

Abstract
The anti-apoptotic effect of PGE2 was examined in Jurkat cells (human T-cell leukemia) by incubation with PGE2 (5nM) prior to treatment with the cancer chemotherapeutic agent camptothecin. Apoptosis was evaluated by caspase-3 activity in cell extracts and flow cytometry of propidium iodide-labeled cells. Pre-incubation with PGE2 reduced camptothecin-induced caspase activity by 30% and apoptosis by 35% respectively. Pharmacological data demonstrate that the EP4 receptor is responsible for mediating the protection from camptothecin–induced apoptosis. Pre-treatment of the cells with the EP4 antagonist (EP4A) prior to PGE2 and camptothecin abolished the increased survival effect of PGE2. Specific inhibition of the downstream of PI3 kinase or AKT/protein kinase but not protein kinase A prevents the observed increase in cell survival elicited by PGE2. These findings have critical implications regarding the mechanism and potential application of PGE2 receptor specific inhibition in cancer therapy.
Keywords: Prostaglandin, EP4 receptor, Apoptosis, Survival, PI3 Kinase
1. INTRODUCTION
Prostaglandin E2 has wide-ranging effects on many tissues (1). One response that PGE2 elicits in many different cell types is an increase in survival following stimuli that normally lead to apoptosis (2-4). Over expression or inhibition of cyclooxygenase (COX) and the resulting changes in PGE2 levels, directly influence the sensitivity of cells to apoptotic stimuli (5). Prostaglandin dependent survival is an important cellular defense mechanism and also plays an important role in carcinogenesis(6). There is an abundance of evidence implicating PGE2 functioning in tumor initiation and progression as well as being a mechanism of resistance to radiation or chemotherapy. The PGE2 specific signaling events that contribute to neoplastic transformation in mammalian cells remain difficult to elucidate, although the regulation of apoptosis has been identified as a central mechanism underlying tumorigenesis in cancers over-expressing Cox-2 (5, 7). PGE2 exerts its biological effects by binding to four distinct G-coupled receptors, termed EP receptors (EP1-EP4) (8). While the EP1 receptor couples to Gαq and elevates intracellular calcium, EP3 has multiple splice variants which may couple through Gαs, GαI or Gαq (9). EP2 and EP4 couple to the Gαs and stimulate cAMP production. The data available suggest that the PGE2 dependent survival response is mediated by either or both EP2 and EP4 receptors. Hishino et. al. reported that specific agonists to EP2 and EP4 protected gastric mucosal cells from alcohol induced apoptosis (10). The protection mediated through either receptor was blocked by the protein kinase A inhibitor, H89, indicating that the survival signal is mediated by cAMP dependent Protein Kinase A. Protection of T24 bladder and T84 intestinal (11, 12), epithelial cells by PGE2 correlated with and was mimicked by elevation of cAMP, suggesting that EP2 and/or EP4 receptors mediate the protective response. We have previously reported that protection of the gut from γ-irradiation in mice was mediated by the EP2 receptor based upon decreased survival rates in EP2−/− knockout mice (4). Tessner et. al. similarly concluded that PGE2 protection from γ-irradiation in HCT-116 cells was mediated by EP2 although indirectly through transactivation of EGF receptors (13). Machwate et. al. however reported that PGE2 dependent protection of PR-1 periosteal cells from apoptosis induced by low serum was reversed by an EP4 specific antagonist (14). Since EP4 can transduce the PGE2 signal through either elevation of cAMP or activation of PI3 kinase, a detailed examination of how EP4 mediates PGE2 -dependent protection is of interest. Jurkat cells provide an excellent model system in which to analyze the mechanisms underlying EP4 mediated PGE2 survival response. The EP4 receptor was originally described by DeVries in Jurkat cells (15) which are reported to have limited expression of additional EP receptors (16),(17). The induction of apoptosis by the chemotherapeutic agent camptothecin has been well characterized in these cells. In the present study, we investigated the role of EP4 mediated signal transduction in the regulation of camptothecin-induced apoptosis.
2. METHODS AND MATERIALS
2.1. Materials
Prostaglandin E2, Prostaglandin E1-alcohol, sulprostone, and butaprost were obtained from Cayman Chemical. Camptothecin and H89 were from Sigma Chemical. EP4A was a gift from Kathleen Metters (Merck-Frosste). Stock solutions of all compounds were made in DMSO and diluted in media. Final DMSO concentration in treated wells and vehicle controls were matched and less than 0.5%. LY294002 and Wortmannin were obtained from Cell Signaling Technology. AKTi (1L-6-Hydroxymethyl-chiro-inositol 2-(R)-2-O-methyl-3-O-octadecylcarbonate) 4-Cyano-3-methylisoquinoline, and cell permeable, Protein Kinase A Inhibitor 14-22 Amide were obtained from Calbiochem. Antibodies to prostaglandin E2 receptors (EP1-4, Catalog #101740; 101750; 101760 and 101775) and corresponding blocking peptides were from Cayman Chemical. Antibodies to pBad -Ser136 (sc-12969); AKT (sc-8312) and pAKT-Ser473 (sc-7985R) were from Santa Cruz.
