

Abstract
Time-resolved UV resonance Raman (UVRR) spectroscopic studies of WT and mutant myoglobin were performed to reveal the dynamics of protein motion after ligand dissociation. After dissociation of carbon monoxide (CO) from the heme, UVRR bands of Tyr showed a decrease in intensity with a time constant of 2 ps. The intensity decrease was followed by intensity recovery with a time constant of 8 ps. On the other hand, UVRR bands of Trp residues located in the A helix showed an intensity decrease that was completed within the instrument response time. The intensity decrease was followed by an intensity recovery with a time constant of ≈50 ps and lasted up to 1 ns. The time-resolved UVRR study of the myoglobin mutants demonstrated that the hydrophobicity of environments around Trp-14 decreased, whereas that around Trp-7 barely changed in the primary protein response. The present data indicate that displacement of the E helix toward the heme occurs within the instrument response time and that movement of the FG corner takes place with a time constant of 2 ps. The finding that the instantaneous motion of the E helix strongly suggests a mechanism in which protein structural changes are propagated from the heme to the A helix through the E helix motion.
Keywords: hemeprotein, protein dynamics, resonance Raman spectroscopy, time–resolved spectroscopy
Proteins are endowed with both stiff and flexible properties; hence, their dynamics are closely associated with structure and function. Because allosteric proteins, in general, propagate conformational changes over considerable distances, how these conformational changes are generated and transmitted is of major interest for understanding the regulatory, kinetic, and recognition properties of proteins (1–3). A variety of experimental evidence suggests that rapid and long-range propagation of conformational changes through the core of protein plays a vital role in allosteric communication. For example, the cooperative oxygen-binding properties of hemoglobin (Hb) result from a change in quaternary structure, which is initiated by ligand binding/release at the heme (ligand binding site). Therefore, if the pathway by which one quaternary structure is converted to the other quaternary structure is structurally characterized, our understanding how a protein performs its function will be greatly advanced. The ligand-induced dynamics of myoglobin (Mb) are a basic subject for studying such features in proteins. Although Mb is a monomeric protein, the three-dimensional structure of Mb is closely similar to that of a subunit of Hb. Thus, the structural changes of Mb can be regarded as a model for the tertiary structural events that cause the quaternary structural change of Hb.
In Mb, the Fe2+ ion contained in the heme is bound to the proximal histidine (His-93) as the sole covalent link to the protein. The magnitude of atomic displacement at the heme required for ligand binding/release is estimated from x-ray crystal structures of unligated (deoxyMb) and CO bound forms (MbCO) (4). The ligated heme has a planar structure with a low-spin iron atom. Upon ligand dissociation, the iron atom is converted from low to high spin states and moves out of the porphyrin plane by 0.3 Å, which transforms the structure of the heme into a domed form.
The crystal structures of deoxyMb and MbCO show significant differences in the positions of the E and F helices, which hold the heme group as shown in Fig. 1. In the deoxyMb, backbone segments near His-64 in the E helix and His-93 in the F helix are shifted in the same direction by 0.3 Å compared with that in MbCO. Transient grating studies suggested the initial motions of the protein are collective in nature (5, 6). Time-resolved crystallography suggested that global conformational change of the protein propagates from the heme throughout the entire protein within 150 ps after CO dissociation (7). Although experimental information on the ultrafast motion of Mb is available, it was not known what specific motions of the protein portion contribute to the early time response, and only qualitative statements about the possible motions could be derived from stationary structures. Time-resolved spectroscopic studies are expected to give an answer to this question for a protein in solution, which cannot be given by x-ray crystallography. However, most of the early spectroscopic studies have been limited to observation of the structural changes within the heme group. Studies to monitor protein motions with high time resolution are required to elucidate the primary protein dynamics induced by ligand association and dissociation (5, 8–13).
Fig. 1.

Diagram showing the arrangement of the E and F helices that hold the heme group. This figure was produced with PyMOL (http://pymol.sourceforge.net) by using a structure from the Protein Data Bank (PDB ID code 1DWR).
