

Abstract
Because of recent high-yield native ligation techniques, chemical synthesis of larger multidomain bioactive proteins is rapidly coming within reach. Here we describe the total chemical synthesis of a designed “microprotein S,” comprising the γ-carboxyglutamic acid-rich module, the thrombin-sensitive module, and the first epidermal growth factor-like module of human plasma protein S (residues 1–116). Synthetic microprotein S expressed anticoagulant cofactor activity for activated protein C in the down-regulation of blood coagulation, and the anticoagulant activity of microprotein S was not neutralized by C4b-binding protein, a natural inhibitor of native protein S in plasma. The correct folding of this complex multidomain protein was enhanced compared with individual modules because the γ-carboxyglutamic acid-rich module and the thrombin-sensitive module markedly facilitated correct folding of the first epidermal growth factor-like module compared with folding of the first epidermal growth factor-like module alone. These results demonstrate that total chemical synthesis of proteins offers an effective way to generate multidomain biologically active proteins.
Theoretical and real risks of pathogen transmission (1) during therapy with protein preparations derived from human plasma or biological expression systems have triggered exploration of novel safer routes for noninfectious protein production. Total chemical synthesis of therapeutic proteins would offer an attractive alternative (2). However, a presumed inability of complex proteins to fold correctly after synthesis is considered a critical deficiency of the total chemical protein synthesis method. Although this skepticism was greatly overcome by chemical synthesis and correct folding of more than 200 proteins so far (2), experimental data on the in vitro folding of chemically synthesized complex multidomain proteins is currently very limited.
Here we describe the chemical synthesis of a multidomain anticoagulant protein, “microprotein S,” through modular assembly of synthetic peptide segments by native chemical ligation. The native chemical ligation technique uses the principle of a chemoselective reaction between two unprotected peptides in aqueous solution in which a C-terminal thioester undergoes a thiol exchange with an N-terminal cysteine sulfhydryl side chain. A subsequent, rapid, intramolecular rearrangement yields a native peptide bond at the site of ligation (3, 4).
Human protein S is a nonenzymatic cofactor for the serine protease enzyme “activated protein C” (APC) in the proteolytic inactivation of blood coagulation factor Va (5, 6). Protein S is a multidomain protein consisting of the γ-carboxyglutamic acid (Gla)-rich module containing 11 Gla residues (the “Gla module”), the thrombin-sensitive module (TSR module), four successive epidermal growth factor (EGF)-like modules, and the sex hormone binding globulin-like region (SHBG). Protein S cofactor activity for APC is inhibited by proteolytic cleavage in the TSR (7, 8) or by binding of C4b-binding protein to residues 413–433 in the SHBG region of protein S (9, 10). Hereditary deficiencies of protein S or protein C are associated with increased risk of thrombosis (11–16), emphasizing the importance of the protein C/protein S anticoagulant pathway in maintaining the hemostatic balance.
A chemically synthesized EGF1 module of human protein S inhibited the protein S cofactor activity for human APC in a human plasma system, apparently because of competition between protein S and the EGF1 for binding to APC (17, 18).
Like the other vitamin K-dependent coagulation proteins, the Gla module of protein S contains a Ca2+-dependent binding site for negatively charged phospholipids (19). When this observation was combined with the requirement of an intact TSR module for expression of protein S cofactor activity for APC (7, 8), the intriguing question arose as to whether the covalent attachment of the TSR and the Gla module to the EGF1 module would generate a functional protein S-like cofactor. To access this construct comprising residues 1–116 of protein S, we designed a multiple native chemical ligation approach in which sequential ligation of three peptide segments representing the respective Gla, TSR, and EGF1 modules of human protein S resulted in the target sequence designated “microprotein S” (Fig. 1). Correct folding of the construct was demonstrated by assessment of biological anticoagulant cofactor activity for APC in the attenuation of blood coagulation, and by ablation of cofactor activity of microprotein S by thrombin.
Figure 1.

Amino acid sequence of microprotein S, which consists of a Gla-rich module (residues 1–46), the TSR module (residues 47–75), and the EGF1 module (residues 76–116). The gray residues at positions 46–47 and 79–80 represent the Gly-Cys and Gln-Cys native chemical ligation sites. Bold Asp residues represent putative cleavage sites for endoproteinase Asp-N. A solid line joins disulfide-bonded Cys residues. γ denotes γ-carboxyglutamic acid.
Materials and Methods
Materials.