2.2. Cell and Culture conditions
Jurkat cells (TIB-152), Clone E6-1, were obtained from the ATCC. Jurkat cell were maintained between a density of 0.15-1.5× 106 cells /ml in RPMI1640 media with 10%Fetal Bovine Serum supplemented with 2mM glutamine and antibiotics. For FACS or caspase analysis of apoptosis, prostaglandins were added 1hr prior to addition of camptothecin. Pathway specific inhibitors were added 30 minutes prior to prostaglandin addition. Inhibitors were added at a concentration between 2 and 10 times their reported inhibition constants.
2.3. FACS analysis
1.5 × 106 cells in 2.5mls were aliquoted to 6 well plates and were treated for 12 hours with 25uM camptothecin. Cells were washed with PBS, fixed with 70% ethanol at 4° for 1 hour, washed 2 times with PBS and then resuspended in PBS containing propidium iodide, 50ug/ml and RNAse A, 0.25mg/ml.
2.4. Caspase assays
Cells were diluted to a density of 5×105cells/ml in fresh media and 2.5 × 104 cells aliquoted to a 96 well opaque plate. Caspase activity was assayed 6 hours after camptothecin addition by measuring fluorescence at 481/521 nm of Promega's Apo-One Caspase 3/7 reagent (Z-DEVD-R-110) using Synergy HT fluorometer. Camptothecin at 25uM has been previously shown to induce apoptosis within 6 hrs in Jurkat cells (18).
2.5. Western Blotting
Western analysis was performed on cells harvested in 50uL RIPA buffer containing 1mM NaF, 1mM NaVO4 and 0.1mM PMSF. Twelve micrograms of soluble protein was loaded per lane. Proteins were transferred to nitrocellulose membranes, blocked with 2% dried mild/0.5% Tween-20 and incubated with primary antibodies. Primary antibodies to the EP receptors used at a dilution of 1:1000 and incubated overnight at 4o. Blocking peptides to EP3 and EP4 were preincubated with diluted antibody at a concentration of 0.5ug/ml. The primary antibody to pBAD 136 was used at a dilution of 1:500. The primary antibody to AKT and pAKT were used at a dilution of 1:1000. Antibodies to phosphorylated proteins were incubated for 1 hour at room temperature with NaF present.
2.6. Real-time Quantitative PCR
Total RNA samples were extracted from cells with Trizol. To prepare cDNAs, these samples were reverse transcribed with SuperScript III, T7 Oligo (dT) primer, and Random Primers. Reaction conditions were: 25° for 10 min., 50° for 50 min., 70° for 15 min. The cDNA samples were then amplified using iQ SYBR Supermix at 95° for 3 min., then 95° for 15 sec. and 62° for 15 sec. for a total of 40 cycles. All PCR reactions were performed in triplicate in an iCycler iQ real-time PCR detection system Relative mRNA levels were assessed by standardization β-actin). The EP real time primers were confirmed using HCT116 cells (not shown). Results were expressed as an n-fold difference in gene expression. Sequences of the PCR primers pairs used were: huEP1 ( forward ) 5′-GTG GAG ATG GTG GGC CAG-3′ ;huEP1 ( reverse ) 5′-GCC ACC AAC ACC AGC ATT G-3′; huEP2 ( forward ) 5′-GAC CAC CTC ATT CTC CTG GCT A-3′; huEP2 ( reverse ) 5′-TCC TTT CGG GAA GAG GTT TCA-3′; huEP3 ( forward ) 5′-GAT CAT GTG CGT GCT GTC G-3′; huEP3 ( reverse ) 5′-CCG TGT GTG TCT TGC AGT GC-3′; huEP4 ( forward ) 5′-CAT CTG CTC CAT CCC GCT-3′; huEP4 ( reverse ) 5′-GGA TGG CCT GCA AAT CTG G-3′
3. RESULTS
3.1. Prostaglandins are protective
Camptothecin treatment of Jurkat cells result in an induction of caspase activity (Figure 1A) leading to apoptosis (Figure 1B). Caspase activity was markedly increased when assayed 6 hours after camptothecin addition to the media. When cells were harvested 12 hours after camptothecin treatment and stained with propidium iodide for FACS analysis, ∼40% of the cells contain subdiploid DNA content, indicative of apoptosis. Treatment of the cells with 5nM PGE2 prior to the addition of camptothecin reduced the level of caspase activity by ∼30% and diminished apoptosis by 35%, respectively. The dose response curve for PGE2 (Figure 1C) was maximal between 2 and 10 nM, similar to published receptor binding data for the EP receptors (19-21).
