fig. 1. Met-Tyr-Trp adduct in Mtb KatG.
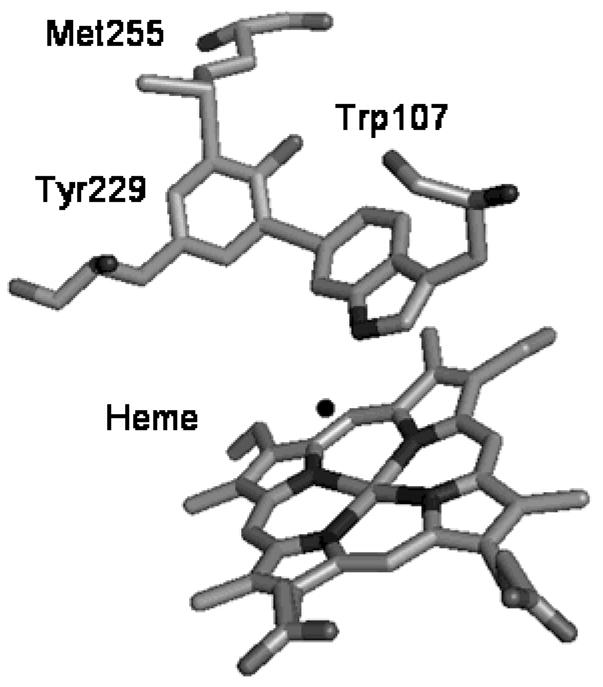
The figure was constructed using the coordinates deposited in the Protein Data Bank (accession code 2CCA) and displayed using PyMOL software.
Amino acid based radicals play roles in those mechanistic changes. The formation of the covalent bonds within the distal side adduct is also proposed to involve Tyr and Trp radicals (14). No direct insights into the post-translational modification reactions have emerged to date. Interestingly, a related Tyr52-Trp51 modification is found in the structure of the H52Y mutant of CCP. (Trp 51, conserved in all Class I peroxidases, is conserved at position 107 in Mtb KatG.) The reaction leading to that modification has also been shown to require turnover of the enzyme and presumably, radical recombination at these sites (17).
In earlier reports (10), tyrosyl radical(s) formation in Mtb KatG was probed using RFQ-EPR spectroscopy. Here evidence is provided for at least two different residues stabilizing tyrosyl radicals in this enzyme, presenting the challenge of identifying and confirming both sites. HF EPR spectroscopy identified a tryptophanyl radical and confirmed tyrosyl radical as the majority species both at early time points and after several seconds of reaction with peroxide. Examination of new mutant enzymes KatG[W107F], KatG[W91F] and KatG[Y229F/W107F] using X-band RFQ-EPR sprectroscopy also assisted in assignments. The effect of INH in a new approach, demonstrated changes at the earliest time points during initial turnover of resting WT KatG with peroxide. Analysis using a theoretical approach to redox coupling (18–20) along with the recently solved X-ray crystal structure provided additional electron transfer and structural information for this work. The results overall are consistent with radicals formed initially on residues Y229 and W107 closest to the heme, followed by appearance of additional radical(s) at more distant sites.
Experimental Procedures
Chemicals and Reagents
All standard chemicals and reagents were purchased either from Fisher Scientific or from Sigma-Aldrich. PAA (32%) was diluted to 10 mM in potassium phosphate buffer and was incubated with 780 units/ml bovine liver catalase for 4 hours at 37 °C to remove hydrogen peroxide, followed by removal of the enzyme by ultrafiltration.
Construction, expression, and purification of the W107F, W91F and [W107F][Y229F] mutants of KatG
The plasmid pKAT II was used as an overexpression vector for KatG (21) and for mutagenesis. E. coli strain UM262 (recA katG::Tn10 pro leu rpsL hsdM hsdR endl lacY) (22) was used for overexpression of both wild-type and mutated KatG proteins. UM262 and pKAT II were both gifts from Stewart Cole (Institut Pasteur, Paris). Mutagenesis was performed using the QuickChange site-directed mutagenesis kit from Stratagene (La Jolla, CA). The pairs of complementary oligonucleotides (synthesized and purified by Qiagene, CA) were designed to introduce the required mutation. The oligonucleotide pairs (mutated codons are in bold) were:
W107F 303GTTTATCCGGATGGCGTTCCACGCTGCC GGCACC336-3′ and 5′-336GGTGCCGGCAGCGTGGAACGCCATCCGG ATAAAC303-3′
W91F 256CCTCGCAGCCGTGGTTTCCCGCCGACTA CG286-3′ and 5′-286CGTAGTCGGCGGGAAACCACGGCTGCGA GG256-3′
Y229F 672GATGGGGCTGATCTTCGTGAACCCGGAG G700-3′ and 5′-700CCTCCGGGTTCACGAAGATCAGCCCCAT C672-3′
Mutagenesis was performed according to the manufacturer’s protocol and the reaction products were transformed into the E. coli XL1-Blue strain. Sequencing (Gene Wiz, Inc.) of the mutated katG gene confirmed that only the desired nucleotide substitutions occurred, and the mutated plasmid was transformed into E. coli strain UM262 for protein overexpression. Recombinant WT KatG and mutated enzymes were purified as previously described (23), in potassium phosphate buffer, pH 7.2. The pure enzymes had optical purity ratios (A407/A280) greater than or equal to 0.5.
RFQ-EPR sample preparation
The RFQ-EPR samples were prepared using an Update Instrument, Inc. Model 1000 chemical-freeze-quench apparatus as described previously (10). Solutions of enzyme (typically 100 μM heme) and peroxyacetic acid (300 μM) in 20 mM potassium phosphate buffer, pH = 7.2, were mixed in 1:1 ratio and the mixture was incubated for the indicated time periods followed by freeze-quenching in isopentane at −130 °C. PAA solutions were freshly prepared before each experiment from stock solution (H2O2-free) stored at −80 °C.
For RFQ-EPR experiments in the presence of INH, KatG (100 μM) was reacted with a mixture of peroxyacetic acid (300 μM) and isoniazid (10 mM) introduced from the second syringe in a typical RFQ-EPR protocol. This concentration of INH was appropriate to ensure rapid reaction with enzyme-based radicals forming and decaying on the millisecond timescale (10). The frozen samples were then examined by EPR spectroscopy as described above. An RFQ-EPR experiment was performed using KatG pre-treated with ten-fold excess PAA for one hour at 4 °C. The enzyme was then exchanged into pure phosphate buffer and was re-reacted with PAA in a typical RFQ-EPR protocol.
Frozen sample powders collected from the rapid freeze-quench apparatus were packed into precision-bore quartz EPR tubes immersed in the isopentane bath and after removal of excess isopentane were transferred to liquid nitrogen and finally examined using a Bruker E500 EPR spectrometer operating at X-band. A finger Dewar inserted into the EPR cavity was used for recording spectra at liquid nitrogen (77 K) temperature. The other experimental conditions were: modulation amplitude, 4 G; microwave power, 1 mW; modulation frequency, 100 kHz; microwave frequency, 9.491 GHz. EPR data acquisition and manipulation was performed using XeprView software (Bruker). CuSO4 in 50% ethylene glycol was used as a standard for spin quantification by double integration of EPR signal intensities. The signal-to-noise ratio in EPR spectra was improved by signal averaging when necessary. The estimation of spin concentration in freeze-quenched samples included application of a packing factor of 0.5 to account for sample dilution by isopentane.