UV resonance Raman (UVRR) spectroscopy is a powerful tool for protein structural studies because it enables selective observation of the Raman bands of aromatic amino acid residues by tuning the excitation wavelength (14, 15). Because several vibrational bands of aromatic residues serve as structural markers of proteins, time-resolved UVRR spectroscopy can provide site-specific information about protein motion (16, 17). Previously, we had reported preliminary results using picosecond time-resolved UVRR spectroscopy and proved its advantages to study ultrafast protein motion (18). Accordingly, we carried out more detailed study using mutants and succeeded in detecting primary structural changes after CO dissociation in Mb. The present results provide a concrete picture of the primary protein response after ligand dissociation and insights into the pathway by which protein structural changes propagate from the heme to the sites far from the heme.
Results
UVRR Spectral Changes in WT Mb.
Fig. 2A shows picosecond time-resolved UVRR difference spectra at delay times from −5 ps to 1,000 ps. The procedure for calculating the UVRR difference spectra from unprocessed time-resolved UVRR spectra is described elsewhere [see supporting information (SI) Text and SI Fig. 7]. UVRR bands of Trp and Tyr residues in the spectra were identified by comparison with UVRR spectra of aqueous amino acid solutions. Mode assignments made by Harada and coworkers (14) were adopted. Immediately after CO dissociation, several negative bands appeared in the difference spectra. The negative bands at 759, 1,011, 1,357, and 1,559 cm−1 result from the intensity changes of the W18, W16, W7, and W3 bands of Trp residues, whereas the weak negative band at 1,179 cm−1 results from the intensity change of the Y9a band of Tyr residues. The negative band at 1,620 cm−1 arises from intensity changes of both Y8a and W1 bands. On the basis of the intensity ratio of W3 to W1 bands of an aqueous Trp solution observed with 232-nm excitation, the contribution from W1 to the negative band at 1,620 cm−1 was estimated to be approximately one-third of the total intensity. Because the time-resolved difference spectra were obtained by subtracting the spectrum of MbCO from that of photodissociated MbCO, the appearance of the negative bands indicates that band intensities of photodissociated MbCO are weaker than those of MbCO. Fig. 2B shows overlaid difference spectra at early time delays. The spectrum for each time delay is normalized in terms of intensity of the W3 negative band for ease of comparison of the 1,620-cm−1 band intensities. It is evident that the intensity decrease of the 1,620-cm−1 band is delayed relative to the decrease of the W3 band.
Fig. 2.

Picosecond UVRR difference spectra of the photodissociated WT horse skeletal MbCO. (A) The difference spectra in the range of 650–1,800 cm−1. The top trace is a probe-only spectrum corresponding to the UVRR spectrum of MbCO divided by a factor of 30. The bottom trace is a deoxyMb-minus-MbCO difference spectrum. Spectra have been offset for clarity. (B) The overlaid difference spectra at early time delays showing slower intensity decrease of the 1,620-cm−1 band relative to that of W3 band. The spectrum for each time delay is normalized in terms of intensity of the W3 negative band.
Temporal behaviors of integrated intensity of W18, W16, and W3 bands relative to those of MbCO are shown in Fig. 3. The intensity of the W16 band showed a decrease within the instrument response time that was followed by an exponential increase with a time constant of 45 ps. After the increase to −2.1%, the relative intensity of the W16 band remained unchanged up to 1,000 ps. The temporal evolutions of intensity of the W18 and W3 bands were similar to that of W16. Fig. 3 Lower shows a close-up of the time region from −7 to 35 ps.
Fig. 3.

Temporal changes of integrated intensity of W18, W16, and W3 bands relative to the integrated intensity in the probe-only spectrum. The solid lines are fits using an exponential function of the form A[1 + B exp(−t/τrise)] convoluted with an instrument response function (dashed line). The fit line for each band was obtained with the parameters of τrise = 49 ± 16 ps and B = 0.62 ± 0.06 for W18, τrise = 45 ± 15 ps and B = 0.48 ± 0.05 for W16, and τrise = 59 ± 18 ps and B = 0.34 ± 0.03 for W3, respectively. The lower panel shows a close-up of the curve in the early time region of the upper panel.