Figure 1.
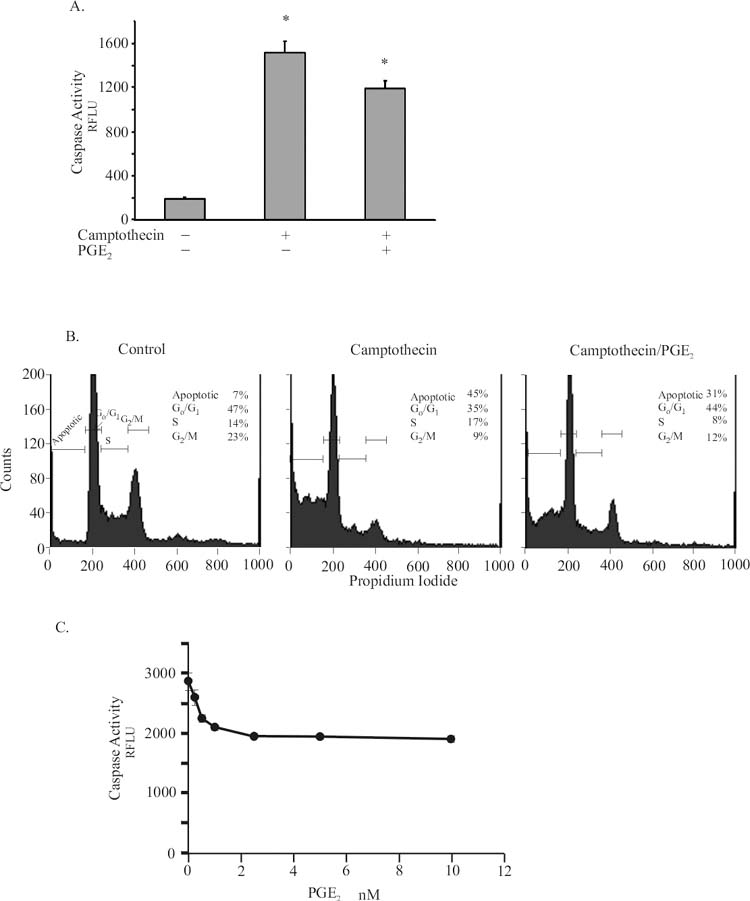
a. Jurkat cells were treated with 25 uM Camptothecin for 6 hours with or without 1 hour pretreatment with 2 nM PGE2. Caspase-3/7 activity was assayed using a fluorescent substrate as described in Methods. Activity is expressed as Relative Fluorescence Units (RFLU). Data represent the mean ± standard error of 5 experiments in triplicate. B. Cells were treated as above and harvested after 12 hours, stained with propidium iodide for FACS analysis of apoptosis as described in Methods. Data are from representative experiment, N=3.
3.2. EP receptor expression in Jurkat cells
Characterization of EP receptor expression in Jurkat cells by real-time RT PCR (Figure 2A) and immunoblotting (Figure 2B) indicate that the cells express EP3 and EP4 prostaglandin receptors. By real-time PCR analysis, the EP4 mRNA level is approximately 4 times greater than the level of EP3, although quantitation of Western blots indicate that EP3 protein levels are twice as great as EP4. Pre-incubation of the receptor antibodies with their respective blocking peptide confirmed the specificity of receptors (Figure 2C). Real-time PCR analysis indicated no expression of either EP1 or EP2 mRNA in these cells.
Figure 2.

EP receptor expression was assayed by (a) real time PCR or (b) Western blot as described in Methods. Western blots were quantitated by densitometry. Values for RNA and protein are expressed relative to EP4. Antibody specificity was confirmed (c) by pre-incubating diluted antibody with its corresponding blocking peptide. Data are from representative experiment, N=3.