The 130 GHz EPR experiments were performed on a spectrometer assembled at the Albert Einstein College of Medicine, which uses a bridge designed and built at the Donetsk Physico-Technical Institute of the Ukrainian National Academy of Sciences. The magnetic field is generated using a 7 T Magnex superconducting magnet equipped with a 0.5 T sweep/active shielding coil. Field swept spectra were obtained in the two pulse echo-detected mode with the following parameters: temperature 7 K; repetition rate 30 Hz; 400 averages per point; 90 degree pulse 40 ns, time τbetween pulses 170 ns. The magnetic field was calibrated to an accuracy of ~ 3 G using a sample of Mn doped into MgO (24). The temperature of the sample was maintained to an accuracy of approximately +/− 0.3 K using an Oxford Spectrostat continuous flow cryostat and ITC503 temperature controller. The spectra presented in Fig. 4 are derivatives of the echo-detected spectra.
Fig. 4. High-field (130 GHz) EPR spectra of Mtb KatG.
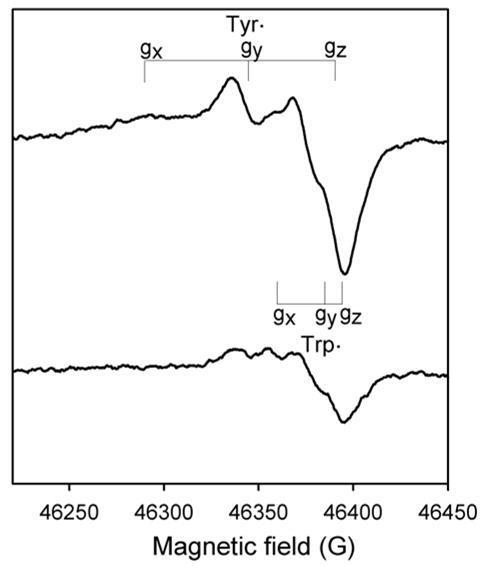
The 800 ms sample (bottom spectrum) was prepared using the RFQ-EPR apparatus modified to load the sample into a quartz capillary. Final concentrations: KatG, 300 μM, PAA, 900 μM. The 10-sec reaction sample (top spectrum) was mixed and loaded into the capillary manually. The g-tensor components of the tyrosyl radical and the tryptophanyl radical are indicated. The spectra are derivatives of echo-detected spectra obtained at 7 K. Other acquisition parameters are given in the Experimental Procedures.
The RFQ sample (~ 800 msec) used to obtain the D-band spectrum was prepared as follows. The 0.55 mm OD quartz capillary tube was fitted inside the end of the 700 ms reactor tube of the RFQ instrument. The capillary was immersed in liquid nitrogen, and the reaction mixture was then injected into the immersed tube. Calculations estimate the freeze time of the solution once it enters the tube to be under 100 msec. With the delay time set at 700 msec by choice of the aging loop, the total incubation time for this sample is estimated close to 800 ms.
Simulation of EPR data used Simfonia software (Bruker). Determination of the ring rotation angles, θ, in tyrosyl and tryptophanyl radicals was found from the dihedral angle formed between the phenoxyl (indolyl) ring plane and the R – Cβ-C1 plane by examination of the three-dimensional structure of Mtb KatG (PDB code 2CCA). Software and algorithms by Svistunenko were used for calculation of hyperfine coupling values based on the structure of particular residues (25,26).
Electron-transfer couplings were calculated used PATHWAYs analysis (18) in HARLEM programs (19,20) and were based on the WT KatG crystal structure.
RESULTS
RFQ-EPR spectroscopy of ferric KatG plus peroxide and INH
The RFQ-EPR approach used here has been described previously; briefly, resting (ferric) enzyme is mixed with a 3-fold molar excess of alkyl peroxide (hydrogen peroxide free) and sample mixtures are frozen in liquid isopentane at −130 °C in precision bore EPR tubes. This small excess of peroxide is sufficient to generate Cmpd I in the enzyme and maximize the yield of radicals (10). Spectra are then recorded at 77 K. The time regime available here ranges from 6.4 ms to any longer period of incubation after rapid mixing.
Previously reported RFQ-EPR results of experiments in which INH was added to pre-formed radical species in WT KatG (10) demonstrated quenching of EPR signals. Here, reaction of the resting enzyme with peroxyacetic acid in the presence of INH elicited a new EPR signal (Fig. 2) different from that found previously. A doublet was detected (~ 17 G linewidth) at the earliest time points (for example, 6.4 ms) and after 50 ms reaction time was only barely detectable. Without INH, a broad doublet (~ 29 G) was consistently observed (10). The new narrow doublet (ND) signal is reasonably assigned to a radical on the same residue as that giving the broad doublet in WT KatG that has undergone a structural rearrangement due to the binding of INH (see description below of how these signals depend on the orientation of the phenoxyl ring plane in tyrosyl radicals). It is important to note that the narrow doublet signal is not due to a hydrazyl or other radicals derived from INH (27) which is a peroxidase substrate of KatG (28) known to react with Cmpd I and with protein-based radicals (10). A hydrazyl radical would exhibit a more complicated spectrum analogous to that of amine radical powder pattern spectra at low temperature, due to the nitrogen and hydrogen nuclear hyperfine couplings (29); other radicals missing the hydrazyl group formed upon breakdown of an INH-hydrazyl radical (27) would also exhibit more complex spectra due to hyperfine couplings to the ring hydrogens as well as the pyridine nitrogen nucleus. A peroxyl radical also identified during the breakdown of INH radicals may also be ruled out because they exhibit axial g-tensors with EPR features around g = 2.035 and 2.006.
Fig. 2. RFQ-EPR spectra of Mtb KatG.
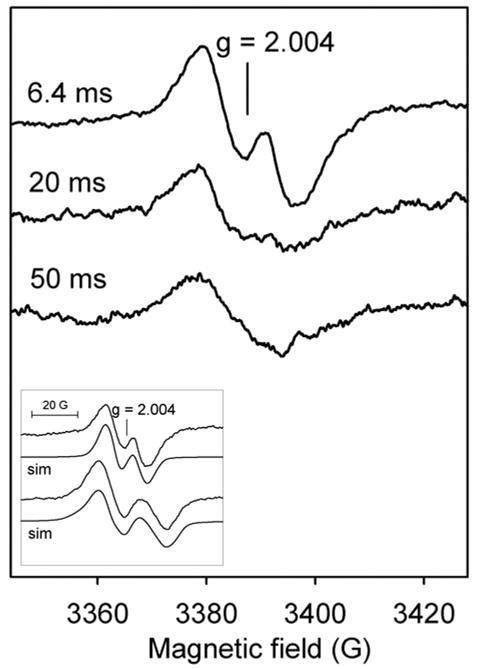
Resting enzyme (wild-type KatG, 50 μM final) was reacted with a mixture of INH (10 mM) and PAA (150 μM final) at 25 °C (narrow doublet). Incubation times before freezing are indicated. Inset: the experimental narrow and wide doublets with simulated spectra based on the parameters in Table 1.
