

Abstract
Peroxisome proliferator-activated receptor α (PPARα) is responsible for peroxisome proliferator-induced pleiotropic responses, including the development of hepatocellular carcinoma in rodents. However, it remains to be determined whether activation of PPARα only in hepatocytes is sufficient to induce hepatocellular carcinomas. To address this issue, transgenic mice were generated that target constitutively activated PPARα specifically to hepatocytes. The transgenic mice exhibited various responses that mimic wild-type mice treated with peroxisome proliferators, including significantly decreased serum fatty acids and marked induction of PPARα target genes encoding fatty acid oxidation enzymes, suggesting that the transgene functions in the same manner as peroxisome proliferators to regulate fatty acid metabolism. However, the transgenic mice did not develop hepatocellular carcinomas, even though they exhibited peroxisome proliferation and hepatocyte proliferation, indicating that these events are not sufficient to induce liver cancer. In contrast to the transgenic mice, peroxisome proliferators activate proliferation of hepatic nonparenchymal cells. Thus, activation of hepatic nonparechymal cells and/or associated molecular events is an important step in peroxisome proliferators-induced hepatocarcinogenesis.
Keywords: VP16PPARα, LAP, carcinogenesis, hepatocytes, tet-off, doxycycline
Introduction
Peroxisome proliferator chemicals (PP) represent a large and structurally diverse group of agents including fibrate hypolipidemic drugs, phthalate ester plasticizers and herbicides, with a high likelihood of clinical, occupational and environmental exposure to humans [1]. Exposure of rodents to PPs results in pleiotropic responses including hepatomegaly, proliferation of peroxisomes, and induction of genes encoding mitochondrial, peroxisomal, and microsomal fatty acid oxidation enzymes [2–5]. In addition, although PPs have not been shown to act as genotoxic carcinogens, long-term treatment of rats and mice with PPs results in 100% incidence of hepatocellular carcinomas [5–7].
An increased understanding of the mechanism underlying the effects of PPs was initiated by identification of the nuclear receptor known as peroxisome proliferator-activated receptor α (PPARα) [8]. Binding and activation of PPARα by PPs are essential for induction of peroxisome proliferation and fatty acid metabolizing enzymes, hepatomegaly and ultimately liver cancer after prolonged exposure of PPs to rodents. PPARα is a member of the nuclear hormone receptor superfamily and three PPAR (α, β and γ) genes were identified that are expressed in different tissues or organs. Among the three, PPARα is mainly expressed in liver, kidney and heart. PPARs function as transcription factors through the classic ligand-dependent nuclear hormone receptor mechanism. Upon binding to their ligands, PPARs undergo conformational changes that allow co-repressor release and co-activator recruitment, heterodimerization with retinoid X receptor (RXR), and selective binding to specific DNA sequences termed peroxisome proliferators response elements (PPREs) in the promoters of target genes. PPARα serves a fundamental role in mammals by acting as a central modulator of signaling molecules that mediate changes in gene expression to maintain lipid homeostasis. In addition, PPARα has also been linked to the regulation of genes important in cell growth and differentiation [9,10].
Use of the Pparα-null mouse model revealed that PPARα is responsible for PP-induced pleiotropic responses, including the development of hepatocellular carcinoma in mice, as these null mice were refractory to all known PP-induced pleiotropic responses including peroxisome proliferation, hepatomegally and hepatocellular carcinomas [11,12]. While carcinogenesis is a complicated multistage process, the induction of oxidative stress and lipid peroxidation, increase in hepatocyte proliferation and/or decrease in apoptosis are central components of the cascade of molecular events leading to liver tumors in rodents fed PPs [5]. However, the link between PPARα and hepatocarcinogenesis in rodents at the molecular and cellular level is not fully understood. To clarify whether activation of PPARα only in hepatocytes is sufficient to the induction of hepatic neoplasia and whether nonparenchymal hepatic cells contribute to the carcinogenesis process, the potent viral transcriptional activator VP16 was fused to the mouse PPARα cDNA to create a transcription factor that constitutively activates PPARα-responsive genes in the absence of ligands. Transgenic mice were produced whereby inducible expression of the VP16PPARα transgene was targeted to hepatocytes using the tetracycline regulatory system under the control of the liver-enriched activator protein (LAP or cEBPβ) promoter [13,14] to determine whether activation of PPARα in hepatocytes can produce same hepatic responses as PPARα ligand treatment in mouse. In comparison with wild-type mice treated with the potent PPARα ligand Wy-14,643, LAP-VP16PPARα mice exhibited significantly decreased serum triglycerides and free fatty acids, peroxisome proliferation and marked induction of PPARα target genes encoding fatty acid βoxidation enzymes. However, the VP16PPARα transgenic mice at one year of age exhibited no gross or microscopic evidence of either preneoplastic hepatic lesions or hepatocellular neoplasia.
Materials and methods
Generation of transgenic mice
The tetracycline response element (TRE) promoter-driving expression of the VP16PPARα fusion protein were recently generated [15]. Briefly, the potent viral transcriptional activator VP16 was fused to the mouse PPARα cDNA to create a transcription factor that constitutively activates PPARα-responsive genes in the absence of ligand. The single transgenic mice were generated with the VP16PPARα fused to the tetracycline response element (TREVP16PPARα) [14]. Subsequently, double transgenic (LAP-VP16PPARα) mice were produced by breeding with LAP/tTA transgenic mice expressing the tetracycline-controlled transactivator (tTA) transgene under the control of the liver-enriched activator protein (LAP or cEBPβ) promoter [13,14] to reconstitute the tetracycline responsive regulatory system [16,17]. TREVP16PPARα mice and LAP/tTA transgenic mice behaved similar to wild-type (Wt) mice throughout the study. Therefore, mice with these three genotypes were grouped together as control littermates unless otherwise specified. Mice expressing both transgenes were subsequently bred into the 129/Sv strain background at least four generations. The transgenic animals were screened by southern blot analysis or polymerase chain reaction (PCR) of tail DNA. The primers for the transgene VP16PPARα, tTA and mouse endogenous PPARα were described previously [14,15,18].
Treatment
Mice were maintained under a standard 12-h light/12-h dark cycle with water and chow provided ad libitum. Handling was in accordance with animal study protocols approved by the National Cancer Institute Animal Care and Use Committee. Some mice were administered doxycycline (dox; 200 mg/kg; Bio-Serv, Frenchtown, NJ) in the diet to regulate expression of the transgene. To compare the effects caused by the transgene and PPARα ligands, wild-type mice (129Sv background) were also administered Wy-14,643 (0.1% (w/w); Bio-Serv, Frenchtown, NJ) in the diet for indicated time.