3.3. Protection is mediated by EP4
Pharmacological characterization of the survival effects of PGE2, as measured by the attenuation of camptothecin-induced caspase activity, indicates that EP4 is responsible for mediating the protection of Jurkat cells (Figure 3). Prostaglandin E1-alcohol, an EP3/4 agonist, was as effective as PGE2 in protecting the cells, but sulprostone, an EP1/3 agonist elicited no protection. The dose response curve for prostaglandin E-1 alcohol indicated that when PGE1-alcohol was added at concentrations above 1uM to Jurkat cells prior to camptothecin, caspase activity was reduced by 30 percent. The EC50 for PGE1-alcohol was approximately 200nM. Butaprost, an exclusively EP2 agonist had no protective effect. Pre-treatment of the cells with the EP4 antagonist EP4A(14) prior to PGE2 (or Prostaglandin E1-alcohol,data not shown) prevented the increased survival response (Figure 4a). PGE2 dose response curves in the presence of increasing concentrations of EP4A (Figure 4b) resulted in comparable decrease in caspase activity but at corresponding higher concentrations of PGE2. Schilds analysis of the inhibition by EP4A indicated that the compound displayed an IC50 of ∼20nM.
Figure 3.
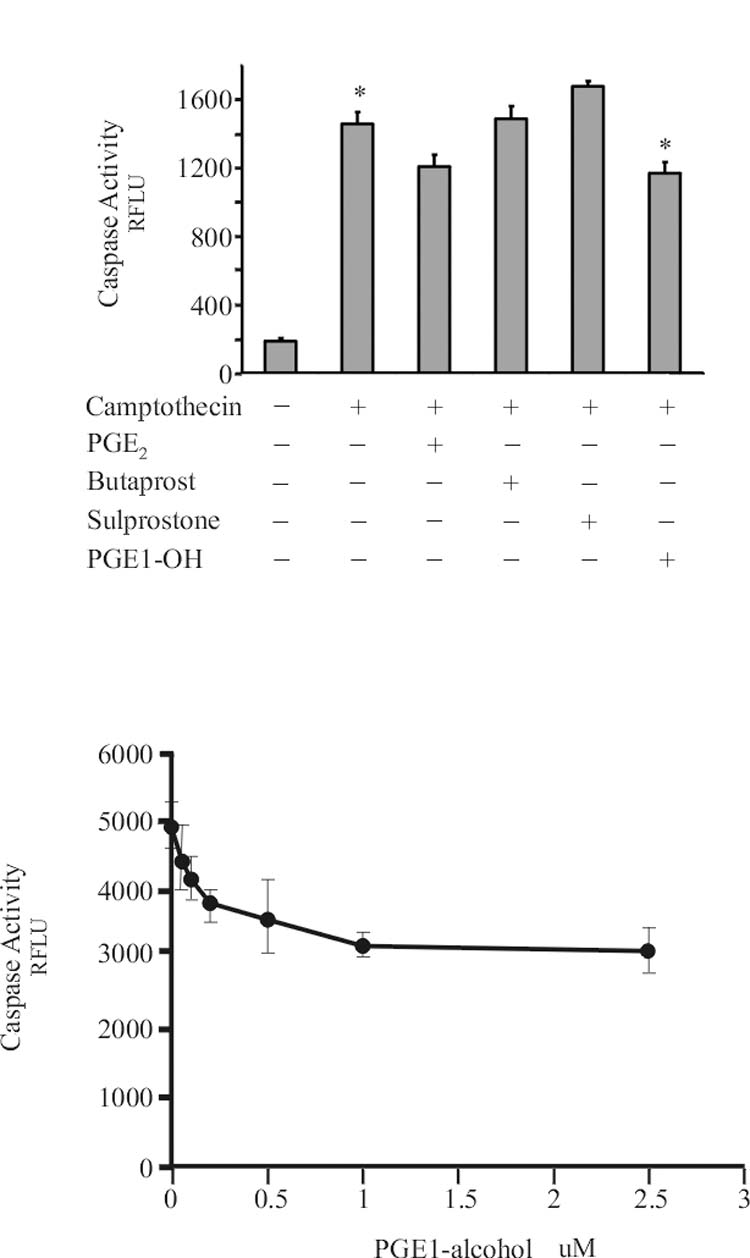
Cells were pretreated with EP agonist as indicated 1 hour before addition of 25uM camptothecin. The concentration of prostanoid added were: PGE2, 5nM; Butaprost, 5uM; Sulprostone, 0.1uM; PGE1-alcohol, 5uM. Caspase activity was assayed 6 hours later as described in Methods. Caspase activity is expressed in relative fluorescence units. Data represent the mean ± standard error of 3 experiments in triplicate
Figure 4.

Cells were treated and caspase activity assayed as in Figure1. When added, cells were treated with 0.1 uM EP4A 1 hour before addition of 5nM PGE2. Levels are expressed as percent caspase activity elicited by camptothecin. Data represent the mean ± standard error of 5 experiments in triplicate.