Simulation of the new ND signal was performed, according to the parameters for a tyrosyl radical given in Table 1. A satisfactory simulation was obtained with Aβ1iso ~ 9 G and Aβ2iso ~ 2 G (Fig. 2, inset). No satisfactory simulation was produced assuming the ND signal is due to a neutral tryptophanyl radical using reasonable values for Aβ1,2iso, A5′,7′ and A14N (30)2.
Table 1.
EPR parameters used for simulation of tyrosyl radicalsa.
| Wide doublet (Fig. 2, inset) θ = 33° | Narrow doublet (Fig. 2) θ = 52° | Narrow singlet (Fig. 3) θ = 60° |
|---|---|---|

| ||
| Aβ1b:
Ax = 15.00 Ay = 15.00 Az = 16.00 |
Aβ1:
Ax = 8.00 Ay = 10.00 Az = 10.00 |
Aβ1:
Ax = 6.00 Ay = 6.00 Az = 6.00 |
| Aβ2:
Ax = 1.20 Ay = 1.20 Az = 1.50 |
Aβ2:
Ax = 1.80 Ay = 1.80 Az = 2.00 |
Aβ2:
Ax = 6.00 Ay = 6.00 Az = 6.00 |
| Ac3′,5′H:
Ax = 9.00 Ay = 2.50 Az = 5.50 |
A3′,5′H:
Ax = 6.00 Ay = 2.50 Az = 5.00 |
A3′,5′H:
Ax = 9.00 Ay = 2.50 Az = 5.50 |
The g - tensor values were taken from the 130-GHz EPR spectrum and assumed to be the same for all species: gx = 2.0064, gy = 2.0042, gz = 2.0022.
Absolute values for hyperfine coupling constants are given in G and were varied to attain the best fit to the experimental spectrum.
Hyperfine coupling constants for the 3′ and 5′ hydrogens (G) adopted from ENDOR results (37).
The hyperfine splittings in EPR spectra of tyrosyl (and tryptophanyl) radicals are determined by the known dependence of β-methylene hyperfine couplings on the phenoxyl ring plane orientation, and on the relatively constant hyperfine coupling interactions for the ring protons and has been extensively described elsewhere (25,30–33). The orientation dependence allows prediction of the angle formed between the ring plane and the Cβ - Hβ1 and Hβ2 hydrogens; alternatively, when the structure of a particular tyrosine residue is known, prediction of the hyperfine coupling constants can be made. Svistunenko has presented a user-friendly approach relied upon here for the analysis of EPR spectra (25,34).
For the wide doublet (WD) signal (Fig. 2, inset), the hyperfine coupling values are consistent with an angle, θ, close to 30 degrees. For the ND signal found in the presence of INH, the reduced splitting indicates an angle around 52 degrees. The most interesting aspect of the effect of INH is that its binding, reported to be within the heme pocket, provides clues for the identity of the residue forming the initial tyrosyl radical doublets (35,36) (see below).
Other RFQ-EPR experiments were performed using wild type KatG in the absence of the drug in order to get more information about the principal radical species forming on the millisecond to seconds time scale. For example, the initial WD signal is replaced by an apparent wide singlet (WS) (20 G) over 10 s (Fig. 3, spectrum A) (10). Here, incubation times were extended up to several minutes and further transition to a singlet with a linewidth of ~ 15 G (spectra B and C) was found. No hyperfine splitting was detectable for this signal even under optimized spectrometer conditions. The narrow singlet (NS) can be simulated using hyperfine couplings of 6 G for two β-methylene hydrogens in tyrosyl radical (33,37) (Fig. 3, dotted trace) (Table 1) consistent with a phenoxyl ring rotation angle, θ, close to 60 degrees (25).
Fig. 3. RFQ-EPR spectrum of KatG[W107F].
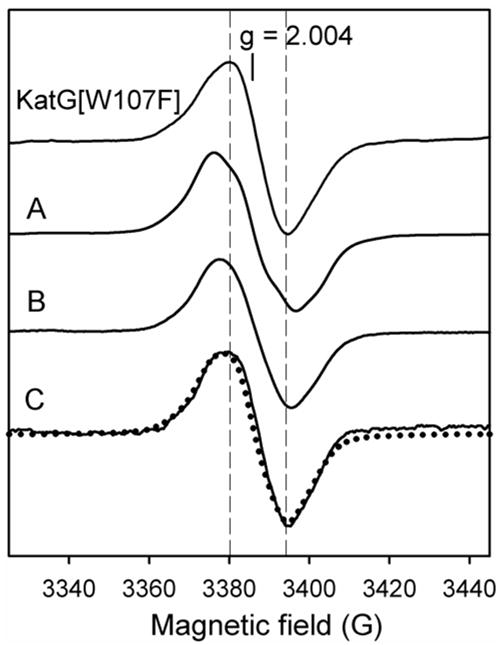
Resting enzyme (50 μM final) was reacted with PAA (150 μM final) at 25 °C and frozen after 6.4 ms reaction (top spectrum, narrow singlet). Also shown are spectra of wild type KatG frozen at the indicated reaction times after manual mixing (same concentrations): A – 10 sec (wide singlet), B – 2 min, C – 5 min (narrow singlet). The dotted spectrum is a simulation of the narrow singlet based on the parameters given in Table 1.
Do these observations help identify the residue(s) on which the radicals are formed? The NS signal is addressed first. The recently reported X-ray crystal structure of M. tuberculosis KatG (26,35) provides the opportunity to predict β-methylene hydrogen hyperfine couplings for any tyrosine residue, an approach greatly facilitated using the software designed by Svistunenko (25). A tyrosyl radical had been identified on residue Y353 in WT KatG (11) using a nitric oxide trapping approach. This finding coupled with the observation that Y353 has a ring orientation angle of 60 degrees in the Mtb KatG crystal structure, substantiates the assignment to this residue of the NS signal in wild-type KatG. Additional evidence lending credence to this assignment comes from the RFQ-EPR results for KatG[Y353F] (11).
For completeness, however, and based solely on the conformation of phenoxyl rings observed for other tyrosines in the crystal structure of Mtb KatG, residues Y210 and Y339 may also yield a narrow singlet EPR signal. Both of these are more than 20 Å from heme iron, while Y353 is 14 Å away. The PATHWAYs analysis predicts poor redox coupling to the heme for residues Y210 and Y339 compared to Y353 (see supplementary material); other candidates (Y155, Y113) are ruled out because KatG[Y155S] and KatG[Y113F] mutants exhibit close similarity to WT KatG in their RFQ-EPR spectra (8,10). Other tyrosines are predicted either to give a doublet EPR signal or are beyond 20 from the heme and poorly coupled for electron transfer. The fact that KatG[Y353F] still exhibits some narrow singlet EPR signal demonstrates the possible involvement of other residues, and also highlights the difficulty in making assignments based solely on EPR results for a single mutation.
Additional analysis of RFQ-EPR results (the doublet signals) follows below.