2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and tert-butoxycarbonyl (Boc)-protected amino acids were obtained from NovaBiochem (San Diego). Boc-Arg(p-toluenesulfonyl)-OH and Boc-Asn(xanthyl)-OH were obtained from Bachem Bioscience (King of Prussia, PA). S-Tritylmercaptopropionic acid was obtained from Peptides International (Louisville, KY). N,N-Diisopropylethylamine (DIEA) was obtained from Applied Biosystems (Foster City, CA). Methylbenzhydrylamine (MBHA) resin was obtained from Peninsula Laboratories (Belmont, CA). 2-(Methylsulfonyl)ethyl-4-nitrophenyl carbonate was obtained from Fluka (Milwaukee). N,N-Dimethylformamide (DMF) and HPLC-grade acetonitrile were purchased from Fisher. Trifluoroacetic acid (TFA) was obtained from Halocarbon (River Edge, NJ). HF was purchased from Matheson Gas (Cucamonga, CA).
Peptide Synthesis.
Peptides were prepared by manual solid-phase peptide synthesis (SPPS), typically on a 0.25-mmol scale, by using the in situ neutralization/HBTU activation procedure for Boc chemistry as previously described (20). After each coupling step, yields were determined by measuring residual free amine with the quantitative ninhydrin assay (21). Side-chain-protected amino acids were Boc-Arg(p-toluenesulfonyl)-OH, Boc-Asn(xanthyl)-OH, Boc-Asp(O-cyclohexyl)-OH, Boc-Cys(4-methylbenzyl)-OH, Boc-Glu(O-cyclohexyl)-OH, Boc-Lys(2-Cl-Z)-OH, Boc-Ser(benzyl)-OH, Boc-Thr(benzyl)-OH, Boc-Trp(formyl)-OH, and Boc-Tyr(2-Br-Z)-OH. The derivatized Gla amino acid, Nα-Boc-γ,γ-dicyclohexyl-l-γ-carboxyglutamic acid, necessary for SPPS using Boc chemistry, was synthesized as described (22). After chain assembly, the peptides were deprotected and cleaved from the resin by treatment with anhydrous HF for 1 h at 0°C with 4% anisole (Gla module, residues 1–46; TSR, residues 47–79) or p-cresol (EGF1, residues 80–116) as a scavenger. Before HF treatment, the formyl group from Trp108 was removed by a 2-h treatment with 10% piperidine in dimethylformamide at 0°C. Trityl-associated mercaptopropionic acid leucine (TAMPAL) resin was prepared as described (4). Native chemical ligation of unprotected peptides was performed as described (3, 4).
Microprotein S Module Polypeptide Syntheses.
The synthesis of Gla module-(1–46) and TSR-(47–79) peptides used a thioester-generating linker on MBHA resin (TAMPAL) so that cleavage of the Gla and TSR peptides from their resins by HF treatment yielded a C-terminal mercaptopropionic acid-leucine (MPAL)-activated thioester moiety (COSR) that could be used directly for native chemical ligation (4). Peptides Gla module (1–46: A N S L L γ γ T K Q G N L γ R γ C I γ γ L C N K γ γ A R γ V F γ N D P γ T D Y F Y P K Y L G) and TSR (47–79: C L R S F Q T G L F T A A R Q S T N A Y P D L R S C V N A I P D Q) were synthesized on TAMPAL resin to yield C-terminal MPAL activated thioesters. Peptide EGF1 (80–116: C S P L P C N E D G Y M S C K D G K A S F T C T C K P G W Q G E K C E F D) was synthesized on MBHA resin to yield a C-terminal amide group after deprotection and cleavage of the peptide resin. The naturally occurring Val46 residue in native protein S was changed to glycine because of slow valine-to-cysteine native chemical ligation rates (4). The alteration did not affect protein S-cofactor activity for APC (23). Peptide TSR (47–79) was N-terminally protected with a 2-(methylsulfonyl)ethyl carbonate (Msc) group by a 2-h incubation with a 10-fold excess of activated Msc nitrophenyl ester in a minimal volume of dimethylformamide/5% diisopropylethylamine (24). After deprotection and cleavage from the resin, the polypeptides were HPLC-purified, lyophilized, and stored at −20°C until use.
HPLC.