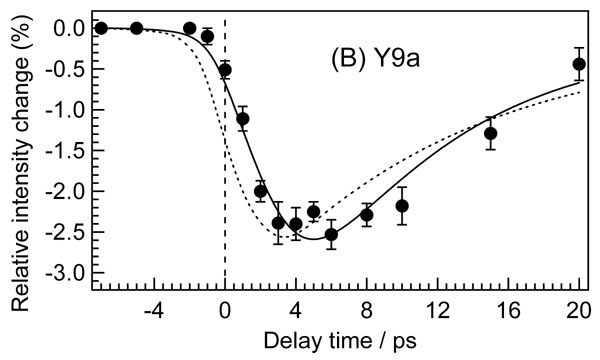
Fig. 4 shows the temporal behavior of integrated intensities of the band at 1,620 cm−1 and the Y9a band. In contrast to the time evolutions of the band intensity of Trp modes, the decay of the intensity did not match the instrument response function as shown in Fig. 4 Lower. Initial intensity change of the 1,620-cm−1 band is delayed relative to that of the W3 band (see SI Fig. 8). After the intensity decrease, initial band intensity recovered within tens of picoseconds. A good fit to the intensity was obtained by convoluting the instrument response with a 1.9-ps exponential decay and a 7.3-ps exponential rise. It should be noted that the initial decay component was necessary to obtain a good fit (see SI Fig. 9). The intensity change of the Y9a band can be fitted by a convolution of the instrument response with a 2.0-ps exponential decay and a 8.0-ps exponential rise. Although the 1,620-cm−1 band is composed of the Y8a band of Tyr and the W1 band of Trp, the temporal behavior of its intensity is similar to that of the Y9a band, indicating that the intensity change of the 1,620-cm−1 band arises mainly from the Y8a component.
Fig. 4.

Temporal changes of integrated intensity of the 1620-cm−1 and Y9a bands relative to the intensity in the probe-only spectrum. It should be noted that the intensity change of the band at 1620 cm−1 mainly arises from intensity change of Y8a band with a smaller contribution from intensity change of W1 band. The solid lines are fits using a sum of two exponential functions of the form A[1 − exp(−t/τdecay)] + B[exp(−t/τrise) − 1] convoluted with the instrument response function (dashed line). The fit line for the 1,620-cm−1 band was obtained with the parameters of τdecay = 1.9 ± 0.9 ps, τrise = 7.3 ± 1.4 ps, A = 0.21 ± 0.03, and B = 0.19 ± 0.03. The fit line for the Y9a band was obtained with the parameters of τdecay = 2.0 ± 0.8 ps, τrise = 8.0 ± 3.6 ps, A = 0.19 ± 0.03, and B = 0.18 ± 0.03. The lower panel shows a close-up of the curve in the early time region of the upper panel.
UVRR Spectral Changes in Single Trp Mutants.
Horse Mb contains two Trp residues: Trp-7 and Trp-14. To identify which Trp residue contributes to the intensity changes shown in Fig. 3, we performed time-resolved UVRR measurements for single Trp mutants in which one of the two Trp residues was substituted for Phe. Because the mutants of horse Mb were not available, we used sperm whale Mb instead. Sperm whale Mb contains two Trp residues in the same locations as in horse Mb, and its amino acid sequence is very similar to that of horse Mb. The temporal evolutions of Trp band intensities observed for WT sperm whale Mb were similar to those observed for WT horse Mb (see SI Fig. 10). Accordingly, environmental changes in the Trp residues in sperm whale Mb are predicted to be similar to those of Trp residues in horse Mb.
Fig. 5 A–C shows the time-resolved difference spectra of WT, W7F, and W14F mutants of sperm whale Mb, respectively. Fig. 5D compares the temporal profiles of the intensity change relative to the band intensities in the probe-only spectra. In both time-resolved difference spectra of WT Mb and of W7F mutant, negative bands of Trp appeared at 0-ps delay after CO dissociation. In contrast, a negative band was not observed in the difference spectrum of the W14F mutant. The time evolution of intensity changes of Trp bands in W7F mutant (Trp-14) resembled those in WT Mb. In contrast, at each time delay, the intensity loss of Trp bands in the W14F mutant (Trp-7) was considerably smaller than those in WT Mb and W7F mutant. Intensity changes relative to the corresponding band intensity in the probe-only spectra are larger for the W7F mutant than those for the WT Mb (Trp-7 + Trp-14), because the changed portion of UVRR intensity is larger for Trp-14 than for Trp-7. Based on these observations, we conclude that the instantaneous intensity decrease of Trp bands observed for WT photodissociated MbCO is mainly due to a change of Trp-14.
Fig. 5.