High-field EPR spectroscopy
The use of HF-EPR spectroscopy allows resolution of g-tensor elements that differentiate tryptophanyl and tyrosyl radicals. Here, the application of rapid freeze-quench sample preparation and HF-EPR data, first of all, confirmed the presence of tyrosyl radical as the majority species and also revealed a signal reasonably assigned to a Trp radical in WT KatG, in samples frozen 800 ms or 10 s after mixing resting enzyme with PAA (Fig. 4). The rhombic signal characteristic of tyrosyl radical signal is indicated by the g-tensor values (gx = 2.0064, gy = 2.0042 and gz = 2.0022) (33,37) while the features characteristic of tryptophanyl radical (gx = 2.0035, gy = 2.0024 and gz = 2.0020) (30,38,39) are not fully resolved in the data at 130 GHz. Overlap also occurs between the components of the tryptophanyl radical signal in the gmin region of the tyrosyl radical. The g-values for these signals are similar to those reported for tryptophanyl and tyrosyl radicals in Synechocystis KatG (13). The majority species for samples frozen at the two chosen time points was tyrosyl radical with the contribution of tryptophanyl radical giving only an apparently comparable intensity in derivative spectra because of its reduced g-tensor anisotropy compared to the Tyr radical signal. Estimation of the contribution of tryptophanyl radical suggests an upper limit of 30% of the total intensity (Fig. 4, top spectrum).
A reexamination of the earlier X-band RFQ-EPR results (10) for corresponding evidence for Trp radical was warranted, given also that HF-EPR data is not available at very early time points (impracticable due to the very low signal intensities). A reasonable approach was based on the fact that spectra recorded for L-3,3-[2H]2-tyrosine labeled KatG, in which the tyrosyl radical doublet appears as a singlet due to the change in hyperfine couplings upon isotope substitution (10), contains some intensity not previously accounted for and assumed here to arise from a tryptophanyl radical doublet (Fig. 5, spectrum A). (A doublet, was assumed because the signal in unlabeled WT KatG at similar time points is a doublet, meaning that a contribution from a tryptophanyl radical likely overlaps the tyrosyl radical signal.) Therefore, a neutral tryptophanyl radical doublet EPR spectrum was simulated to give a linewidth that would produce the wings of intensity; β-methylene hydrogen hyperfine coupling values of 14 – 17 G and typical hyperfine coupling values for the 5′, 7′ – indolyl ring hydrogens and the indole nitrogen (Fig. 5, spectrum B) were used. These values are very similar to those reported for the neutral tryptophanyl radical in the W177Y mutant of ribonucleotide reductase (30). To simulate the tyrosyl radical component of the spectrum for the L-3,3-[2H]2 tyrosine labeled KatG sample, hydrogen hyperfine coupling parameters from the corresponding wide doublet in unlabeled enzyme (Fig. 5, spectrum C) were adjusted by the ratio gn(2H)/gn(1H) = 0.1535 (Table 1). A sum composed of 80% deuterated tyrosyl (Fig. 5, spectrum C) and 20% tryptophanyl radical (spectrum B) provided a reasonable fit to the experimental spectrum (spectrum D). This result accounts for a small contribution of a Trp radical in the X-band RFQ-EPR experiments as early as 200–250 ms. (Recall that HF-EPR spectrum confirmed the tryptophanyl radical at 800 ms reaction time.) A Trp radical is also found in the HF-EPR results at 10 s, but at this time point, no evidence for its contribution in X-band EPR spectra can be detected most likely due to its very low intensity. Factors contributing to this difference are the inherent greater sensitivity of the HF-EPR experiment and the improved resolution compared to X-band measurements.
Fig. 5. RFQ-EPR spectra and simulations of the protein-based radical signals in Mtb KatG.
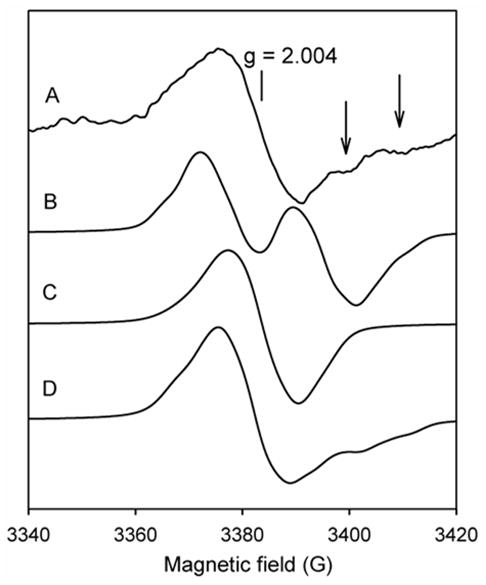
L-3,3-[2H]2 tyrosine-labeled KatG (50 μM final) mixed with PAA (150 μM final) and frozen after 250 ms reaction at 25 °C (A). (The spectrum was taken from (10), Fig. 3e). Simulation of the tryptophanyl radical signal (wide doublet) (B). Simulation of the deuterium-substituted tyrosyl radical spectrum using adjusted hyperfine coupling parameters obtained from simulation of the doublet for unlabeled KatG (C). The simulated sum spectrum composed of 80% deuterium-substituted tyrosyl radical and 20% tryptophanyl radical signal intensities (D); g- tensor and hyperfine coupling values are given in Tables 1 and 2.
The same approach was used to simulate a spectrum of unlabeled WT KatG data and in this case also, a small contribution of Trp radical included in a simulation sum improves the fitting compared to a simulation of tyrosyl radical alone (not shown). The hyperfine coupling values that give reasonable simulations for the tryptophanyl radical contribution in the data. (indole ring orientation angle around 86°) correspond to the structure of residue W107 in the X-ray crystal structure though other possibilities exist. Residue W107 is assigned the best redox coupling factor for electron transfer to the heme in Mtb KatG (see supplemental data). W321, which also has a good electron-transfer coupling value, had been ruled out for stabilization of a radical in both the Mtb and the Synechocystis KatG enzymes (23,40).
Rapid freeze-quench EPR spectroscopy of KatG[W107F] and KatG[W91F] mutants
RFQ-EPR experiments using two KatG mutants, W107F, chosen because of the evidence above and its proximity to the heme (~ 5 Å) and W91F, because it has been identified as an important site for radical formation in Synechocystis KatG, were also performed for some confirmation of assignment.
KatG[W107F] was mixed with peroxyacetic acid and samples were frozen at incubation times from 6.4 ms to 30 s. These conditions are known from optical stopped-flow measurements to generate Compound I (not shown) and to also result in cycling of the enzymes back to the resting state, consistent with endogenous electron transfer processes (the second order rate constant for Compound I formation was found to be 1.43 × 106 M−1.s−1, which is 120-fold faster than WT KatG while the spontaneous return to the resting state was somewhat slower for the mutant (50 sec compared to 40 sec for the wild type (10)). Interestingly, a singlet with linewidth ~ 15 G, centered at g = 2.0040 appeared at all time points accessible (Fig. 3, the top spectrum). The maximum yield of the radical (calculated as spins/mol heme) was found at the earliest time points, equivalent to 0.06 spins/mol heme, which is less than one third the maximum radical yield reported for WT KatG (10). The EPR signal intensity decayed slowly and the same signal could still be observed after 30 sec reaction time without any significant change, suggesting that a single radical species was formed. According to reasoning above, the disappearance of the doublet upon mutation of W107 should not be taken as evidence that the doublet signal arises exclusively from a radical on this Trp residue because the principal signal at the early time points is due to tyrosyl radical. The lack of doublet through the entire reaction time frame here suggests that either the electron-transfer pathway leading to a radical giving the WD is disrupted in the mutant, or the radical is localized only on the secondary site, Y353, both ideas being consistent with the results from PATHWAYs analysis; coupling between residue Y229 and the heme requires W107 but for Y353 this tryptophan is not required (see supplementary material). Alternatively, the structure of the tyrosyl radical giving the WD in WT KatG is altered such that it now appears as a singlet by mutation of W107. Both possibilities would be consistent with the initial doublet assigned to residue Y229.