Analytical reversed-phase HPLC was performed on a Hewlett Packard HPLC 1050 system using Vydac C-18 columns (5 μm, 0.46 × 15 cm). Semipreparative reversed-phase HPLC was performed on a Rainin HPLC system using a Vydac C-18 column (10 μm, 1.0 × 25 cm). Linear gradients of acetonitrile in water/0.1% trifluoroacetic acid were used to elute bound peptides. The flow rates used were 1 ml/min (analytical), and 5 ml/min (semipreparative).
Electrospray Ionization Mass Spectrometry (ESI-MS).
ESI-MS was performed on an API-III triple quadrupole mass spectrometer (PE-Sciex, Concord, ON, Canada). Peptide masses were calculated from the experimental mass-to-charge (m/z) ratios from all of the observed protonation states of a peptide by using MacSpec software (Sciex). Theoretical masses of peptides and proteins were calculated by using MacProMass software (Beckman Research Institute, Duarte, CA).
Disulfide Mapping of Microprotein S.
Qualitative disulfide mapping of microprotein S was performed with endoproteinase Asp-N (Sigma–Aldrich). Microprotein S (1 mg/ml) was incubated with endoproteinase Asp-N (10 μg/ml) in 50 mM Tris⋅HCl/1 mM EDTA, pH 8.0 at room temperature. After 90 min and 150 min, 10-μl samples were applied to HPLC (C-18: 0–60% acetonitrile, 2%/min). Peaks were collected and analyzed by ESI-MS.
Circular Dichroism (CD) Spectroscopy.
CD spectra were recorded on an Aviv Associates (Lakewood, NJ) CD spectrophotometer model 62A DS at 25°C. Microprotein S was dissolved in 25 mM boric acid at pH 7.4, and its concentration was determined by quantitative amino acid analysis. The percentage of α-helix was calculated as a function of the mean residue ellipticity at 222 nm (Eq. 1).
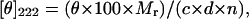 |
1 |
where [θ]222 is the mean residue ellipticity (deg⋅cm2⋅dmol−1), θ is the observed ellipticity at 222 nm, Mr is the molecular weight of the peptide, c is the concentration in mg/ml, d is the pathlength in cm, and n is the number of residues (25). The maximal theoretical mean residual ellipticity at 222 nm was determined with Eq. 2 (26).
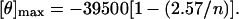 |
2 |
The percentage α-helical structure in microprotein S was determined by using Eq. 3.
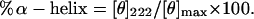 |
3 |
Results and Discussion
To access synthetic microprotein S (Fig. 1), optimized Boc chemistry solid-phase peptide synthesis protocols were used to generate three separate polypeptides comprising the three human protein S modules: Gla, Ala1–Gly46-α-thioester; TSR, Msc-Cys47–Gln79-α-thioester; and EGF1, Cys80–Asp116 (Fig. 2A). Because of the three-segment native chemical ligation strategy involved in the generation of microprotein S, the middle TSR piece contained both reactive moieties, namely an N-terminal cysteine together with a C-terminal thioester moiety. To prevent cyclization (27) or polymerization of the bifunctional TSR polypeptide under native chemical ligation conditions, the sulfhydryl moiety of the N-terminal cysteine in the TSR module was reversibly blocked with Msc (24). After syntheses, all peptides were HPLC purified and used in native chemical ligation reactions.
Figure 2.

Chemical synthesis of microprotein S by modular synthesis and multistep native chemical ligation. (A) Synthetic scheme leading to the 116-residue microprotein S polypeptide chain. To avoid polymerization or cycliza- tion reactions of the middle peptide segment corresponding to the TSR (residues 47–79), the N-terminal Cys residue of the TSR peptide-thioester was reversibly protected with an Msc group (24, 27). After ligation to the EGF1 module peptide (residues 80–116) was completed, the N-terminal Msc was removed, and the Gla-rich peptide (residues 1–46) was ligated to the TSR-EGF1 polypeptide (residues 47–116). ESI-MS of the reduced 1–116 polypeptide chain of microprotein S revealed a mass of 13,618 Da, corresponding to the theoretical mass of microprotein S containing 11 Gla residues in the Gla module. (B) After folding of the 1–116 polypeptide in 1 M guanidine at pH 8.0, microprotein S was obtained with an apparent molecular mass of 13,608 Da, the decrease of 10 Da caused by loss of 10 protons as a result of the formation of 5 disulfide bonds. Note the change in charged state distribution of the folded microprotein S to higher m/z ratios, which is characteristic behavior of folded proteins. The three-dimensional microprotein S structure shown is based on molecular homology modeling (28).