Comparison of intensity change of Trp Raman bands in WT Mb and in Trp mutants of Mb. (A–C) Time-resolved difference spectra of WT Mb, W7F-Mb and W14F-Mb, respectively. The top trace in each panel is the UVRR spectrum of MbCO. Spectra in each panel have been offset for clarity. (D) The changes of integrated intensity of W18 and W16 bands in WT and the two mutant Mbs relative to the intensity in the individual probe-only spectra against a delay time.
Discussion
In this study, the intensity changes of Trp and Tyr UVRR bands were observed after CO dissociation, indicating conformational changes of the side chains and/or environmental changes around those residues (19, 20). As a factor that can affect the UVRR intensity other than conformational changes and the environmental changes, a heating effect and a through-space electronic coupling between the amino acid residues and the heme should be taken into account. The contribution of the former is supposed to be very small, as discussed previously (18). The contribution of the latter is considered to be negligible for the following reason. A static UVRR study on sperm whale Mb showed that the differences in the Tyr bands between MbCO and deoxyMb are predominantly due to Tyr-151 (21), although Tyr-151 is farthest from the heme among the Tyr residues. If the coupling was significant, Tyr residues closer to the heme should contribute to the spectral changes more upon the change in the electronic state of the heme. Therefore, the intensity change of the UVRR bands due to the electronic coupling is negligible. Thus, we have concluded that the intensity changes observed in the UVRR spectra reflect conformational changes and/or the environmental changes around the residues.
Structural Changes of Protein Around the Tyr Residues.
Horse Mb has two Tyr residues, Tyr-103 and Tyr-146. The former is involved in the G helix, whereas the latter is in the C-terminal region of the H helix. Fig. 6 presents the crystallographic structure of horse Mb (22). Because Tyr-103 is exposed to solvent in both deoxyMb and MbCO structures, the contribution of Tyr-103 to the UVRR intensity changes upon CO dissociation is unlikely. On the other hand, the hydroxyl group of Tyr-146 forms a hydrogen bond to the backbone carbonyl of Ile-99 in the FG corner and is adjacent to the backbone carbonyl of His-93 in the F helix. In addition, the aromatic ring of Tyr-146 contacts the side chain of Ala-94. An analysis of high-resolution x-ray crystal structures showed that the positions of main-chain atoms in the F helix and FG corner in deoxyMb were distinctly different from those in MbCO (4). These structural differences suggested that, upon CO dissociation, the F helix moves away from the heme and that the FG corner moves toward the heme group in opposition to the motion of F helix (see Fig. 6). These movements of the F helix and FG corner were also found crystallographically in a transient structure at 150 ps after CO dissociation (7, 23). The reorientations of the F helix and the FG corner indicated by the static and transient x-ray crystal structures can affect the hydrogen bond and nonbonded contacts of Tyr-146 with neighboring residues. Therefore, it is reasonable that these structural changes are responsible for the UVRR intensity change of Tyr modes.
Fig. 6.

Crystallographic structure of horse Mb (22). (A) Close-up of the region near the two Tyr residues. (B) Close-up of the region near the two Trp residues. The orange arrows indicate the directions of motions of the F helix and FG corner after CO dissociation that are expected from crystallographic studies. This figure was produced with PyMOL (http://pymol.sourceforge.net) by using a structure from the Protein Data Bank (PDB ID code 1DWR).
Previous picosecond time-resolved visible resonance Raman studies showed that heme doming concomitant with iron out-of-plane movement is nearly completed within 2 ps (12, 24). The heme doming induces the F helix movement, which should cause the movement of the FG corner and shift position of His-93 and Ala-94, which results in an environmental change around Tyr-146. In addition, the heme doming can directly move the side chain of Ile-99 adjacent to the heme pyrrole ring. Therefore, the intensity decrease of the Tyr bands with a time constant of 2 ps is attributed to the structural change of the FG corner responding to the fast movement of the F helix and/or the heme doming. After the intensity decrease, the intensity of Tyr bands recovered to an initial value with a time constant of 8 ps. This means that further structural changes take place to alter the environment around Tyr-146. The temporal evolution of the amide I IR absorption after CO photolysis indicates that conformational relaxation of the polypeptide skeleton occurs on a 6- to 8-ps time scale (9). The time constant of the intensity recovery of Y9a is close to that of the IR absorption change of amide I band. If the conformational change of the polypeptide skeleton occurs in the F helix and FG corner, it should cause environmental changes around Tyr-146. Therefore, it is likely that the UVRR and IR experiments observe the common structural change of the polypeptide skeleton that takes place in the F helix and FG corner. The time constant of 8 ps is close to that reported for the spectral change of the photoexcited heme because of vibrational cooling (25). But the intensity recovery is not due to the heme cooling, because the contributions of the heating effect and of the electronic coupling on the UVRR intensity are considered to be negligible as described above.