In Synechocystis KatG, a contribution from a radical on W122, the residue corresponding to W107 in the Mtb enzyme, was ruled out for radicals detected after 5 or 10 s incubations and instead, W106 (W91 in Mtb KatG) was assigned as the most likely residue for a tryptophanyl radical in that time frame (13,41). Figure 6 shows RFQ-EPR results for Mtb KatG[W91F]. The spectra for this mutant were nearly identical to those of WT KatG, including the linewidth and signal intensity, as well as the kinetics of evolution from a doublet signal (at milliseconds) to a singlet (after 500 ms) (10). The figure also shows a difference spectrum of normalized data from WT KatG and the mutant, for samples frozen at 200 ms (Fig. 6, the dashed spectrum). This difference spectrum does not reveal any significant EPR signal intensity, ruling out a contribution from a radical on residue W91 in the millisecond time scale. For the 10 s time point, the WT and mutant enzyme spectra are perfectly overlapped (Fig. 6, spectra C). Note that PATHWAYs analysis suggests 10 – 100 fold weaker coupling between residue W91 and the heme compared to residues Y229 and W107, respectively. However, the analysis predicts better coupling between W91 and the heme compared to Y353; therefore, a barely detectable contribution from a W91 radical, which according to the X-ray crystal structure would produce a doublet signal, remains a possibility.
Fig. 6. RFQ-EPR spectra of WT KatG (solid lines) and KatG[W91F] mutant (dotted lines).
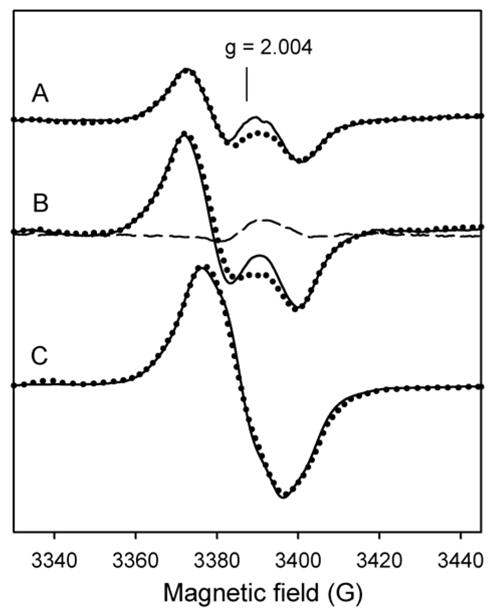
Resting enzymes (50 μM final) were mixed with peroxyacetic acid (150 μM final) at 25 °C. Reaction mixtures were freeze-quenched after (A) 50 ms; (B) 200 ms (wide doublet); (C) 10 s (wide singlet). The difference spectrum (dashed line) obtained by subtraction of the normalized experimental spectra of the wild type and KatG[W91F] is also shown.
RFQ – EPR spectroscopy of KatG[Y229F] and KatG[W107F][Y229F] mutants
For KatG[Y229F], previously reported results (8,10) showed a singlet signal in RFQ-EPR samples frozen at or after 10 ms reaction with peroxide (in one sample out of three separate experiments using enzyme from three preparations, a very short lived doublet signal was detected only below 10 ms, but could not be reproduced; this is noteworthy because it may be evidence for a short-lived radical on W107 in the Y229F mutant.) The EPR signal intensity also showed a progressive decrease after the maximum of 0.09 spins/mol heme reached at 100 ms, which was less than 50% of the yield in WT KatG. The most straightforward suggestion is that the principal doublet signal in WT KatG arises from a tyrosyl radical on residue Y229. PATHWAYs analysis (18–20) predicts the strongest coupling between that residue and the heme among all tyrosines (supplemental data). It does not show reasonable coupling between other tyrosine candidates and the heme requiring Y229 for electron transfer, evidence that in this mutant a primary radical site is lost.
Simulation of the EPR spectrum of the doublet here could make use of an experimentally determined value for the spin density on C1 (0.42), which was not available for our previous report. This value was calculated according to the gmax value found by HF-EPR data (see above and (25)). β-Methylene hydrogen hyperfine coupling values consistent with a ring rotation angle θ ~ 30° were suitable for this simulation (Table 1). This is the angle found for the phenoxyl ring of residue Y229 in the Mtb KatG crystal structure. A doublet spectrum matching the experimental one, however, can only be obtained if hyperfine interactions with the 3′ and 5′ hydrogens are included (simulations without these protons do not come close to reproducing the data). This result rules out a radical localized in the phenoxyl ring of Y229 within the three amino acid adduct. In any case, such a radical would exhibit significantly reduced hyperfine couplings because of significantly reduced spin density due to the methionyl-sulfur modification of the ring. Preliminary DFT calculations showed ~ 35% reduction in spin density on C1 of Y229 within a hypothetical adduct radical compared to the generally reported spin densities for free tyrosyl radicals (A. Jarzecki, personal communication). This means that if a radical is being detected on Y229 in WT KatG, it occurs on the free side chain. Nevertheless, the ring orientation derived here is close to that found for Y229 in the crystal structure.
To continue addressing issues relating to the assignment of the doublet signal in WT KatG, rapid-freeze quench EPR experiments were performed as above using the double mutant KatG[W107F][Y229F] (Fig. 7, spectrum A). Here, the major contribution after 250 ms reaction time came from a new, axial EPR signal with gII = 2.035 and g⊥ = 2.006. These features are characteristic of peroxyl radicals. A small contribution of another signal is also present. The maximum total yield was found to be 0.08 spins/heme (40% of typical yield compared to wild type). Interestingly, abundant peroxyl radical signal was also observed in RFQ-EPR samples prepared using wild type KatG that had been pre-treated with excess PAA (Fig. 7, spectrum B). Peroxyl radicals all have very similar EPR spectra and whether these are protein-based radicals cannot be confirmed at this time. Tryptophan peroxyl radical(s) are however commonly found in heme proteins treated with peroxide (34,42) under aerobic conditions. Therefore, the rapid freeze-quench results for the double mutant [W107F][Y229F] are taken as evidence for rapidly formed tryptophan peroxyl radical(s) that does not form abundantly in WT KatG. For both the double mutant and the peroxide pre-treated WT KatG, these observations suggest that removal of residues W107 and Y229, or ensuring their complete incorporation in the distal side adduct by pre-cycling with peroxide, eliminates them as primary radical sites.