Starting with the C-terminal NH2-Cys80–Asp116-CONH2 segment, native chemical ligation with the Msc-NH-Cys47–Gln79-α-thioester peptide was performed as described (4). For this Gln-to-Cys ligation, a 90% yield of product was obtained within 8 h, consistent with model peptide studies of ligation rates (4). After Msc removal and purification of the ligation product, the N-terminal NH2-Ala1–Gly46-α-thioester peptide segment was ligated to the NH2-Cys47–Asp116-CONH2 intermediate. As expected for a Gly-Cys ligation, which is among the fastest of Xxx to Cys ligations (4), a yield greater than 95% of ligation product was generated within 4 h. HPLC purification resulted in pure protein S 1–116 polypeptide. ESI-MS revealed a molecular mass of 13,617.9 ± 1.9 Da, in agreement with the theoretical mass of 13,616.8 Da for the reduced protein S 1–116 polypeptide with 11 Gla residues (Fig. 2A).
After folding of the 1–116 polypeptide by stirring in air at a concentration of 0.2 mg/ml in 1 M guanidine at pH 8 for 48 h at 4°C, the observed molecular mass by ESI-MS decreased to 13,607.9 ± 0.1 Da, reflecting the loss of 10 protons because of formation of 5 disulfide bonds (Fig. 2B). The folded disulfide-containing protein product quantitatively eluted 2.5 min earlier on analytical HPLC (C-18: 22.5–41% acetonitrile/15 min). By ESI-MS, a shift in charged-state distribution of the folded microprotein S compared with the reduced polypeptide chain was also observed (Fig. 2). This is characteristic of the native, folded conformation of proteins, which typically carry fewer charges under ESI-MS ionization conditions than the unfolded, extended polypeptides (29). The presence of 5 mM CaCl2 did not influence the folding of microprotein S compared with the absence of CaCl2.
The EGF1 module contains 6 cysteines that form 3 disulfide bonds, and when the fully unprotected synthetic EGF1 module alone was folded, the yield of correct disulfide linkages was very low, with many isoforms observed on analytical HPLC (17). To overcome folding difficulties with the isolated chemically synthesized EGF1 module of protein S, a two-step folding procedure was used in which two of six cysteines were blocked with acetamidomethyl (Acm) groups (30). Spontaneous formation of the first two disulfide bonds was followed by Acm removal and generation of the third disulfide bond (17, 30). Strikingly, covalent attachment of the TSR and Gla modules to the fully unprotected EGF1 overcame this misfolding problem, as only one peak of the folded microprotein S was obtained on HPLC (data not shown). The apparent correct folding of EGF1 within the microprotein S construct favors a view that structural modules in a multidomain protein can act as templates for correct folding of adjacent modules, supporting the hypothesis that sequences of sufficiently long polypeptides contain information to determine the precise three-dimensional structure of a protein (31).
Proteolysis at Asp38 and Asp78, each conveniently located between modules of microprotein S (Fig. 1), was used to investigate possible misfolding of microprotein S resulting in incorrect disulfide cross-linking of protein S modules. Table 1 shows that microprotein S was quantitatively cleaved in three pieces, representing the Gla, TSR, and EGF1 modules. The absence of cross-linked modules within microprotein S indicates that the internal disulfide bonds in the Gla and TSR modules were correctly generated during folding of microprotein S, and that three internal disulfides were present in the EGF1 module. Although the precise orientation of the disulfides within the EGF1 could not be determined by using this technique, the absence of multiple peaks due to misfolded EGF1 (17), as well as the functional data described below, suggests a major part of microprotein S to be in a native-like structure.
Table 1.
Endoproteinase Asp-N proteolysis of microprotein S: Identification of fragments
| Digestion time, min | Retention time, min | Peak area, % | Mass, Da
|
Identity | |
|---|---|---|---|---|---|
| Observed | Calculated | ||||
| 0 | 17.13 | 100 | 13,608 | 13,607 | Ala1 –Asp116 |
| 90 | 15.08 | 30 | 4,792 | 4,793 | Ala1 –Thr37 |
| 15.58 | 70 | 4,302* | 4,303 | Asp78 –Asp116 | |
| 9,341* | 9,340† | Ala1 –Pro77‡ | |||
| 4,565* | 4,565† | Asp38 –Pro77 | |||
Microprotein S (1 mg/ml) was cleaved with endoproteinase Asp-N (10 μg/ml) at pH 8.0. Digests were applied to HPLC and peaks were analyzed by using ESI-MS.