Structural Changes of Protein Around the Trp Residues.
Two Trp residues are located in the A helix. Crystallographic data showed that Trp-7 is partially exposed on the protein surface and contacts Lys-79 in the EF corner. On the other hand, Trp-14 is buried within the protein and contacts hydrophobic side chains of Leu-69 and Leu-72 in the E helix. Hydrogen bonds are formed between the Trp residues and neighboring water molecules (22). The frequency of W17 band (875 cm−1), which serves as a hydrogen bond marker of Trp indole ring, suggests the formation of medium-strength hydrogen bonds in solution (26). Although the W16 and W18 bands showed frequency shift concomitant with the intensity decrease, the W17 band did not shift upon CO dissociation (Fig. 2A). Therefore, it is concluded that the intensity change of Trp bands does not reflect the change of the hydrogen bonds but does reflect the change in environment around Trp-14. This environmental change would also cause the shift of the W16 and W18 bands because these are ring-breathing modes sensitive to an electronic state of the aromatic ring.
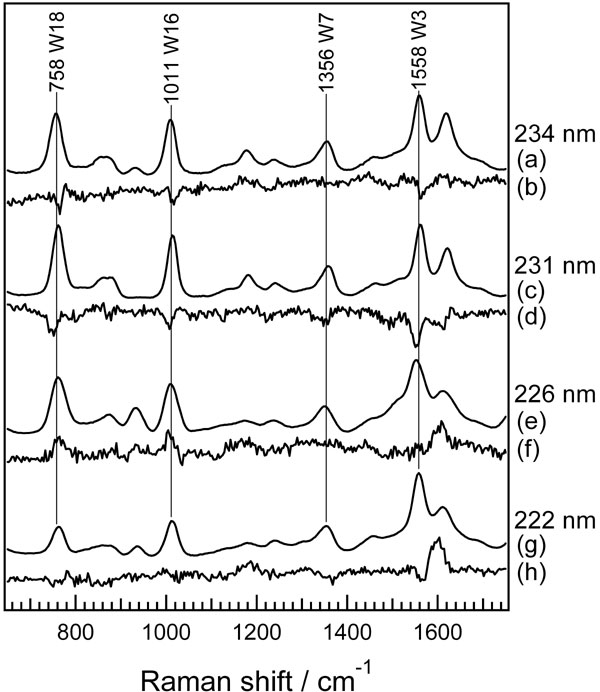
The results of UVRR studies of the Trp mutants strongly suggest that the instantaneous intensity decrease of Trp bands in WT photodissociated MbCO is mainly attributable to the change in the Trp-14 environment. Thus, the structural change is significant at the interface between the E helix and A helix at the AB corner, whereas a structural change is relatively small at the interface between the EF corner and the A helix on the N-terminal side (Fig. 6B). UVRR studies of Trp in various solvents found the correlation between the Raman intensities and solvent polarizability or hydrophobicity (19). Studies of structurally well defined proteins showed the correlation is applicable to Trp residues in protein (27, 28). If a Trp residue is buried into more polarizable or hydrophobic environments, the Bb absorption band shifts to lower energy and, concomitantly, the Raman excitation profile is expected to exhibit a red shift. The red shift should result in the enhancement of Raman intensity when the excitation wavelength is on the red side of the excitation profile. In fact, the deoxyMb-minus-MbCO difference spectra measured at various excitation wavelengths showed that the difference features of the Trp bands changed from negative to positive when the excitation wavelength shifted shorter from 230 to 226 nm (see SI Fig. 11). This indicates that the maximum of the excitation profile is present between 226 and 230 nm and the present excitation wavelength at 232 nm lies on the red side of the excitation profile. Therefore, the instantaneous intensity decrease of the Trp bands can be attributed to a decrease of polarizability or hydrophobicity around Trp-14.