3.4. EP4 dependent protection is mediated by PI3 kinase pathway
Downstream signaling by the Gαs coupled EP4 receptor may be through either the Protein Kinase A or PI3 kinase pathways. Pretreatment of the cells with a protein kinase A inhibitor, H89, shown in Figure 5 (or 4-Cyano-3-methylisoquinoline or the cell permeable, myristolated peptide PKA Inhibitor 14-22 Amide, not shown) failed to block the pro-survival effect of PGE2. Addition of PI3 kinase inhibitors, Ly294002 or Wortmannin, however, completely blocked the PGE2 response (Figure 5). Specific inhibition of the downstream AKT/protein kinase B by the AKT inhibitor (AKTi) also prevents the observed increase in cell survival elicited by PGE2. Additionally, actinomycin, an inhibitor of protein synthesis, did not alter the ability of PGE2 to protect the cells from camptothecin induced apoptosis indicating that de novo protein synthesis is not required for the prostaglandin dependent protection.
Figure 5.
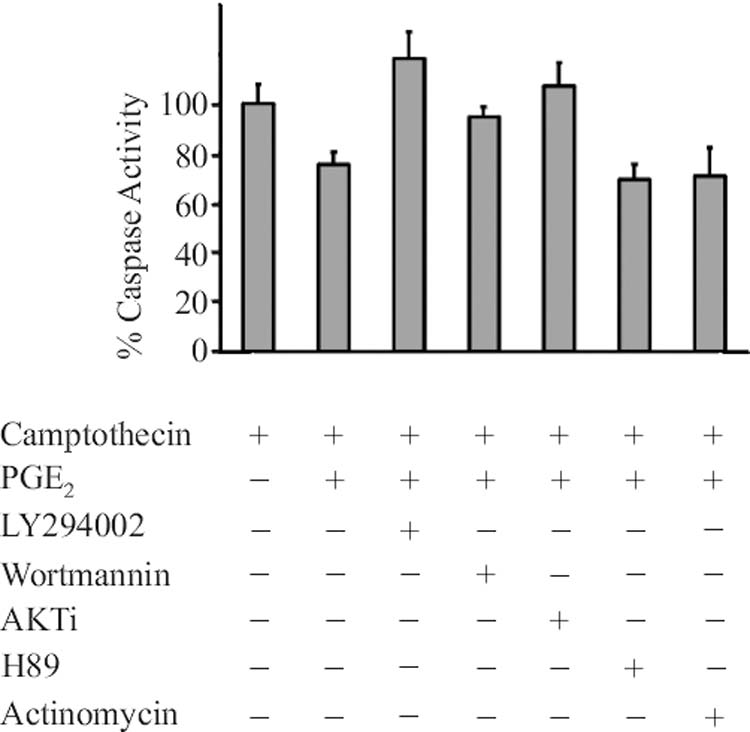
Cells were treated and caspase activity assayed as in Figure1. When added, cells were treated with inhibitors 1 hour before addition of 5nM PGE2. The concentration of inhibitors added were: LY294002, 10uM; Wortmannin, 0.1uM; AKTi, 10uM; H89, 0.1uM; actinomycin, 10ug/ml. Levels are expressed as percent caspase activity elicited by 25 uM camptothecin alone. Data represent the mean ± standard error of 3-5 experiments in triplicate.
Analysis of the AKT/protein kinase B phosphorylation status demonstrated that the treatment of the cells with PGE2 results in a 4-fold increase in phosphorylation of serine 473 (Figure 6). Activation of AKT has been shown to decrease apoptosis through many different mechanisms. The specific mechanism by which PGE2 stimulation of the EP4 receptor leads to survival is unclear, however, evaluation of the phosphorylation state of BAD after stimulation with PGE2 indicates an increased level of phosphorylation at serine 136 (∼3.5 fold by densitometry). The pharmacology of the phosphorylation of BAD is identical to that of the PGE2-mediated survival response. PGE2 and Prostaglandin E1-alcohol are effective while sulprostone and butaprost are not. Furthermore, PGE2 dependent phosphorylation of pBAD136 is also blocked by pretreatment with EP4A (data not shown).
Figure 6.

Cells (2× 106) were treated with 5nM PGE2 or 5 uM PGE1-alcohol as in caspase experiments, washed with PBS and lysed with 50ul RIPA buffer. Proteins were isolated by PAGE using Invitrogen NU-PAGE system and blotted to nitrocellulose membrane. The blot was incubated and developed with phospho-specific antibody to either pBAD (ser-136) or pAKT (ser-473), stripped and reprobed with antibody to AKT protein. Data are from representative experiment, N=3.