These fragments all co-eluted in a heterogeneous peak at 15.58 min.
Calculated mass is based on hydrolysis of the peptide bond at Asp68 (+18) in the TSR module (Fig. 1).
The Ala1–Pro77 fragment was detectable during the early phase of proteolysis but disappeared after prolonged incubation (150 min).
To investigate the native properties of microprotein S, qualitative Ca2+ binding of microprotein S was studied by using CD spectroscopy. A typical protein spectrum was observed, consisting of α-helix, β-structure, and random coil (Fig. 3). When CaCl2 was added to a sample of microprotein S at 0.1 mM and 1 mM, an increase in mean residual ellipticity at 222 nm was observed with increasing CaCl2 concentrations (Fig. 3), indicating an increase of α-helicity of microprotein S upon binding of Ca2+ ions. At 10 mM CaCl2, the microprotein S self-associated. It was calculated that the amount of α-helicity of microprotein S increased by 25% on addition of CaCl2 (1 mM), from 18 residues in the absence of CaCl2 to 23 residues in the presence of 1 mM CaCl2. Ca2+ binding to Gla modules of the vitamin K-dependent family members factor II (32) and factor IX (33) also showed an comparable increase in helical structure of the respective Gla modules, resulting in structural ordering of the Ca2+-bound conformation.
Figure 3.

Effect of Ca2+ ions on the CD spectrum of synthetic microprotein S. The spectrum for microprotein S at 25°C is shown in the absence of CaCl2 (●) or in the presence of 0.1 mM CaCl2 (○) or 1 mM CaCl2 (▾). MRW, mean residue weight.
The functional anticoagulant cofactor activity of synthetic microprotein S was studied in a plasma milieu with a typical assay (34) that is based on the enhancement of APC-dependent prolongation of the activated partial thromboplastin time. Addition of purified protein S or of normal pooled plasma to protein S-deficient plasma in the presence of APC resulted in a dose-dependent prolongation of the clotting time (Fig. 4A), reflecting the anticoagulant potential of the APC/protein S pathway. The synthetic microprotein S showed a dose-dependent increase of the prolongation of clotting time of protein S-deficient plasma by APC that indicated at least 30% bioactivity compared with native plasma-derived protein S (Fig. 4A). Addition of protein S or microprotein S to protein S-deficient plasma in the absence of APC under these conditions failed to induce the dose-dependent prolongation of clotting time observed in the presence of APC, confirming that microprotein S functioned as an APC cofactor. Ablation of anticoagulant cofactor activity by proteolysis of the TSR in protein S and microprotein S by thrombin resulted in total loss of cofactor activity for APC (Fig. 4A).
Figure 4.

Biological anticoagulant cofactor activity of microprotein S and the effect of plasma inhibitor C4b-binding protein. (A) Prolongation of the clotting time of protein S-deficient plasma in the presence of APC because of addition of microprotein S (●), native plasma-derived protein S (▾), standard plasma calibrator (■), thrombin-cleaved microprotein S (⊙), and thrombin-cleaved protein S (▿·). In controls in the absence of APC, no significant anticoagulant effect of microprotein S (○) or protein S (▿) was observed. Protein S unicalibrator lyophilized plasma containing protein S at a level of 115% was used as a calibration curve. Molecular concentrations of plasma protein S and microprotein S were determined with an ELISA and amino acid analysis, respectively. (B) The effect of C4b-binding protein on the anticoagulant APC-cofactor activity of protein S (300 nM, ▾) and microprotein S (900 nM, ●). In controls, C4b-binding protein alone was added to protein S deficient plasma (▵).
These observations show that microprotein S, a chemically synthesized multidomain protein, acts as a cofactor to APC in the down-regulation of blood coagulation.
When C4b-binding protein, a component of complement, is bound to protein S (9), the cofactor activity for APC is lost. Because C4b-binding protein binds to protein S at residues 413–433 in the sex hormone binding globulin-like (SHBG) region of protein S (10), microprotein S would be expected to be insensitive to this natural inhibitor of protein S. Indeed, in contrast to native protein S, microprotein S bioactivity was not neutralized by addition of C4b-binding protein (Fig. 4B). This finding is consistent with the observation that the anticoagulant cofactor activity of a truncated recombinant protein S construct containing residues 1–242 is not neutralized by C4b-binding protein (35).