Serum lipids
For serum analysis, mice were deprived of food for 16 h, blood collected, and then returned to the appropriate diet for an additional 3 days before they were killed. Serum total triglycerides and free fatty acid were measured using a commercial kit (Sigma, St. Louis, MO).
Pamitoyl-CoA oxidation
The liver homogenates in 20% sucrose were centrifuged at 600g for 10 min. The resulting supernatant was centrifuged at 10,000g for 10 min to yield a subcellular fraction containing primarily mitochondria, peroxisomes, and lysosomes. Pamitoyl-CoA oxidation was measured in this subcellular fraction as the reduction of NAD+ at 340 nm in the presence of KCN as an inhibitor of mitochondrial β-oxidation [2,19].
Determination of liver lipid content
Liver triglyceride and cholesterol contents were determined using the previously described procedures [20].
Hepatocyte proliferation and apoptosis
For determination of cell proliferation using BrdU, mice were anesthetized with 2.5% avertin and implanted subcutaneously with an osmotic pump (Alzet model 2001, 1 μl/h DURECT Corporation, Cupertino, CA) containing 200 μl of 16 mg/ml BrdU (Sigma, St. Louis, MO). After 1 week of receiving the respective diet, the mice were killed by overexposure to carbon dioxide, livers removed and weighed, and sections from both the left and median lobes were obtained and fixed in 10% phosphate-buffered formalin (PBF) (Fisher scientific, Fair Lawn, NJ). Sections from the duodenum were also obtained and fixed in PBF. The duodenum sample was used to verify uniform flow of BrdU up to the time the animals were killed. Apoptosis in hepatocytes was analyzed by using terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) (Promega Corporation, Madison, WI) as previously described (14).
Histological Analyses
Mice were killed by over-exposure to carbon dioxide and the livers were excised, fixed in 10% neutral buffered formalin (Fisher Scientific, Fair Lawn, N.J.), embedded in paraffin, and 4- to 6-μm sections were prepared. Sections were stained with hematoxylin and eosin, and were evaluated by light microscopy.
Immunochemistry
Immunostaining for the VP16 tag was performed using polyclonal antibodies (Abcam, Cambridge, MA), and detected by use of the ABC-kit (Vector Laboratory Inc., Burlingame, CA). Immunostaining for BrdU (DakoCytomation, Carpinteria, CA) was performed using monoclonal antibody, which was labeled with biotin by ARK kit (DakoCytomation, Carpinteria, CA) prior to application to tissues. In brief, sections were washed in PBS and incubated in 0.3% hydrogen peroxide in 100% methanol for 30 min at room temperature. The sections were then incubated in citrate buffer (pH 6.0) at 100°C for 10 min. After washing in PBS, the sections were blocked in PBS containing 5% skim milk at room temperature for 30 min. The sections were then rinsed and incubated sequentially with primary antibody (diluted 1: 100 in PBS containing 1% BSA) for 2 h at room temperature, biotinylated goat anti-rabbit IgG (diluted 1: 50 in PBS containing 1% BSA) for 30 min (when polyclonal antibody was used as the primary antibody), avidin-biotinylated peroxidase complex (Vector Laboratory, Burlingame, CA) in PBS for 30 min. The bound antibody was visualized by 3,3′-diaminobenzidine (DAB) as a peroxidase substrate. Sections were rinsed in water, counterstained with hematoxylin (Sigma), dehydrated, and mounted in permanent mounting medium. The BrdU labeling index was determined by counting at least 1500 nuclei/slide (at random high power fields; magnification, 300 ×) and calculated as 100% × (number of stained hepatocyte nuclei/total number of stained + unstained hepatocyte nuclei).
Northern blot analysis
Total RNA was isolated by mechanical disruption of liver tissue in Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. The concentration of RNA was determined by spectrophotometry. Northern blot analysis was carried out as described previously [21]. Briefly, 10 μg of total RNA was electrophoresed on a 1.0% agarose gel containing 0.22 M formaldehyde, transferred to a nylon membrane, and cross-linked by ultraviolet light exposure. Membranes were hybridized in ULTRAhyb buffer (Ambion, Austin, TX) with random primer 32P-labeled cDNA probes [21,22] following the manufacturer’s protocol, and washed with salt/detergent solution using standard procedures.
Quantitative real-time PCR
Real-time PCR reactions were carried out using SYBR Green PCR master mix (AB Applied Biosystems, Warrington, UK) and the ABI PRISM 7900 HT sequence detection system (AB Applied Biosystems). The sequence and GenBank accession number for the forward and reverse primers used to quantify mRNA were: ACOX (AB340914), forward: 5′-CCAGTCTGAAATCAAGAGAAGCGAG-3′ and reverse: 5′-AAAGTGGAAGGCATAGGCGGTG-3′; Cyclin D1 (NM_007631) forward: 5′- ACCCTGACACCAATCTCCTCAAC-3′ and reverse: 5′-TGGATGGCACAATCTCCTTCTG-3′; CDK4 (NM_009870) forward: 5′-TTGTGCAGGTAGGAGTGCTG-3′ and reverse: 5′- TGCCAGAGATGGAGGAGTCT-3′. The following conditions were used for PCR: 95 °C for 15 sec, 94 °C for 10 sec, 60 °C for 30 sec, and 72 °C for 30 sec, in 45 cycles. Relative expression levels of mRNA were normalized to GAPDH and analyzed for statistical significance with the Student’s t test.
Immunoblot Analysis
Immunoblot analysis was carried out on liver homogenates. Proteins were subjected to SDS-PAGE and immunoblotting using monoclonal anti-Catalase (Sigma) or rabbit anti-PMP70 polyconal antibodies (Abcam Inc., Cambridge, MA) as primary, anti-rabbit IgG horseradish peroxidase as secondary antibodies (Sigma), and an enhanced chemiluminescence detection kit (Pierce). Immunoblotting with goat anti-β-actin or anti-GAPDH antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used as loading controls.
Data analysis
All data are presented as the mean ± S.E.M. The differences between groups were assessed by ANOVA. Differences were considered statistically significant at p < 0.05.