4. DISCUSSION
Using a well-characterized model of apoptosis, we have demonstrated that PGE2 is capable of eliciting a survival response in Jurkat cells, and that the response in these cells is mediated by the EP4 receptor. Our data also demonstrates that PGE2 signaling through the EP4 receptor results in activation of PI3Kinase /AKT signaling pathways that regulate cell survival. These data confirm a previous observation that EP4, in addition to EP2, can mediate a survival response and suggest that different downstream effector pathways are able to mediate cytoprotection based upon the particular EP receptor activated.
Camptothecin is a topoisomerase I inhibitor that is highly cytotoxic and is a potent anticancer chemotherapeutic agent. Jurkat cells have been shown to be particularly sensitive to camptothecin and cells that come in contact with this agent undergo a complete cessation of DNA synthesis followed by an increase in apoptosis(18). The data presented demonstrate that treatment of Jurkat cells with PGE2 is able to protect cells from the apoptotic stimuli caused by camptothecin. This observation is consistent with a large body of evidence describing the pro-survival, anti-apoptotic effects of PGE2. Our data demonstrate an 8-fold increase in caspase activity when compared to untreated cells and 40% apoptosis as judged by the population of cells with subdiploid DNA content by FACs analysis. Addition of PGE2 prior to camptothecin treatment of the cells reduced apoptosis by 30-35%.
PGE2 acts via four specific transmembrane G protein-coupled receptors designated EP1-4 (22). EP1 signals via increased intracellular Ca2+. EP3 inhibits the generation of cAMP, whereas EP2 and EP4 stimulate an increase in cAMP levels. Multiple EP3 isoforms have been described, which couple to different signaling pathways. Using EP-specific agonists and antagonists, we have addressed the involvement of EP receptors in PGE2-induced signaling and protection from camptothecin-induced apoptosis expression in Jurkat cells. We found that neither the EP2 agonist, butaprost nor the EP3 agonist, sulprostone were able to substitute for PGE2 in conferring protection from camptothecin-induced apoptosis. This is consistent with an earlier report suggesting that Jurkat cells lacked functional EP2 receptors, as butaprost was unable to elicit an elevation of cAMP. (16,17). Using Real-Time RT PCR we did not detect EP2 receptor mRNA as did a previous report that EP2 was undetectable in Jurkat cells by RT-PCR (16). The functional expression of EP3 receptors on Jurkat cells has been previously documented by a study that examined the role of PGE2 in prolactin secretion(17). In that study, the EP1/3 agonist 17-phenol trinor PGE2 was able to stimulate prolactin secretion. Our data confirm the presence of EP3 mRNA by Real-time PCR and western blotting in these cells; but demonstrate the inability of sulprostone to mimic the protective response to PGE2, thus indicating that these receptors do not play a role in the cytoprotective effects of PGE2. Our data however also confirm EP4 receptor expression, and that the specific antagonist EP4A blocked PGE2-induced cytoprotection.
In many cell types, the protective PGE2 response is mediated through the elevation of cAMP levels by activation of the EP2 receptor. Both Gαs coupled EP2 and EP4 are capable of elevating cAMP in cells and elevation of intracellular cAMP levels by addition of exogenous cAMP analogs or treatment with forskolin is able to mimic the PGE2 protective response in many cell types. The maximal level of PGE2-stimulated cAMP formation in EP4 receptor-transfected cells was only ∼20% that achieved in EP2 receptor-transfected cells, even though both receptors were expressed to nearly the same extent prior to agonist pretreatment (23). Recent studies suggest that not only are EP4 receptors less efficiently coupled to adenylate cyclase, but they have additional pathways of signal transduction that do not involve the activation of cAMP/PKA signaling (24). Moreover, there is an increasing body of evidence indicating that the Gs coupled EP4 receptor transduces signals through the PI3 kinase pathway to a greater degree than through activation of adenylate cyclase. In HEK cells transfected with EP4 receptor, PGE2 activated Tcf/Lef signaling in a PI3 kinase -dependent manner(24). Additionally, PGE2 stimulation of EP4 led to phosphorylation of the extracellular signal-regulated kinases (ERKs) via PI3 kinase dependent pathway and stimulation of EP4 receptors in LS-174 cells increased cell motility and proliferation in a PI3K-dependent manner(25). Our data suggest that the PGE2 dependent survival response in Jurkat cells is mediated by activation of the EP4 receptor and downstream PI3/AKT kinase pathways. We have demonstrated that the PI3 kinase inhibitors, LY294002 and Wortmannin block the PGE2 dependent increase survival in Jurkat cells whereas treatment with H89 a PKA specific inhibitor did not. Furthermore PGE2 treatment of the cells results in increased phosphorylation of AKT kinase at serine 473. Inhibition of AKT activation (AKTi) also blocks the PGE2 mediated increase in cell survival. The exact mechanism by which EP4 is linked to PI3 kinase remains unclear. Murga et. al. have demonstrated the activation of both PI3Kβ and PI3Kγ through the βγ heterodimer of the G-protein complex of the β-adrenergic receptor(26). Activation of PI3 kinase has also been shown to be mediated through the alternate cAMP exchange protein, EPAC (27). Recently Fujino and Regan have reported that EP4 can activate PI-3 kinase through an inhibitory G-protein (Mol Pharmacol 69:5-10, 2006).