The high-resolution structures of MbCO and deoxyMb showed that the main chain atoms of His-64 in the E helix are closer to the heme by 0.3 Å in deoxyMb than in MbCO (4). In addition, the E helix movement toward the heme was observed crystallographically in the transient structure at 1 ns after CO photodissociation (29). If the E helix displaces toward the heme group, it should cause weakening of the nonbonded contacts of Trp-14 with Leu-69 and Leu-72 that result in a decrease of hydrophobicity around Trp-14. Accordingly, we conclude that the hydrophobicity decrease of the Trp-14 environment reflects the E helix movement toward the heme that takes place within the instrument response time.
Our observation suggests that the environmental change around Trp-14 occurs in the primary protein response. On the other hand, static UVRR and time-resolved fluorescence studies suggested that the environment difference between MbCO and deoxyMb is found not around Trp-14 but around Trp-7 (21, 30). This means that the environmental change around Trp-14 is transient. The UVRR intensity decrease of Trp-14 does not recover completely up to 1,000 ps. Therefore, further structural relaxation that changes the environment of both Trp residues would take place later than a nanosecond. This structural change may be attributable to the A helix rearrangement in response to the fast E helix motion, because a change in the relative orientation between the A and E helices should influence the contacts of Trp-7 with the EF corner and of Trp-14 with the E helix.
Picture of the Primary Protein Response.
Transient phase grating measurements suggested that Mb changes its shape within 500 fs after CO dissociation (5). The present data imply the occurrence of ultrafast motion of the E helix within the instrument-limited time, suggesting its involvement in the shape change.
Transient grating studies suggested that photodissociation of Fe–CO bond activates the collective motions of the protein to drive the protein from its ligated to deligated conformation (5, 6). A molecular dynamics simulation showed that, as the Fe–CO bond stretches, the protein structure evolved in the direction predicted from the comparison of equilibrium structures (31). The normal mode analysis of the oxy- and deoxyMb found that the functionally important conformational change can well be expressed in terms of a relatively small number of collective low-frequency modes (32). These results provide the view that the reaction forces at the heme become channeled very efficiently into the spatially extended collective motions of the globin. The present study experimentally identified what specific motions of protein portion contribute to the primary protein response: we demonstrated that primary protein response involves not only the FG corner motion responding to the F helix motion and/or the heme doming but also the E helix motion, which is impulsively driven upon the ligand dissociation, consistent with the view described above.
Propagation of Structural Changes from the Heme to the E Helix.
It is believed that structural changes are propagated from the heme to globin through the sole covalent linkage between the heme and protein (Fe–His-93). However, the instantaneous displacement of the E helix strongly suggests a pathway other than that through the Fe–His-93 bond. There must be a mechanism that directly drives the E helix following the CO dissociation. A recent static UVRR study of the H93G mutant Mb suggests that the Fe–His-93 bond is not responsible for transmitting the structural changes from the heme to the N terminus of A-helix (33). Thus, the structural changes at the heme proximal side induced by a change of the Fe–His-93 bond would only be a partial element of the protein response. Despite the relatively weak interaction between the heme and the E helix, the E helix moves very quickly in response to the heme doming. The ligand dissociation and/or heme doming would drive collective protein motions involving a segmental motion of the E helix. Some parts of the protein should mediate the force for the collective mode.
The most likely mediator is Val-68 whose side chain is in contact with the oxygen atom of the bound CO molecule in the heme (4). Studies on distal heme pocket indicated that the side chain of Val-68 gives rises to a steric hindrance for the bound CO (34–36). The time-resolved x-ray crystallographic data suggested that movements of hydrophobic residues near the heme including Val-68 occur by 100 ps to accommodate doming of the heme and translocation of the CO after photolysis (37). Therefore, the dissociation-induced relief of the contact of Val-68 with CO could trigger the movement of the E helix toward the heme. It should be noted that Guallar et al. (31) had attributed the loss of this contact to the rapid E helix movement toward the heme observed in their molecular dynamics simulation. Seno and Go (32, 38) demonstrated that protein conformational changes are transmitted from the heme to the A helix in the direction from Leu-69 and Leu-72 to Val-13 and Trp-14. Therefore, we propose that the mechanism in which the structural changes are transmitted to the A helix is the dissociation-induced movement of the E helix and concomitant change of the nonbonded contacts of Trp-14 with Leu-72 and Leu-69. The propagation of structural changes from the heme to the E and A helices through the nonbonded contact of Val-68 with CO appears to be plausible. The validity of the pathway through the contact of CO with Val-68 remains to be further tested in future studies by time-resolved UVRR using mutant Mb, substituting Val-68 for residues with less bulky side chains.