The level of microprotein S anticoagulant cofactor activity compared with native protein S was approximately 30% (Fig. 4A). Several contributions of residues 117–635 of protein S might be considered. First, it has been reported that the SHBG region of protein S contains a binding site for factor Va (36, 37), which might contribute to a more favorable orientation of the enzyme–cofactor APC–protein S complex toward the substrate factor Va. Second, the third and fourth EGF modules that contain high affinity Ca2+-binding sites (38) might contribute to protein S activity by one or more mechanisms. However, a recombinant construct including the Gla module, the TSR module, and all four EGF modules (protein S, residues 1–242) (35) had less bioactivity (±10% of native protein S) in a plasma bioassay than synthetic microprotein S described here.
In conclusion, we have designed and constructed by native chemical ligation synthetic microprotein S, a multidomain protein that exhibits the anticoagulant properties of the native parent plasma protein. Significantly, the anticoagulant activity of synthetic microprotein S was unaffected by the natural plasma inhibitor of protein S, C4b-binding protein. Specifically, as shown by this example, incorporation of the EGF1 module into its protein S “context” facilitated correct folding of the EGF1 module, thereby expressing its native structural determinants.
In the “postgenome era” of biomedical research, rapid chemical synthesis of biologically active multidomain proteins, as exemplified by microprotein S, will contribute in a unique way to functional studies of new multidomain proteins whose sequences are discovered in human and other genomes.
Acknowledgments
We thank Dr. J. K. Judice for his expert advice in the synthesis of Nα-Boc-γ,γ-dicyclohexyl-l-γ-carboxyglutamic acid. This work was in part supported by National Institutes of Health grants to P.E.D. (GM 59380), S.B.H.K., and J.H.G. (HL 21544) and a fellowship of the Royal Netherlands Academy of Arts and Sciences to T.M.H.
Abbreviations
- APC
activated protein C
- ESI-MS
electrospray ionization mass spectrometry
- Gla and γ
three- and one-letter codes for γ-carboxyglutamic acid
- Gla module
Gla-rich module
- TSR module
thrombin-sensitive module
- EGF1 module
first epidermal growth factor-like module
- Boc
tert-butoxycarbonyl
- MBHA
methylbenzhydrylamine
- TAMPAL
trityl-associated mercaptopropionic acid leucine
- Msc
2-(methylsulfonyl)ethyl carbonate.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.260239797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.260239797
References
- 1.Teitel J M. Haemophilia. 1998;4:393–401. doi: 10.1046/j.1365-2516.1998.440393.x. [DOI] [PubMed] [Google Scholar]
- 2.Wilken J, Kent S B H. Curr Opin Biotechnol. 1998;9:412–426. doi: 10.1016/s0958-1669(98)80016-5. [DOI] [PubMed] [Google Scholar]
- 3.Dawson P E, Muir T W, Clark-Lewis I, Kent S B H. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 4.Hackeng T M, Griffin J H, Dawson P E. Proc Natl Acad Sci USA. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kisiel W, Canfield W M, Ericsson L H, Davie E W. Biochemistry. 1977;16:5824–5831. doi: 10.1021/bi00645a029. [DOI] [PubMed] [Google Scholar]
- 6.Walker F J. J Biol Chem. 1981;256:11128–11131. [PubMed] [Google Scholar]
- 7.Dahlbäck B. Biochem J. 1983;209:837–846. doi: 10.1042/bj2090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker F J. J Biol Chem. 1984;259:10335–10339. [PubMed] [Google Scholar]
- 9.Dahlbäck B, Stenflo J. Proc Natl Acad Sci USA. 1981;78:2512–2516. doi: 10.1073/pnas.78.4.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernández J A, Heeb M J, Griffin J H. J Biol Chem. 1993;268:16788–16794. [PubMed] [Google Scholar]