Fig. 7. RFQ-EPR spectra of KatG[W107F][Y229F].
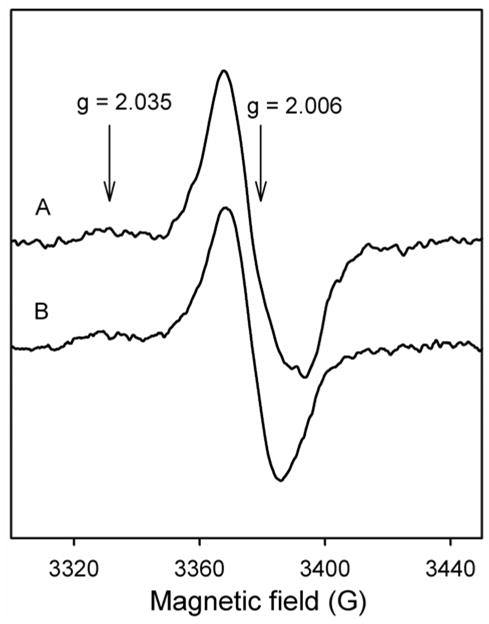
Resting enzyme (50 μM final) was mixed with PAA (150 μM final) and freeze-quenched after 250 ms (A). The WT KatG had been pre-treated with a 10-fold excess of PAA (B) (see text). g-values of peroxyl radical are indicated.
DISCUSSION
In this work, new insights are presented into radical formation in Mtb KatG both on the millisecond time scale and at longer intervals during turnover of the enzyme with alkyl peroxide. The approach was designed to optimize endogenous radical formation, while the issue of identification of physiologically relevant peroxide substrates for KatG remains to be addressed. INH is activated by KatG in vivo under conditions allowing the enzyme to function as a peroxidase and in such reactions, Compound I may be reduced by electron transfer producing a protein-based radical no matter which substrate initiates the reaction cycle. Evidence points to residues Y229 and W107 as sites for the initially formed radicals.
While radicals form and persist well beyond the time intervals accessed using the RFQ-EPR spectroscopy approach, it has become clear that they do not represent the same species produced on the millisecond time scale in Mtb KatG. Previous work provided good evidence for a radical on residue Y353 in Mtb KatG based on formation of nitrotyrosine only on that residue after NO trapping. This residue is not the site giving the initial doublet signal observed in WT KatG as KatG[Y353F] still forms the same doublet species (11). Instead, Y353 is a reasonable site for the narrow singlet reported and analyzed here. The crystal structure of Mtb KatG allowed prediction of this singlet EPR signal for a Y353 radical. Therefore, Y353 must be considered a secondary radical site, consistent with its distance from the heme and the weak PATHWAYs analysis coupling factor. Earlier results considered whether a single tyrosine could give rise to both the doublet and the singlet signals in WT KatG, as a result of a reorientation of the phenoxyl ring after radical formation. This possibility is now ruled out.
The ND signal as well as the WD are considered here to be localized on the same residue, Y229. This suggestion is based on the observation that the initial WD spectrum is altered in the presence of INH without appearance of any other EPR signal, on information about an INH binding site in the enzyme, and on mutagenesis results. Also, the EPR doublet signal intensity at 6.4 ms in the presence of INH (~ 0.002 spins/heme) is similar to that of the WD signal in the absence of INH. This kinetic similarity also argues in favor of assignment of both signals to the same residue. The linewidth changes observed by EPR spectroscopy may be explained by phenoxyl ring re-orientation caused by INH binding. Alternatively, INH addition could have completely quenched the WD species and allowed a radical to form on a different residue. In either case, the effect of INH provides a clue that the radical and the INH binding site are near one another. (The finding that INH not only produces a new EPR signal but quenches the radicals (10) raises the possibility of a role for the radicals in activation of the antibiotic, known to proceed by oxidation producing drug-based radical intermediates (27). Arguing against an activation mechanism requiring amino-acid based radicals is the direct reduction of Compound I by INH (28). However, this remains an intriguing complexity in explaining the role of KatG in INH activation.)
NMR relaxation studies suggested a binding site for INH over the δ-meso heme edge in KatG (43). Other drug binding information comes from a more theoretical analysis (36,44) implicating residues W107 and H108, located in the distal pocket of KatG, for favorable interactions with the hydrazide moiety of INH (35). Thus, two approaches predict INH binding within the distal pocket. Resonance Raman data for WT KatG and the INH-resistant mutant KatG[S315T] also suggested that INH could be bound within the pocket (45,46). While a more remote binding site has been proposed based on electron density for a small unidentified molecule in the crystal structure of Bp KatG in the region close to a propionate side chain (47), it has not been confirmed. If binding at this remote site is assumed to occur, its proximity to Y304 would lead to the suggestion that the initial doublet signal arises from a tyrosyl radical on this residue. However, Y304 was ruled out by RFQ-EPR data of KatG[Y304F] (11) as the site giving the initial WD signal.
INH binding then, may be considered to have altered the orientation of the phenoxyl ring of Y229 through H-bonding to the W107 indole within the distal pocket (41) but only if these side chains are not covalently attached to one another. This then means that the initial radicals are formed in enzyme lacking the covalent bonds between Cε1 of Y229 and the Cη2 of the indole ring of W107.
Independent evidence for assignment of the doublet EPR signals to Y229 comes from PATHWAYs analyses (19) based on coordinates from the crystal structure of Mtb KatG, which shows that the optimal electron-transfer pathway for the tyrosine donor/heme acceptor path in this enzyme is: (Y229 → W107 → Fe) (electron-transfer coupling factor is 1.89 × 10−3 across a distance of 5.1 Å). Note that this analysis predicts that residue W107 is required for coupling to Y229 and these two residues in particular are the most efficiently coupled to heme for electron transfer.
Other tyrosine residues that would exhibit a doublet EPR signal if they formed radicals include: Y28, Y337 and Y413 (with other candidates, including Y197, ruled out by mutagenesis) (11). Arguing against these as sites for tyrosyl radical formation is their poor electron-transfer coupling to the heme (supplemental data). Also, INH binding in the heme pocket (or even at the potential remote binding site) is not expected to elicit effects on any of these four residues in Mtb KatG. Based on this collection of observations, the most likely candidate for the residue producing the initial doublet signal in Mtb KatG is residue Y229, while a contribution from W107 is also reasonable and accounted for in our analysis.
It is also evident that different radical centers are present in different time regimes in Mtb KatG and that radical formation may not involve the same residues in all KatG enzymes. As no specific catalytic function for a radical on a particular residue has been assigned in a catalase-peroxidase, there is no known requirement for conservation of function for radicals in KatG. Therefore, the lack of evidence for a Trp radical on W91 in Mtb KatG and confirmation of a radical on this site (W106) in Synechocystis KatG is an interesting difference between these two enzymes.