Results
Generation and characterization of the LAP-VP16PPARα transgenic mice
Northern blot analysis of liver samples revealed the expression of both endogenous PPARα and transgenic LAP-VP16PPARα mRNA in adult liver samples. The high molecular weight bands corresponding to the endogenous mouse PPARα were detected in all samples, whereas the mRNA corresponding to the transgenic VP16PPARα only appeared in LAP-VP16PPARα mice in the absence of dox (Figure. 1A). As expected, dox repressed the expression of VP16PPARα in LAP-VP16PPARα mice as noted by the absence of VP16PPARα mRNA in dox-treated LAP-VP16PPARα mice. Immunohistochemical analysis of liver sections showed that VP16PPARα proteins (detected by a VP16 antibody) was expressed only in the hepatocytes of LAP-VP16PPARα mice in the absence of dox (Figure 1B), in contrast to the absence of VP16PPARα proteins in Wt or LAP-VP16PPARα mouse liver in the presence of dox (data not shown). Thus, by employing the tetracycline operon regulation system, constitutive expression of the LAP-VP16PPARα transgene in mouse hepatocytes was achieved.
Figure 1. Generation and characterization of the LAP-VP16PPARα mice.
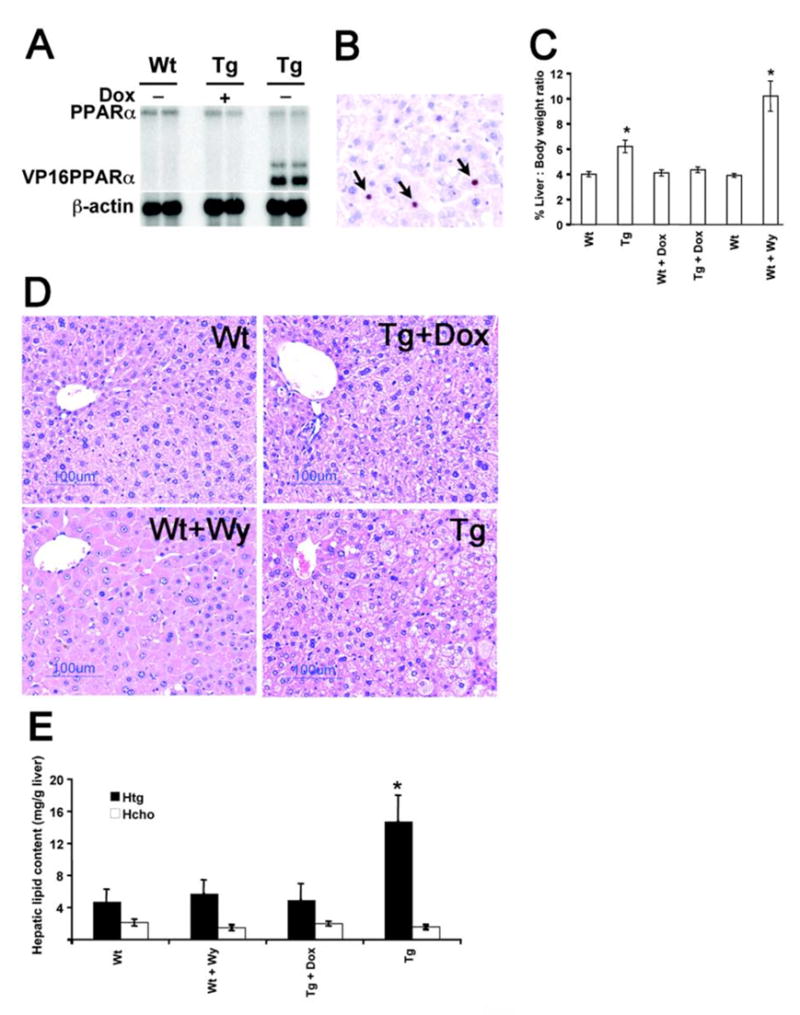
(A) Northern blot analysis of RNA extracted from 8- to10-week-old mouse livers. Expression of the VP16PPARα transgene was observed only in LAP-VP16PPARα (Tg) mice in the absence of doxycycline (dox), whereas expression of endogenous PPARα is found in both lines. As expected, dox represses the expression of VP16PPARα. (B) VP16 immunostaining in 8- to10-week-old mouse livers. Sections of Wt and LAP- VP16PPARα mice in the absence or in the presence of dox were stained with anti-VP16 antibody. Positive cells were visualized with DAB (brown, shown by arrows). Nuclei were counter-stained with hematoxylin (blue). Bars: 125 μm. (C) Increases in percentage of liver:body weight ratio in 8- to10-week-old mice. WY, Wy-14,643. Values are mean ± S.E.M. (n = 4–6); *p < 0.05 compared with Wt control. (D) Histological analysis of 8- to10-week-old mouse livers by H&E staining. Note that the increased hepatocyte size and the eosinophilic cytoplasm were observed in Wt mice treated with Wy-14,643 and fat accumulation was observed in LAP-VP16PPARα mice in the absence of dox. (E) Hepatic triglycerides (Htg) and total cholesterol (Hcho) levels were determined in the liver from 8- to 10-month-old mice. Values are mean ± S.E.M. (n = 4–6); *p < 0.05 compared with Wt control.
Hepatomegaly is one of pleiotropic effects caused by PPs in rats and mice. Hepatomegaly was observed in LAP-VP16PPARα mice in the absence of dox as revealed by the increased liver to body weight ratio compared to Wt mice. As expected, dox treatment abolished this effect, indicating that it is due to expression of the transgene (Figure 1C). However, the extent of hepatomegaly in LAP-VP16PPARα mice was lower than the hepatomegaly induced by two weeks Wy-14,643 treatment in Wt mice. Histological examination of liver sections revealed that Wt mice treated with Wy-14,643 for two weeks had hepatocyte hypertrophy with obvious eosinophilic cytoplasm (Figure 1D). In contrast, no hypertrophy and eosinophilic cytoplasms were observed in LAP-VP16PPARα mice in the absence of dox (Figure 1D). In addition, lipid accumulation was observed in the liver of LAP-VP16PPARα mice in the absence of dox (Figure 1D). Analysis of liver lipid content indicated that the increased lipid accumulation is due to triglycerides, but not cholesterol (Figure 1E). As expected, dox treatment abolished this phenotype, indicating that it is due to expression of the VP16PPARα transgene.