- 11.Griffin J H, Evatt B, Zimmerman T S, Kleiss A J, Wideman C. J Clin Invest. 1981;68:1370–1373. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comp P C, Nixon R R, Cooper M R, Esmon C T. J Clin Invest. 1984;74:2082–2088. doi: 10.1172/JCI111632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz H P, Fischer M, Hopmeier P, Batard M A, Griffin J H. Blood. 1984;64:1297–1300. [PubMed] [Google Scholar]
- 14.Branson H E, Katz J, Marble R, Griffin J H. Lancet. 1983;ii:1165–1168. doi: 10.1016/s0140-6736(83)91216-3. [DOI] [PubMed] [Google Scholar]
- 15.Seligsohn U, Berger A, Abend M, Rubin L, Attias D, Zivelin A, Rapaport S I. N Engl J Med. 1984;310:559–562. doi: 10.1056/NEJM198403013100904. [DOI] [PubMed] [Google Scholar]
- 16.Mahasandana C, Suvatte V, Marlar R A, Manco-Johnson M J, Jacobson L J, Hathaway W E. Lancet. 1990;335:61–62. doi: 10.1016/0140-6736(90)90201-f. [DOI] [PubMed] [Google Scholar]
- 17.Hackeng T M, Dawson P E, Kent S B H, Griffin J H. Biopolymers. 1998;46:53–63. doi: 10.1002/(SICI)1097-0282(199808)46:2<53::AID-BIP1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 18.Hackeng T M, Yegneswaran S, Johnson A E, Griffin J H. Biochem J. 2000;349:757–764. doi: 10.1042/bj3490757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stenflo J. Crit Rev Eukaryot Gene Expr. 1999;9:59–88. [PubMed] [Google Scholar]
- 20.Schnölzer M, Alewood P, Jones A, Alewood D, Kent S B H. Int J Pept Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 21.Sarin V K, Kent S B H, Tam J P, Merrifield R B. Anal Biochem. 1981;117:147–157. doi: 10.1016/0003-2697(81)90704-1. [DOI] [PubMed] [Google Scholar]
- 22.Nishiuchi Y, Nakao M, Nakata M, Kimura T, Sakakibara S. Int J Pept Protein Res. 1993;42:533–538. doi: 10.1111/j.1399-3011.1993.tb00361.x. [DOI] [PubMed] [Google Scholar]
- 23.He X, Dahlbäck B. Eur J Biochem. 1993;217:857–865. doi: 10.1111/j.1432-1033.1993.tb18314.x. [DOI] [PubMed] [Google Scholar]
- 24.Canne L E, Winston R L, Kent S B H. Tetrahedron Lett. 1997;38:3361–3364. [Google Scholar]
- 25.Schmid F X. In: Protein Structure, a Practical Approach. Creighton T E, editor. Oxford: IRL Press; 1991. pp. 251–285. [Google Scholar]
- 26.Chen Y H, Yang J T, Chau K H. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Tam J P. J Am Chem Soc. 1997;119:2363–2370. [Google Scholar]
- 28.Villoutreix B O, Teleman O, Dahlbäck B. J Comput-Aided Mol Design. 1997;11:293–304. doi: 10.1023/a:1007912929828. [DOI] [PubMed] [Google Scholar]
- 29.Chowdhury S K, Katta V, Chait B T. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 30.Yang Y, Sweeney W V, Schneider K, Chait B T, Tam J P. Protein Sci. 1994;3:1267–1275. doi: 10.1002/pro.5560030813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anfinsen C B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 32.Soriano-Garcia M, Padmanabhan K, de Vos A M, Tulinski A. Biochemistry. 1992;31:2554–2566. doi: 10.1021/bi00124a016. [DOI] [PubMed] [Google Scholar]
- 33.Freedman S J, Furie B C, Furie B, Baleja J D. Biochemistry. 1995;34:12126–12137. doi: 10.1021/bi00038a005. [DOI] [PubMed] [Google Scholar]
- 34.Wolf M, Boyer-Neumann C, Leroy-Matheron C, Martinoli J L, Contant G, Amiral J, Meyer D, Larrieu M J. Blood Coagul Fibrinolysis. 1991;2:705–712. doi: 10.1097/00001721-199112000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Van Wijnen M, Stam J G, Chang G T, Meijers J C, Reitsma P H, Bertina R M, Bouma B N. Biochem J. 1998;330:389–396. doi: 10.1042/bj3300389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hackeng T M, van 't Veer C, Meijers J C M, Bouma B N. J Biol Chem. 1994;269:21051–21058. [PubMed] [Google Scholar]
- 37.Heeb M J, Kojima Y, Rosing J, Tans G, Griffin J H. J Biol Chem. 1999;274:36187–36192. doi: 10.1074/jbc.274.51.36187. [DOI] [PubMed] [Google Scholar]
- 38.Dahlbäck B, Hildebrand B, Linse S. J Biol Chem. 1990;265:18481–18489. [PubMed] [Google Scholar]