In conclusion, protein structural changes are propagated from the heme to throughout the entire globin through at least two pathways. In one pathway, changes are transmitted to the C-terminal region of the H helix through the structural changes in the proximal side of the heme as proposed so far: the rearrangement of the F helix and FG corner induced by the heme doming. However, the instantaneous environmental change around Trp-14 in the A helix is not compatible with the transmission of the structural change through the proximal side of the heme. In another pathway, protein structural changes are propagated from the heme to the N-terminal region of the A helix through the E helix.
Materials and Methods
Details of our UVRR apparatus were described previously (18). A Ti/sapphire oscillator–amplifier laser system provided pulses tuned to 816 nm with a repetition rate of 1 kHz. The second harmonic (408 nm) of the laser output was focused into a Raman shifter filled with methane gas to generate first Stokes stimulated scattering at 463 nm. A probe pulse at 232 nm was generated as the second harmonic of the 463-nm output. Another portion of the 408-nm pulse was used as a pump pulse, which passed through an optical delay stage. At the sample point, energies of the probe and pump pulses were 0.4 and 15 μJ, respectively. The cross correlation width between the two pulses was 3.3 ps.
Horse skeletal Mb (Sigma) and sperm whale Mb (Biozyme) were used without further purification. They were dissolved into a 50 mM phosphate buffer at pH 7.4 to make 250 μM solutions. W14F and W7F mutants of sperm whale Mb were expressed and purified as described previously with some modifications (21, 39). The concentration of the Trp mutants was adjusted to 180 μM in 50 mM phosphate buffer at pH 7.4. The sample solution subjected to the UVRR measurement was replaced with a fresh one every 100 min. Sample integrity after exposure to laser irradiation was confirmed with UV–Vis absorption spectra.
Supplementary Material
Acknowledgments
This work was supported by Grant-in-Aid for Specially Promoted Research 14001004 (to T.K. and Y.M.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and Grant-in-Aid for Scientific Research (B) 17350009 to (to Y.M.) from the Japan Society for the Promotion of Science.
Abbreviations
- Hb
hemoglobin
- Mb
myoglobin
- deoxyMb
deoxy form of Mb
- MbCO
carbon monoxide-bound form of Mb
- UVRR
ultraviolet resonance Raman.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611560104/DC1.
References
- 1.Perutz MF, Fermi G, Luisi B, Shaanan B, Liddington RC. Acc Chem Res. 1987;20:309–321. doi: 10.1101/sqb.1987.052.01.063. [DOI] [PubMed] [Google Scholar]
- 2.Dickerson RE, Geiss I. Hemoglobin: Structure, Function, Evolution, and Pathology. Menlo Park, CA: Benjamin Cummings; 1983. [Google Scholar]
- 3.Jain R, Chan MK. J Biol Inorg Chem. 2003;8:1–11. doi: 10.1007/s00775-002-0405-8. [DOI] [PubMed] [Google Scholar]
- 4.Kachalova GS, Popov AN, Bartunik HD. Science. 1999;284:473–476. doi: 10.1126/science.284.5413.473. [DOI] [PubMed] [Google Scholar]
- 5.Goodno GD, Astinov V, Miller RJD. J Phys Chem A. 1999;103:10630–10643. [Google Scholar]
- 6.Miller RJD. Acc Chem Res. 1994;27:145–150. [Google Scholar]
- 7.Schotte F, Soman J, Olson JS, Wulff M, Anfinrud PA. J Struct Biol. 2004;147:235–246. doi: 10.1016/j.jsb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Ansari A, Jones CM, Henry ER, Hofrichter J, Eaton WA. Science. 1992;256:1796–1798. doi: 10.1126/science.1615323. [DOI] [PubMed] [Google Scholar]