Induction of PPARα target genes and fatty acid oxidation in the LAP-VP16PPARα transgenic mice
To examine whether expression of the VP16PPARα transgene induces PPARα target genes, the expression of known hepatic PPARα target genes were examined through northern blot analysis. Compared to Wt control, the induction of genes encoding enzymes involved in peroxisomal (ACOX, BIEN), mitochondrial (LCAD, VLCAD), and microsomal (CYP4A3) fatty acid catabolism was observed in LAP-VP16PPARα mice in the absence of dox, and the extent of induction was comparable to Wt mice treated with Wy-14,643 for two weeks (Figure 2A). Administration of dox to LAP-VP16PPARα mice abolished these effects, indicating the role of VP16PPARα expression in the phenotypes observed. As these genes contain PPRE binding sites in their promoter regions [23], this result demonstrated that expression of the VP6PPARα transgene in LAP-VP16PPARα mice functions in the same manner to regulate hepatic target gene expression as PPARα activated by ligand in Wt mice.
Figure 2. Induction of PPARα target genes and fatty acid oxidation in the LAP-VP16PPARα transgenic mice.
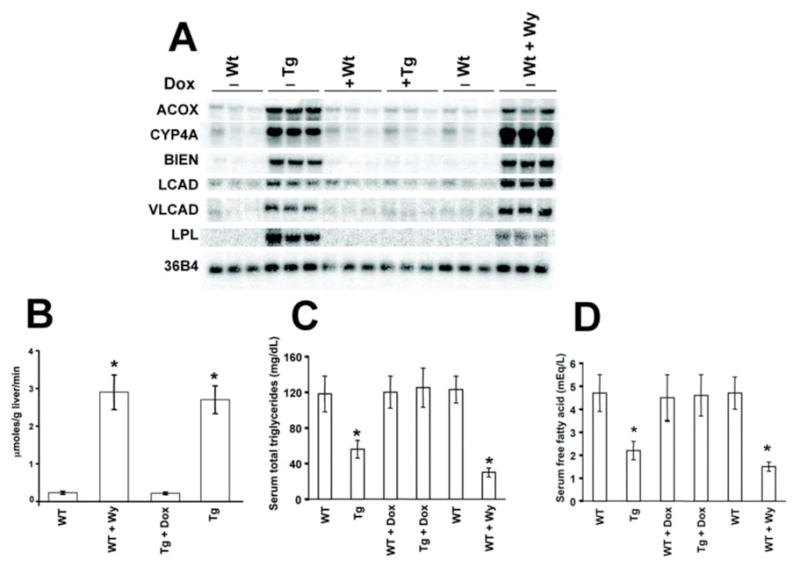
(A) Northern blot analysis of fatty acid oxidation genes in liver total RNA from 8- to10-week-old mice using probes as indicated. Microsomal (CYP4A, cytochrome P450 4A family), peroxisomal (ACOX, acyl-CoA oxidase and BIEN, the bifunctional enzyme), and mitochondrial fatty acid oxidation genes (LCAD, long chain acyl-CoA dehydrogenase and VLCAD, very long chain acyl-CoA dehydrogenase), 36B4 as a loading control. Note that both Wt mice treated with Wy and LAP-VP16PPARα mice in the absence of dox showed the increased induction of these genes. (B) Effects of activation of PPARα on hepatic palmitoyl-CoA oxidation in 8- to10-week-old mouse livers. Values are mean ± S.E.M. (n = 3–4); *P < 0.05 compared with Wt control. Note that both Wt mice treated with Wy-14,643 and LAP-VP16PPARα mice in the absence of dox showed increased palmitoyl-CoA oxidation. (C, D) Serum total triglycerides and free fatty acids in 8–10 weeks old mice. Values are mean ± S.E.M. (n = 4–6); *p < 0.05 compared with Wt control. Note that both Wt mice treated with Wy- 14,643 and LAP-VP16PPARα mice in the absence of dox showed a decreased in these parameters.
To examine whether induction of these genes changed fatty acid metabolism, the palmitoyl-CoA oxidation activity was examined. Compared to Wt control, the induction of palmitoyl-CoA oxidation was observed in LAP- VP16PPARα mice in the absence of dox, and the extent of induction was comparable to Wt mice treated with Wy-14,643 for two weeks (Figure 2B). Administration of dox to LAP- VP16PPARα mice abolished this effect, indicating that it is due to the expression of VP16PPARα. As a result, in the absence of dox, LAP-VP16PPARα mice also had significantly decreased serum triglycerides (Figure 2C) and free fatty acids (Figure 2D) compared to Wt mice. These effects were comparable to Wt mice treated with Wy-14,643 for two weeks (Figure 2C and 2D). Both effects were also abolished upon dox treatment. This indicated that expression of the VP16PPARα transgene in mouse hepatocytes is sufficient to reduce serum lipids.
Induction of peroxisome proliferation in the LAP-VP16PPARα transgenic mice
The hallmark feature of rodents administered with PPs is peroxisome proliferation. To determine whether peroxisome proliferation occurred by expression of the transgene VP16PPARα, the protein levels of the major peroxisomal membrane protein 70 (PMP70), a marker of peroxisome proliferation, were examined by western blot analysis. Compared to Wt control mice, a robust induction of PMP70 was observed in LAP-VP16PPARα mice in the absence of dox; the extent of induction was similar to that observed in the Wt mice treated with Wy-14,643 for two weeks (Figure 3A). As expected, dox treatment abolished this induction, indicating that it was due to expression of the VP16PPARα transgene. In combination with the induction of genes encoding the peroxisomal ACOX and BIEN enzymes in LAP-VP16PPARα mice (Figure 2A), this result indicated that activation of PPARα only in hepatocytes is sufficient to induce peroxisome proliferation. As peroxisome proliferation is always associated with an induction in expression of catalase, catalase protein levels were determined by western blot analysis. An increase in catalase was observed in Wt mice treated with Wy-14,643 as compared with untreated Wt controls (Figure 3B). However, no changes were observed in LAP-VP16PPARα mice in the absence of dox compared to control, suggesting that activation of PPARα only in hepatocytes is not sufficient to induce catalase expression.
Figure 3. Induction of peroxisome proliferation in the LAP-VP16PPARα mice.