The current results and observations are consistent with 1) formation of radicals on residues immediately surrounding the distal pocket on the millisecond time scale, followed by 2) migration or de novo formation on other residues including Y353, during continued incubation with peroxide. Also relevant to this discussion is the very reasonable proposal that adduct formation involves radicals formed on the distal side Tyr and Trp residues during post-translational processing, requiring peroxidase activity of the heme group (also true for Tyr-Trp cross-linkage in CCP[H52Y]), and more importantly, that the stoichiometry of the adduct contained in overexpressed Mtb KatG is less than 100% (6,7,14). Then, the current results are presented as evidence for the radical processing of enzyme lacking the Met-Tyr-Trp adduct. This suggestion must be reconciled with the fact that the x-ray structures of all KatG enzymes reported to date reveal this adduct rather than the free side chains. The discrepancy may be explained by the fact that storage of the enzyme and the low pH conditions required for crystallization both may favor adduct formation. Heme catalysis is required for this processing and during growth, isolation and purification, and storage of KatG, which depends directly on the level of endogenous peroxides available to initiate heme turnover (6,7,17).
Whether KatG enzymes play different physiological roles as a function of the level of hydrogen peroxide and the extent of post-translational modification is worth understanding. For example, KatG may function as a peroxidase under conditions where peroxide levels are low and a physiological substrate is available, but when bacteria are exposed to high levels ofperoxide, the enzyme can complete formation of the adduct which will transform it into a good catalase. This makes good sense since KatG is the only catalase in Mtb (48,49).
Questions still remain to be answered and a battery of multiple mutants may be one approach to better understanding of electron transfer pathways and comprehensive confirmation of radical formation sites in KatG in the future.
Supplementary Material
Table 2.
EPR parameters used for simulation of neutral tryptophanyl radical.
| EPR signal | g - tensora | Aβ1b | Aβ2b | ANa | A5′a | A7′a |
|---|---|---|---|---|---|---|
| Wide doublet (Fig. 5) | gx = 2.0035
gy = 2.0024 gz = 2.0020 |
Ax = 16.00
Ay = 16.00 Az = 16.00 |
Ax < 1
Ay < 1 Az < 1 |
Ax = 0.50
Ay = 0.50 Az = 9.40 |
Ax = 5.60
Ay = 0.80 Az = 4.60 |
Ax = 0.80
Ay = 5.60 Az = 4.60 |
The g -, AN - and A5′,7′ - tensor values were taken from (30)
Absolute values for hyperfine coupling constants are given in G and were varied to attain the best fit to the experimental spectrum.
The abbreviations used are
- KatG
catalase-peroxidase
- Mtb
Mycobacterium tuberculosis
- WT
wild type
- KatG[W107F] and KatG[W91F]
W107F and W91F mutants of KatG, respectively
- INH
isonicotinic acid hydrazide
- PAA
peroxyacetic acid
- EPR
electron paramagnetic resonance
- RFQ-EPR
rapid freeze-quenched EPR
- HF-EPR
high-field EPR
Footnotes
This work was supported by National Institutes of Health Grant AI-43582 and AI-060014 (National Institute of Allergy and Infectious Diseases).
The principal hyperfine coupling in tyrosyl and tryptophanyl radicals arises from the interaction of unpaired electron spin in the π – orbital system of the phenoxyl (indolyl) ring with the methylene hydrogens, Hβ1 and Hβ2 of Cβ. This leads to essentially isotropic hyperfine couplings for the β-methylene hydrogens. The magnitude of the coupling constants is related to the spin density at C1 (or C3 for Trp radicals) and to the angle between the π – orbital axis of the ring and the methylene hydrogens according to the McConnell equation (Heller, C. and McConnell, H. M. (1960) J. Chem. Phys. 32, 1535–1539): Aβiso = ρCn(B’ + B”cos2θ), where Aβ iso is the isotropic hyperfine coupling constant, ρCn is the spin density on C1 or C3, B’ and B” are constants equal to 0 and 58 G (or 0 and 50 G), respectively, for tyrosyl and tryptophanyl radicals (Fessenden, R. W., and Schuler, R. H. (1963) J. Chem. Phys. 39, 2147–2195; Lendzian, F. (2005) Biochim. Biophys. Acta 1707(1), 67–90).
References
- 1.Rouse DA, DeVito JA, Li Z, Byer H, Morris SL. Mol Microbiol. 1996;22 (3):583–592. doi: 10.1046/j.1365-2958.1996.00133.x. [DOI] [PubMed] [Google Scholar]
- 2.Musser JM. Emerg Infect Dis. 1996;2(1):1–17. doi: 10.3201/eid0201.960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marttila HJ, Soini H, Huovinen P, Viljanen MK. Antimicrob Agents Chemother. 1996;40(9):2187–2189. doi: 10.1128/aac.40.9.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakopitsch C, Droghetti E, Schmuckenschlager F, Furtmuller PG, Smulevich G, Obinger C. J Biol Chem. 2005;280(51):42411–42422. doi: 10.1074/jbc.M508009200. [DOI] [PubMed] [Google Scholar]
- 5.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. J Inorg Biochem. 2006;100(4):568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 6.Jakopitsch C, Kolarich D, Petutschnig G, Furtmuller PG, Obinger C. FEBS Lett. 2003;552:135–140. doi: 10.1016/s0014-5793(03)00901-3. [DOI] [PubMed] [Google Scholar]
- 7.Ghiladi RA, Knudsen GM, Medzihradszky KF, Ortiz de Montellano PR. J Biol Chem. 2005;280(24):22651–22663. doi: 10.1074/jbc.M502486200. [DOI] [PubMed] [Google Scholar]
- 8.Yu S, Girotto S, Zhao X, Magliozzo RS. J Biol Chem. 2003;278(45):44121–44127. doi: 10.1074/jbc.M304757200. [DOI] [PubMed] [Google Scholar]
- 9.Ghiladi RA, Medzihradszky KF, Rusnak FM, Ortiz de Montellano PR. J Am Chem Soc. 2005;127(38):13428–13442. doi: 10.1021/ja054366t. [DOI] [PubMed] [Google Scholar]
- 10.Chouchane S, Girotto S, Yu S, Magliozzo RS. J Biol Chem. 2002;277(45):42633–42638. doi: 10.1074/jbc.M207916200. [DOI] [PubMed] [Google Scholar]
- 11.Zhao X, Girotto S, Yu S, Magliozzo RS. J Biol Chem. 2004;279(9):7606–7612. doi: 10.1074/jbc.M311884200. [DOI] [PubMed] [Google Scholar]
- 12.Ivancich A, Dorlet P, Goodin DB, Un S. J Am Chem Soc. 2001;123(21):5050–5058. doi: 10.1021/ja0036514. [DOI] [PubMed] [Google Scholar]
- 13.Ivancich A, Jakopitsch CMA, Un S, Obinger C. J Am Chem Soc. 2003;125:14093–14102. doi: 10.1021/ja035582+. [DOI] [PubMed] [Google Scholar]
- 14.Ghiladi RA, Medzihradszky KF, Ortiz de Montellano PR. Biochemistry. 2005;44(46):15093–15105. doi: 10.1021/bi051463q. [DOI] [PubMed] [Google Scholar]
- 15.Hillar A, Peters B, Pauls R, Loboda A, Zhang H, Mauk AG, Loewen PC. Biochemistry. 2000;39(19):5868–5875. doi: 10.1021/bi0000059. [DOI] [PubMed] [Google Scholar]
- 16.Carpena X, Loprasert S, Mongkolsuk S, Switala J, Loewen PC, Fita I. J Mol Biol. 2003;327(2):475–489. doi: 10.1016/s0022-2836(03)00122-0. [DOI] [PubMed] [Google Scholar]
- 17.Bhaskar B, Immoos CE, Shimizu H, Sulc F, Farmer PJ, Poulos TL. J Mol Biol. 2003;328(1):157–166. doi: 10.1016/s0022-2836(03)00179-7. [DOI] [PubMed] [Google Scholar]
- 18.Beratan DN, Onuchic JN, Winkler JR, Gray HB. Science. 1992;258:1740–1741. doi: 10.1126/science.1334572. [DOI] [PubMed] [Google Scholar]
- 19.Kurnikov IV. HARLEM - Molecular Modeling Program. 1996–2003 http://www.kurnikov.org/harlem_main.html.