Activation of the AKT kinase is now recognized as a critical mechanism, associated with activation of multiple downstream pathways that increase cell survival. AKT dependent phosphorylation of p21, MDM2, BAD, AFX and FKHR all promote cell survival (28). In this report we have demonstrated that phosphorylation of BAD is stimulated by PGE2 in Jurkat cells. BAD is an important proapoptotic member of the Bcl-2 family, which acts by inactivating antiapoptotic Bcl-2 family proteins(29, 30). BAD exists in an inactive complex with the molecular chaperone 14-3-3 via the phosphorylation of four serine residues (Ser-112, -136, -155, and -170) (31). Following apoptotic stimuli, BAD is dephosphorylated, dissociated from 14-3-3, and translocated to the outer membrane of the mitochondria, where it subsequently dimerizes with Bcl-XL and promotes mitochondrial sequestration of BAD in the cytosol, preventing BAD from associating with and inactivating BCL-XL. BCL-XL inhibits the release of cytochrome C from the mitochondria and prevents the subsequent activation of caspase 9 resulting in inhibition of apoptosis.
There is an abundance of evidence supporting PGE2 also functioning in tumor initiation and progression, as well as a mechanism of resistance to radiation treatment or chemotherapy. Increased survival of a transformed cell in response to PGE2 stimulation is one potential mechanism by which prostaglandins may contribute to tumor initiation and progression. Our results demonstrate that EP4 in addition to EP2 can elicit a survival response and that the downstream mechanisms mediating the survival differ. Given the significance of increased COX-2 derived PGE2 synthesis in many cancers, receptor specific targeted inhibition of PGE2 mediated signaling pathways may provide an excellent strategy to enhance the efficacy of chemotherapeutic and chemopreventative agents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work was funded by NIH grants DK066161, DK002822, and the Washington University Digestive Disease Research Cores Center #P30 DK52574 (to C.W. Houchen) and DK62265 (to S. Anant).
REFERENCES
- 1.Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108(1):15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vassiliou E, Sharma V, Jing H, Sheibanie F, Ganea D. Prostaglandin E2 promotes the survival of bone marrow-derived dendritic cells. J Immunol. 2004;173(11):6955–64. doi: 10.4049/jimmunol.173.11.6955. [DOI] [PubMed] [Google Scholar]
- 3.Houchen CW, Stenson WF, Cohn SM. Disruption of cyclooxygenase-1 gene results in an impaired response to radiation injury. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G858–65. doi: 10.1152/ajpgi.2000.279.5.G858. [DOI] [PubMed] [Google Scholar]
- 4.Houchen CW, Sturmoski MA, Anant S, Breyer RM, Stenson WF. Prosurvival and antiapoptotic effects of PGE2 in radiation injury are mediated by EP2 receptor in intestine. Am J Physiol Gastrointest Liver Physiol. 2003;284(3):G490–8. doi: 10.1152/ajpgi.00240.2002. [DOI] [PubMed] [Google Scholar]
- 5.Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83(3):493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 6.Hull MA, Ko SC, Hawcroft G. Prostaglandin EP receptors: targets for treatment and prevention of colorectal cancer? Mol Cancer Ther. 2004;3(8):1031–9. [PubMed] [Google Scholar]
- 7.Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58(2):362–6. [PubMed] [Google Scholar]
- 8.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46(2):205–29. [PubMed] [Google Scholar]
- 9.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79(4):1193–226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 10.Hoshino T, Tsutsumi S, Tomisato W, Hwang HJ, Tsuchiya T, Mizushima T. Prostaglandin E2 protects gastric mucosal cells from apoptosis via EP2 and EP4 receptor activation. J Biol Chem. 2003;278(15):12752–8. doi: 10.1074/jbc.M212097200. [DOI] [PubMed] [Google Scholar]