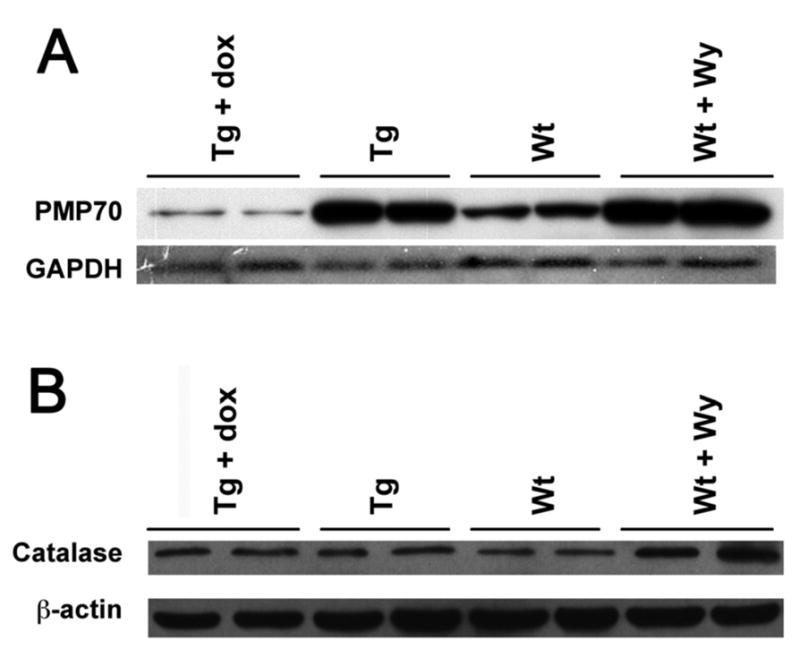
Western bolt analysis of peroxisome proliferation marker enzymes in liver total protein from 8–10 weeks old mice using antibody indicated. (A) PMP70 peroxsiomal membrane protein 70, (B) catalase. Note that both Wt mice treated with Wy-14,643 and LAP- VP16PPARα (Tg) mice in the absence of dox showed the increased of PMP70, however, only Wt mice treated with Wy-14,643 showed increased catalase protein expression.
Induction of hepatocyte proliferation in the LAP-VP16PPARα transgenic mice
Hepatocyte proliferation is considered as a key step in PP-induced hepatocellular carcinomas. Hepatocyte proliferation was determined by measuring BrdU incorporation into hepatocyte nuclei. Immunohistochemical analysis revealed a high degree of incorporation of BrdU in livers of Wt mice after two weeks Wy-14,643 treatment (Figure 4A) with a labeling index average of 20.5 % compared with 1.6% in untreated Wt controls (Figure 4B). In the absence of dox, LAP-VP16PPARα mice also had a high degree of incorporation of BrdU (Figure 4A) with an average labeling index of 20.8% compared with 1.0% in LAP-VP16PPARα mice in the presence of dox (Figure 4B). As expected, dox treatment abolished this induction, indicating that it is due to expression of the VP16PPARα transgene. Interestingly, nonparenchymal cell proliferation was not observed in LAP-VP16PPARα mice in the absence of dox in contrast to the dramatic proliferation of these cells in Wy-14,643-treated Wt mice (Figure 4A). In addition, no significant increases in numbers of apoptotic cells were observed in the Wt mice treated with Wy-14,643 or in LAP-VP16PPARα mice in the absence of dox as determined by the TUNEL assay (data not shown). These data demonstrated that administration of PPs to mice induced both hepatocyte and nonparenchymal cell proliferation and activation of PPARα only in hepatocytes induce hepatocyte proliferation with no evidence for nonparenchymal cell proliferation.
Figure 4. Induction of hepatocyte proliferation in the LAP-VP16PPARα mice.
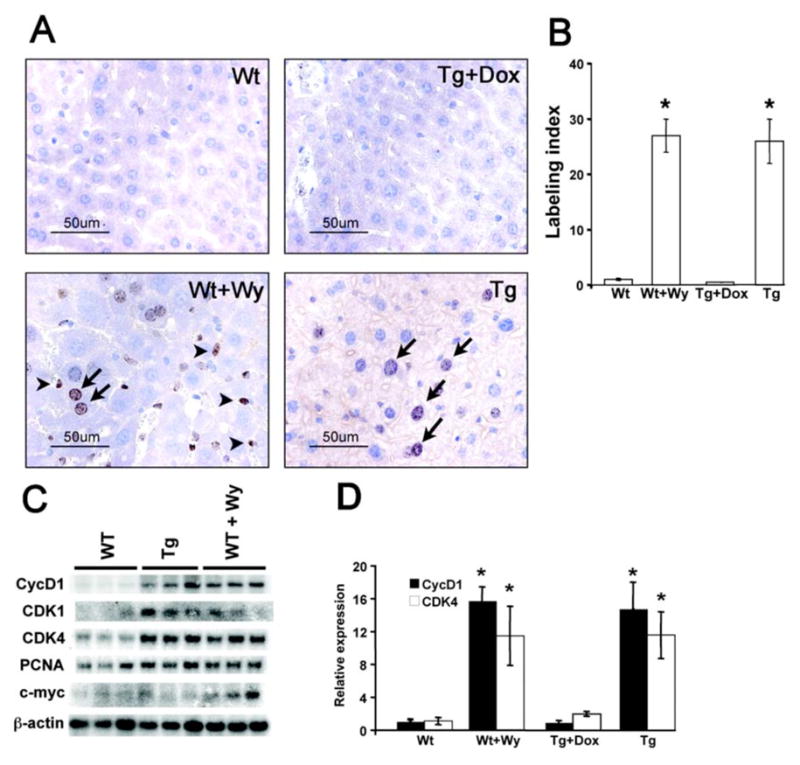
(A) Immunohistochemistry of BrdU incorporation in 8–10 weeks old Wt and LAP- VP16PPARα (Tg) mouse livers. BrdU labeled hepatocytes were showed by arrow and BrdU-labeled nonparenchymal cells were showed by small arrow. Note that Wt mice treated with Wy-14,643 revealed BrdU labeling in nonparenchymal cells. (B) Labeling index of BrdU incorporation into hepatocyte nuclei from (A). Results represent mean ± S.E.M. (n = 3–4), *p < 0.05 compared with Wt control; Note that both Wt mice treated with Wy-14,643 and LAP-VP16PPARα mice in the absence of dox showed increased hepatocyte proliferation. (C) Northern analysis of mRNAs encoding cell cycle control proteins in total liver RNA from 8- to10-week-old mice. PCNA, proliferating cellular nuclear antigen; CDK, cyclin-dependent kinase. (D) Real-time PCR analysis of cyclin D1 and CDK4 mRNAs in total liver RNA from 8- to10-week-old mice. Values are mean ± S.E.M. (n = 4–6); *p < 0.05 compared with Wt control.