- 20.Liang ZX, Kurnikov IV, Nocek JM, Mauk AG, Beratan DN, Hoffman BM. J Am Chem Soc. 2004;126(9):2785–2798. doi: 10.1021/ja038163l. [DOI] [PubMed] [Google Scholar]
- 21.Johnsson K, Froland WA, Schultz PG. J Biol Chem. 1997;272(5):2834–2840. doi: 10.1074/jbc.272.5.2834. [DOI] [PubMed] [Google Scholar]
- 22.Loewen PC, Stauffer GV. Mol Gen Genet. 1990;224(1):147–151. doi: 10.1007/BF00259461. [DOI] [PubMed] [Google Scholar]
- 23.Yu S, Chouchane S, Magliozzo RS. Protein Sci. 2002;11(1):58–64. doi: 10.1110/ps.09902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burghaus O, Rohrer M, Plato M, Mobius K. Meas Sci Tech. 1992;3:765–774. [Google Scholar]
- 25.Svistunenko DA, Cooper CE. Biophys J. 2004;87(1):582–595. doi: 10.1529/biophysj.104.041046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao X, Yu H, Yu S, Wang F, Sacchettini JC, Magliozzo RS. Biochemistry. 2006;45(13):4131–4140. doi: 10.1021/bi051967o. [DOI] [PubMed] [Google Scholar]
- 27.Sipe HJ, Jr, Jaszewski AR, Mason RP. Chem Res Toxicol. 2004;17(2):226–233. doi: 10.1021/tx0341759. [DOI] [PubMed] [Google Scholar]
- 28.Chouchane S, Lippai I, Magliozzo RS. Biochemistry. 2000;39(32):9975–9983. doi: 10.1021/bi0005815. [DOI] [PubMed] [Google Scholar]
- 29.Janovsky I, Knolle W, Naumov S, Williams F. Chem Eur J. 2004;10:5524–5534. doi: 10.1002/chem.200400401. [DOI] [PubMed] [Google Scholar]
- 30.Lendzian F. Biochim Biophys Acta. 2005;1707(1):67–90. doi: 10.1016/j.bbabio.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 31.Rigby SEJ, Nungent JHA, O’Malley PJ. Biochemistry. 1994;33:1734–1742. doi: 10.1021/bi00173a016. [DOI] [PubMed] [Google Scholar]
- 32.O’Malley PJ, MacFarlane AJ, Rigby SEJ, Nugent JHA. Biochim Biophys Acta. 1995;1232:175–179. [Google Scholar]
- 33.Shi W, Hoganson CW, Espe M, Bender CJ, Babcock GT, Palmer G, Kulmacz RJ, Tsai A. Biochemistry. 2000;39(14):4112–4121. doi: 10.1021/bi992561c. [DOI] [PubMed] [Google Scholar]
- 34.Svistunenko DA. Biochim Biophys Acta. 2001;1546(2):365–378. doi: 10.1016/s0167-4838(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 35.Bertrand T, Eady NA, Jones JN, Jesmin, Nagy JM, Jamart-Gregoire B, Raven EL, Brown KA. J Biol Chem. 2004;279(37):38991–38999. doi: 10.1074/jbc.M402382200. [DOI] [PubMed] [Google Scholar]
- 36.Todorovic S, Juranic N, Macura S, Rusnak F. J Am Chem Soc. 1999;121:10962–10966. [Google Scholar]
- 37.Dorlet P, Seibold SA, Babcock GT, Gerfen GJ, Smith WL, Tsai A-l, Un S. Biochemistry. 2002;41(19):6107–6114. doi: 10.1021/bi015871f. [DOI] [PubMed] [Google Scholar]
- 38.DeGray JA, Lassmann G, Curtis JF, Kennedy TA, Marnett LJ, Eling TE, Mason RP. J Biol Chem. 1992;267(33):23583–23588. [PubMed] [Google Scholar]
- 39.Bleifuss G, Kolberg M, Potsch S, Hofbauer W, Bittl R, Lubitz W, Graslund A, Lassmann G, Lendzian F. Biochemistry. 2001;(40):15362–15368. doi: 10.1021/bi010707d. [DOI] [PubMed] [Google Scholar]
- 40.Jakopitsch C, Regelsberger G, Furtmuller PG, Ruker F, Peschek GA, Obinger C. J Inorg Biochem. 2002;91(1):78–86. doi: 10.1016/s0162-0134(02)00374-4. [DOI] [PubMed] [Google Scholar]
- 41.Jakopitsch C, Obinger C, Un S, Ivancich A. J Inorg Biochem. 2006;100(5–6):1091–1099. doi: 10.1016/j.jinorgbio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Kelman DJ, DeGray JA, Mason RP. J Biol Chem. 1994;269(10):7458–7463. [PubMed] [Google Scholar]
- 43.Pierattelli R, Banci L, Eady NA, Bodiguel J, Jones JN, Moody PC, Raven EL, Jamart-Gregoire B, Brown KA. J Biol Chem. 2004;279(37):39000–39009. doi: 10.1074/jbc.M402384200. [DOI] [PubMed] [Google Scholar]
- 44.Henriksen A, Schuller DJ, Meno K, Welinder KG, Smith AT, Gajhede M. Biochemistry. 1998;37(22):8054–8060. doi: 10.1021/bi980234j. [DOI] [PubMed] [Google Scholar]
- 45.Wengenack NL, Todorovic S, Yu L, Rusnak F. Biochemistry. 1998;37(45):15825–15834. doi: 10.1021/bi982023k. [DOI] [PubMed] [Google Scholar]
- 46.Kapetanaki S, Chouchane S, Girotto S, Yu S, Magliozzo RS, Schelvis JP. Biochemistry. 2003;42(13):3835–3845. doi: 10.1021/bi026992y. [DOI] [PubMed] [Google Scholar]
- 47.Deemagarn T, Carpena X, Singh R, Wiseman B, Fita I, Loewen PC. J Mol Biol. 2005;345(1):21–28. doi: 10.1016/j.jmb.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 48.Sherman DR, Mdluli K, Hickey MJ, Barry CE, Kendall Stover C. Biofactors. 1999;10(2–3):211–217. doi: 10.1002/biof.5520100219. [DOI] [PubMed] [Google Scholar]
- 49.Camus JC, Pryor MJ, Medigue C, Cole ST. Microbiology. 2002;148:2967–2973. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.