- 11.Nishihara H, Kizaka-Kondoh S, Insel PA, Eckmann L. Inhibition of apoptosis in normal and transformed intestinal epithelial cells by cAMP through induction of inhibitor of apoptosis protein (IAP)-2. Proc Natl Acad Sci U S A. 2003;100(15):8921–6. doi: 10.1073/pnas.1533221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jabbour HN, Kelly RW, Boddy SC. Autocrine/paracrine regulation of apoptosis in epithelial cells by prostaglandin E2. Prostaglandins Leukot Essent Fatty Acids. 2002;67(5):357–63. doi: 10.1054/plef.2002.0442. [DOI] [PubMed] [Google Scholar]
- 13.Tessner TG, Muhale F, Riehl TE, Anant S, Stenson WF. Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J Clin Invest. 2004;114(11):1676–85. doi: 10.1172/JCI22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Machwate M, Harada S, Leu CT, et al. Prostaglandin receptor EP(4) mediates the bone anabolic effects of PGE(2) Mol Pharmacol. 2001;60(1):36–41. doi: 10.1124/mol.60.1.36. [DOI] [PubMed] [Google Scholar]
- 15.De Vries GW, Guarino P, McLaughlin A, Chen J, Andrews S, Woodward DF. An EP receptor with a novel pharmacological profile in the T-cell line Jurkat. Br J Pharmacol. 1995;115(7):1231–4. doi: 10.1111/j.1476-5381.1995.tb15030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaschke V, Jungermann K, Puschel GP. Exclusive expression of the Gs-linked prostaglandin E2 receptor subtype 4 mRNA in mononuclear Jurkat and KM-3 cells and coexpression of subtype 4 and 2 mRNA in U-937 cells. FEBS Lett. 1996;394(1):39–43. doi: 10.1016/0014-5793(96)00928-3. [DOI] [PubMed] [Google Scholar]
- 17.Gerlo S, Verdood P, Gellersen B, Hooghe-Peters EL, Kooijman R. Mechanism of prostaglandin (PG)E2-induced prolactin expression in human T cells: cooperation of two PGE2 receptor subtypes, E-prostanoid (EP) 3 and EP4, via calcium- and cyclic adenosine 5′-monophosphate-mediated signaling pathways. J Immunol. 2004;173(10):5952–62. doi: 10.4049/jimmunol.173.10.5952. [DOI] [PubMed] [Google Scholar]
- 18.Johnson N, Ng TT, Parkin JM. Camptothecin causes cell cycle perturbations within Tlymphoblastoid cells followed by dose dependent induction of apoptosis. Leuk Res. 1997;21(10):961–72. doi: 10.1016/s0145-2126(97)00077-5. [DOI] [PubMed] [Google Scholar]
- 19.Davis TL, Sharif NA. Pharmacological characterization of [(3)H]-prostaglandin E(2) binding to the cloned human EP(4) prostanoid receptor. Br J Pharmacol. 2000;130(8):1919–26. doi: 10.1038/sj.bjp.0703525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122(2):217–24. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall FH, Patel K, Lundstrom K, Camacho J, Foord SM, Lee MG. Characterization of [3H]-prostaglandin E2 binding to prostaglandin EP4 receptors expressed with Semliki Forest virus. Br J Pharmacol. 1997;121(8):1673–8. doi: 10.1038/sj.bjp.0701332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breyer MD, Breyer RM. G protein-coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001;63:579–605. doi: 10.1146/annurev.physiol.63.1.579. [DOI] [PubMed] [Google Scholar]
- 23.Nishigaki N, Negishi M, Ichikawa A. Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in desensitization and sensitivity to the metabolic inactivation of the agonist. Mol Pharmacol. 1996;50(4):1031–7. [PubMed] [Google Scholar]
- 24.Fujino H, West KA, Regan JW. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem. 2002;277(4):2614–9. doi: 10.1074/jbc.M109440200. [DOI] [PubMed] [Google Scholar]
- 25.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276(21):18075–81. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 26.Murga C, Fukuhara S, Gutkind JS. A novel role for phosphatidylinositol 3-kinase beta in signaling from G protein-coupled receptors to Akt. J Biol Chem. 2000;275(16):12069–73. doi: 10.1074/jbc.275.16.12069. [DOI] [PubMed] [Google Scholar]
- 27.Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem. 2002;277(13):11497–504. doi: 10.1074/jbc.M110856200. [DOI] [PubMed] [Google Scholar]
- 28.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80(2):285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 30.Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci. 2005;1042:203–9. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 31.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87(4):619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]