Cyclins and CDKs regulate the transit of cells through the cell cycle. These proteins were found previously to be markedly up-regulated in Wt mice treated with Wy-14,643 [24]. Consistent with this finding, two weeks Wy-14,643 treatment was shown to cause a marked induction in the expression of various genes involved in cell cycle control (PCNA, CDK1, CDK4, and cyclin D1) in the livers of Wt mice (Figure 4C and 4D). As expected, the expression of these genes was also up-regulated in LAP-VP16PPARα mice in the absence of dox and dox treatment abolished this induction. This indicated that up-regulation of cell cycle control genes may contribute to the induction of hepatocyte proliferation by activation of PPARα only in hepatocytes.
Absence of liver tumors in aged LAP-VP16PPARα transgenic mice
After long-term feeding with Wy-14,643 in the diet, grossly visible lesions were observed in 11-month-old Wt mice as expected from earlier studies [11]. However, grossly visible lesions were not found in LAP-VP16PPARα mice in the absence of dox aged to over one year. Microscopic examination of liver sections was consistent with the gross findings, as hepatocellular carcinomas and hepatic lesions were observed in the long-term Wy-14,643 treated wild-type mice, but not in more than 20 LAP-VP16PPARα mice at age of over one year old in the absence of dox (Figure 5A). Expression of the VP16PPARα transgene in these aged mice maintained induction of the ACOX mRNA to a similar extent as Wt mice under long-term treatmnet with Wy-14,643 (Figure 5B). Furthermore, although aged mice have a lower levels of hepatocyte proliferation, LAP-VP16PPARα mice and Wt mice after long term treatment with Wy-14,643 had a similar induction of hepatocyte proliferation (Figure 5C and 5D). These results demonstrate that constitutive activation of PPARα in mouse hepatocytes induce hepatocyte proliferation, but is not sufficient to induce liver tumors.
Figure 5. Absence of liver tumor in aged LAP-VP16PPARα transgenic mice.
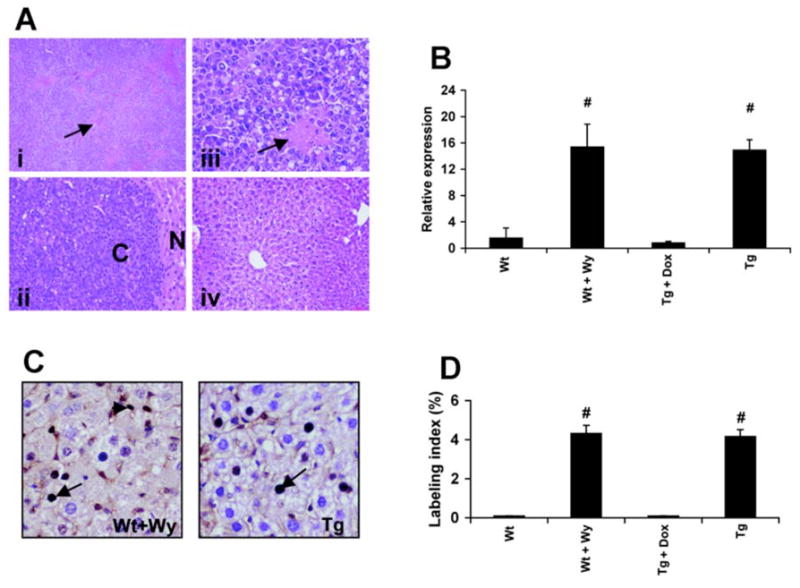
(A) Representative photomicrograph of liver from Wt mice after long-term treatment with Wy-14,643 (11-month-old) or aged LAP-VP16PPARα transgenic mice (11-month-old). (i) Low magnification of Wt mice treated with Wy-14,643 showed toxic lesions (arrow). (ii) High magnification of (i) showed typical hepatocellular carcinoma. ‘C’ indicates carcinoma and ‘N’ indicates the normal tissue surrounding the carcinoma. (iii) High magnification of (ii) showed the toxic lesion (arrow). (iv) LAP-VP16PPARα transgenic mice with same magnification as (ii). Note that no lesions and carcinomas were observed in the transgenic mice. (B) Real-time PCR analysis of ACOX mRNA in liver total RNA from long-term Wy-14,643 treatment or aged LAP-VP16PPARα transgenic mice (11-month-old). Note that long-term treated Wt mice with Wy-14,643 had same extent induction of ACOX mRNA as aged LAP-VP16PPARα mice. (C) Immunohistochemistry of BrdU incorporation in Wt mice after long-term Wy-14,643 treatment or aged LAP- VP16PPARα transgenic mice (11-month-old). BrdU labeled hepatocytes are showed by arrow and BrdU-labeled nonparenchymal cells are showed by small arrow. Note that Wt mice treated with Wy-14,643 exhibited BrdU labeling in nonparenchymal cells. (D) Labeling index of BrdU incorporation into hepatocyte nuclei from (C). Results represent mean ± S.E.M. (n = 3–4); #p < 0.05 compared with Wt control; Note that both Wt mice treated with WY-14,643 and LAP-VP16PPARα mice in the absence of dox showed increased hepatocyte proliferation.
Discussion
Expression of the VP16PPARα transgene in mouse hepatocytes provides a useful strategy for examining PPARα mediated effects in mouse liver. This approach has previously been used to uncover the biological functions of PPARα in skin and mammary gland development [15,25]. LAP-VP16PPARα mice exhibited various peroxisome proliferator-induced responses including significantly decreased serum triglycerides and free fatty acids, peroxisome proliferation and marked induction of PPARα target genes encoding fatty acid β-oxidation enzymes. This indicates that activation of PPARα in only in hepatocytes is sufficient to induce these effects. However, in contrast to a high degree of hepatomegaly and eventual liver tumors after long-term Wy-14,643 feeding, LAP-VP16PPARα mice exhibited only a moderate degree of hepatomagaly and no liver tumors when aged to over one year. This demonstrates that activation of PPARα in mouse hepatocytes is not sufficient to induce liver tumors.
Activation of PPARα is causally related to the induction of liver tumors is susceptible rodent species as revealed by the finding that Pparα-null mice are refractory to hepatocarcinogenesis after feeding prototypical PPs such as Wy-14,643 and bezafibrate [11,12]. The increased oxidative stress and increased hepatocyte proliferation and/or decreased apoptosis have been proposed as key mediators of the hepatocarcinogenic effects by PPs [26]. Production of reactive oxygen species caused by sustained overproduction of H2O2 by increased expression and activity of the peroxisomal enzyme ACOX in response to activation of PPARα was the earliest proposed response to be implicated in the mechanism of carcinogenesis by PPs [27]. LAP-VP16PPARα mice also exhibited a robust expression of ACOX and accompanied peroxisome proliferation and pamitoyl-CoA oxidation, but had no changes in catalase protein expression in contrast to the robust induction of catalase by Wy-14,643 in Wt mice. This suggests that the oxidative stress induced by increased expression of ACOX and peroxisome proliferation is not directly responsible for the development of liver cancer. Indeed, the degree of peroxisome proliferation observed in rodents fed PPs does not always correlate with tumorigenity [28]. Development of liver tumors in mice with a disrupted Acox1 gene suggested the existence of other potential sources oxidative stress [29]. Thus, peroxisome proliferation and induction of ACOX that occurs in response to treatment with PPs appears more associative than causative in the hepatocarcinogenesis response.
Increased hepatocyte proliferation and inhibition of apoptosis was also proposed to mediate the hepatocarcinogenic effects by PPs [26]. Quite surprisingly, the BrdU incorporation study revealed that similar to Wt mice treated with Wy-14,643, LAP-VP16PPARα mice also had marked induction of hepatocyte proliferation. However, no significant changes of apoptosis were observed in both cases. In addition, induction of the cell cycle genes such as CDK1, CDK4, cyclin D1 by Wy-14,643 in Wt mice, which was not observed in Pparα-null mice [30], was also observed in LAP-VP16PPARα mice. This demonstrates that activation of PPARα in hepatocytes only is sufficient to induce hepatocyte proliferation in vivo. To our knowledge, both of these cell cycle genes are not direct PPARα target genes and thus further investigation is required to define how activation of PPARα in hepatocytes induce these genes and hepatocyte proliferation.
The increased liver weight after administration of PPs to rodents is likely the result of both hypertrophy and hyperplasia of hepatocytes. The typical hepatocyte hypertrophy and prominent eosinophilic cytoplasm induced by PPs in Wt mice were not observed in LAP-VP16PPARα mice. Most interestingly, induction of proliferation of nonparenchymal cells was only observed in Wt mice upon Wy-14,643 treatment, but not in LAP-VP16PPARα mice. These results suggest that the differences in hepatocyte morphology and in tumor induction between LAP-VP16PPARα mice and Wt mice treated with WY,14,643 may be associated with the differences in activation of nonparenchymal cells.
Hepatic nonparenchymal cells (NPCs) include Kupffer cells, hepatic stellate cells and sinusoid endothelial cells. However, studies on the involvement of NPCs in peroxisome proliferator-induced liver tumor are limited. NPCs have been implicated in the hepatic proliferative response to PPs. DNA synthesis induced by nafenopin was prevented by the removal of NPCs from normal primary hepatocyte cultures and was restored by returning NPCs to the purified hepatocytes [31,32]. Kupffer cell inhibitors such as methyl palmitate or glycine could prevent the activation of NFκB by reactive oxygen species and induction of hepatocyte proliferation in response to Wy-14,643 [33–35]. This suggests that the involvement of activation of NFκB by Kupffer cells in hepatocyte proliferation caused by PP treatment in vivo. In the present study, induction of hepatocyte proliferation upon expression of activated PPARα only in hepatocytes indicates that hepatocyte proliferation induced by PPs does not require the activation of Kupffer cells, although the existence of Kupffer cells appears to be necessary to support hepatocyte proliferation. On the other hand, PPs appear to directly activate Kupffer cells in vivo [36,37]. The oxygen radicals produced by activated Kupffer cells may contribute to the oxidative stress that in turn may be responsible for the initiation of hepatocarcinogenesis.
Understanding the mechanism of PP-induced carcinogenicity in rodents will allow the evaluation and comparison to possible mechanisms in humans that will be of great value in human risk assessment. The LAP-VP16PPARα mouse model described in this study provides a tool to investigate the carcinogenic mechanism(s) that act through PPARα. In addition, this mouse model should also facilitate examination the pleiotropic effects induced by PPs on various physiological responses during development.
Acknowledgments
This study was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Abbreviations used in this paper
- BrdU
5-bromo-2′-deoxyuridine
- PPAR
peroxisome proliferator-activated receptor
- dox
doxycycline
- TRE
tetracycline response element
References
- 1.Kersten S, Wahli W. Peroxisome proliferator activated receptor agonists. Exs. 2000;89:141–51. doi: 10.1007/978-3-0348-8393-1_9. [DOI] [PubMed] [Google Scholar]
- 2.Lazarow PB, De Duve C. A fatty acyl-CoA oxidizing system in rat liver peroxisomes; enhancement by clofibrate, a hypolipidemic drug. Proc Natl Acad Sci U S A. 1976;73:2043–6. doi: 10.1073/pnas.73.6.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibson GG, Orton TC, Tamburini PP. Cytochrome P-450 induction by clofibrate. Purification and properties of a hepatic cytochrome P-450 relatively specific for the 12- and 11-hydroxylation of dodecanoic acid (lauric acid) Biochem J. 1982;203:161–8. doi: 10.1042/bj2030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy JK, Krishnakantha TP. Hepatic peroxisome proliferation: induction by two novel compounds structurally unrelated to clofibrate. Science. 1975;190:787–9. doi: 10.1126/science.1198095. [DOI] [PubMed] [Google Scholar]
- 5.Cattley RC. Peroxisome proliferators and receptor-mediated hepatic carcinogenesis. Toxicol Pathol, 32 Suppl. 2004;2:6–11. doi: 10.1080/01926230490451680. [DOI] [PubMed] [Google Scholar]
- 6.Reddy JK, Azarnoff DL, Hignite CE. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature. 1980;283:397–8. doi: 10.1038/283397a0. [DOI] [PubMed] [Google Scholar]
- 7.Cattley RC, DeLuca J, Elcombe C, Fenner-Crisp P, Lake BG, Marsman DS, Pastoor TA, Popp JA, Robinson DE, Schwetz B, Tugwood J, Wahli W. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul Toxicol Pharmacol. 1998;27:47–60. doi: 10.1006/rtph.1997.1163. [DOI] [PubMed] [Google Scholar]
- 8.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 9.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–35. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 10.Shearer BG, Hoekstra WJ. Recent advances in peroxisome proliferator-activated receptor science. Curr Med Chem. 2003;10:267–80. doi: 10.2174/0929867033368295. [DOI] [PubMed] [Google Scholar]
- 11.Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–33. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- 12.Hays T, Rusyn I, Burns AM, Kennett MJ, Ward JM, Gonzalez FJ, Peters JM. Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis. 2005;26:219–27. doi: 10.1093/carcin/bgh285. [DOI] [PubMed] [Google Scholar]
- 13.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:10933–8. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung C, Akiyama TE, Ward JM, Nicol CJ, Feigenbaum L, Vinson C, Gonzalez FJ. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 2004;64:3849–54. doi: 10.1158/0008-5472.CAN-04-0322. [DOI] [PubMed] [Google Scholar]
- 15.Yang Q, Yamada A, Kimura S, Peters JM, Gonzalez FJ. Alterations in Skin and Stratified Epithelia by Constitutively Activated PPARalpha. J Invest Dermatol. 2006;126:374–85. doi: 10.1038/sj.jid.5700056. [DOI] [PubMed] [Google Scholar]
- 16.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–51. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–9. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 18.Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115:788–94. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- 19.Gray TJ, Lake BG, Beamand JA, Foster JR, Gangolli SD. Peroxisome proliferation in primary cultures of rat hepatocytes. Toxicol Appl Pharmacol. 1983;67:15–25. doi: 10.1016/0041-008x(83)90240-5. [DOI] [PubMed] [Google Scholar]
- 20.Inoue Y, Inoue J, Lambert G, Yim SH, Gonzalez FJ. Disruption of hepatic C/EBPalpha results in impaired glucose tolerance and age-dependent hepatosteatosis. J Biol Chem. 2004;279:44740–8. doi: 10.1074/jbc.M405177200. [DOI] [PubMed] [Google Scholar]
- 21.Akiyama TE, Ward JM, Gonzalez FJ. Regulation of the liver fatty acid-binding protein gene by hepatocyte nuclear factor 1alpha (HNF1alpha). Alterations in fatty acid homeostasis in HNF1alpha-deficient mice. J Biol Chem. 2000;275:27117–22. doi: 10.1074/jbc.M004388200. [DOI] [PubMed] [Google Scholar]
- 22.Amri EZ, Bonino F, Ailhaud G, Abumrad NA, Grimaldi PA. Cloning of a protein that mediates transcriptional effects of fatty acids in preadipocytes. Homology to peroxisome proliferator-activated receptors. J Biol Chem. 1995;270:2367–71. doi: 10.1074/jbc.270.5.2367. [DOI] [PubMed] [Google Scholar]
- 23.Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters JM, Aoyama T, Cattley RC, Nobumitsu U, Hashimoto T, Gonzalez FJ. Role of peroxisome proliferator-activated receptor alpha in altered cell cycle regulation in mouse liver. Carcinogenesis. 1998;19:1989–94. doi: 10.1093/carcin/19.11.1989. [DOI] [PubMed] [Google Scholar]
- 25.Yang Q, Kurotani R, Yamada A, Kimura S, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha activation during pregnancy severely impairs mammary lobuloalveolar development in mice. Endocrinology. 2006;147:4772–80. doi: 10.1210/en.2006-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- 27.Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat Res. 2000;448:159–77. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 28.Marsman DS, Cattley RC, Conway JG, Popp JA. Relationship of hepatic peroxisome proliferation and replicative DNA synthesis to the hepatocarcinogenicity of the peroxisome proliferators di(2-ethylhexyl)phthalate and [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14,643) in rats. Cancer Res. 1988;48:6739–44. [PubMed] [Google Scholar]
- 29.Fan CY, Pan J, Chu R, Lee D, Kluckman KD, Usuda N, Singh I, Yeldandi AV, Rao MS, Maeda N, Reddy JK. Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol Chem. 1996;271:24698–710. doi: 10.1074/jbc.271.40.24698. [DOI] [PubMed] [Google Scholar]
- 30.Hasmall SC, James NH, Macdonald N, Gonzalez FJ, Peters JM, Roberts RA. Suppression of mouse hepatocyte apoptosis by peroxisome proliferators: role of PPARalpha and TNFalpha. Mutat Res. 2000;448:193–200. doi: 10.1016/s0027-5107(99)00236-5. [DOI] [PubMed] [Google Scholar]
- 31.Hasmall SC, West DA, Olsen K, Roberts RA. Role of hepatic non-parenchymal cells in the response of rat hepatocytes to the peroxisome proliferator nafenopin in vitro. Carcinogenesis. 2000;21:2159–65. doi: 10.1093/carcin/21.12.2159. [DOI] [PubMed] [Google Scholar]
- 32.Parzefall W, Berger W, Kainzbauer E, Teufelhofer O, Schulte-Hermann R, Thurman RG. Peroxisome proliferators do not increase DNA synthesis in purified rat hepatocytes. Carcinogenesis. 2001;22:519–23. doi: 10.1093/carcin/22.3.519. [DOI] [PubMed] [Google Scholar]
- 33.Rose ML, Germolec D, Arteel GE, Schoonhoven R, Thurman RG. Dietary glycine prevents increases in hepatocyte proliferation caused by the peroxisome proliferator WY-14,643. Chem Res Toxicol. 1997;10:1198–204. doi: 10.1021/tx970079u. [DOI] [PubMed] [Google Scholar]
- 34.Rose ML, Germolec DR, Schoonhoven R, Thurman RG. Kupffer cells are causally responsible for the mitogenic effect of peroxisome proliferators. Carcinogenesis. 1997;18:1453–6. doi: 10.1093/carcin/18.8.1453. [DOI] [PubMed] [Google Scholar]
- 35.Rusyn I, Tsukamoto H, Thurman RG. WY-14 643 rapidly activates nuclear factor kappaB in Kupffer cells before hepatocytes. Carcinogenesis. 1998;19:1217–22. doi: 10.1093/carcin/19.7.1217. [DOI] [PubMed] [Google Scholar]
- 36.Bojes HK, Thurman RG. Peroxisome proliferators activate Kupffer cells in vivo. Cancer Res. 1996;56:1–4. [PubMed] [Google Scholar]
- 37.Peters JM, Rusyn I, Rose ML, Gonzalez FJ, Thurman RG. Peroxisome proliferator-activated receptor alpha is restricted to hepatic parenchymal cells, not Kupffer cells: implications for the mechanism of action of peroxisome proliferators in hepatocarcinogenesis. Carcinogenesis. 2000;21:823–6. doi: 10.1093/carcin/21.4.823. [DOI] [PubMed] [Google Scholar]