- 9.Causgrove TP, Dyer RB. J Phys Chem. 1996;100:3273–3277. [Google Scholar]
- 10.Dartigalongue T, Hache F. Chem Phys Lett. 2005;416:313–316. doi: 10.1063/1.2041467. [DOI] [PubMed] [Google Scholar]
- 11.Lim M, Jackson TA, Anfinrud PA. Nat Struct Biol. 1997;4:209–214. doi: 10.1038/nsb0397-209. [DOI] [PubMed] [Google Scholar]
- 12.Mizutani Y, Kitagawa T. J Phys Chem B. 2001;105:10992–10999. [Google Scholar]
- 13.Xie XL, Simon JD. Biochemistry. 1991;30:3682–3692. doi: 10.1021/bi00229a013. [DOI] [PubMed] [Google Scholar]
- 14.Harada I, Takeuchi H. In: Advances in Spectroscopy: Spectroscopy of Biological Systems. Clark RJH, Hester RE, editors. New York: Wiley; 1986. pp. 113–175. [Google Scholar]
- 15.Kitagawa T. Prog Biophys Mol Biol. 1992;58:1–18. doi: 10.1016/0079-6107(92)90009-u. [DOI] [PubMed] [Google Scholar]
- 16.Jayaraman V, Rodgers KR, Mukerji I, Spiro TG. Science. 1995;269:1843–1848. doi: 10.1126/science.7569921. [DOI] [PubMed] [Google Scholar]
- 17.Kim JE, Pan D, Mathies RA. Biochemistry. 2003;42:5169–5175. doi: 10.1021/bi030026d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato A, Mizutani Y. Biochemistry. 2005;44:14709–14714. doi: 10.1021/bi051732c. [DOI] [PubMed] [Google Scholar]
- 19.Chi Z, Asher SA. J Phys Chem B. 1998;102:9595–9602. [Google Scholar]
- 20.Matsuno M, Takeuchi H. Bull Chem Soc Jpn. 1998;71:851–857. [Google Scholar]
- 21.Haruta N, Aki M, Ozaki S, Watanabe Y, Kitagawa T. Biochemistry. 2001;40:6956–6963. doi: 10.1021/bi002640k. [DOI] [PubMed] [Google Scholar]
- 22.Chu K, Vojtchovsky J, McMahon BH, Sweet RM, Berendzen J, Schlichting I. Nature. 2000;403:921–923. doi: 10.1038/35002641. [DOI] [PubMed] [Google Scholar]
- 23.Hummer G, Schotte F, Anfinrud PA. Proc Natl Acad Sci USA. 2004;101:15330–15334. doi: 10.1073/pnas.0405295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizutani Y, Kitagawa T. Chem Rec. 2001;1:258–275. doi: 10.1002/tcr.1012. [DOI] [PubMed] [Google Scholar]
- 25.Lim M, Jackson TA, Anfinrud PA. J Phys Chem. 1996;100:12043–12051. [Google Scholar]
- 26.Miura T, Takeuchi H, Harada I. Biochemistry. 1988;27:88–94. doi: 10.1021/bi00401a015. [DOI] [PubMed] [Google Scholar]
- 27.Efremov RG, Feofanov AV, Nabiev IR. J Raman Spectrosc. 1992;23:69–73. [Google Scholar]
- 28.Takeuchi H, Ohtsuka Y, Harada I. J Am Chem Soc. 1992;114:5321–5328. [Google Scholar]
- 29.Srajer V, Ren Z, Teng TY, Schmidt M, Ursby T, Bourgeois D, Pradervand C, Schildkamp W, Wulff M, Moffat K. Biochemistry. 2001;40:13802–13815. doi: 10.1021/bi010715u. [DOI] [PubMed] [Google Scholar]
- 30.Gryczynski Z, Bucci E. Biophys Chem. 1998;74:187–196. doi: 10.1016/s0301-4622(98)00181-1. [DOI] [PubMed] [Google Scholar]
- 31.Guallar V, Jarzecki AA, Friesner RA, Spiro TG. J Am Chem Soc. 2006;128:5427–5435. doi: 10.1021/ja057318h. [DOI] [PubMed] [Google Scholar]
- 32.Seno Y, Go N. J Mol Biol. 1990;216:111–126. doi: 10.1016/S0022-2836(05)80064-6. [DOI] [PubMed] [Google Scholar]
- 33.Gao Y, El-Mashtoly SF, Pal B, Hayashi T, Harada K, Kitagawa T. J Biol Chem. 2006;281:24637–24646. doi: 10.1074/jbc.M603198200. [DOI] [PubMed] [Google Scholar]
- 34.De Angelis F, Jarzecki AA, Car R, Spiro TG. J Phys Chem B. 2005;109:3065–3070. doi: 10.1021/jp0451851. [DOI] [PubMed] [Google Scholar]
- 35.Egeberg KD, Springer BA, Sligar SG, Carver TE, Rohlfs RJ, Olson JS. J Biol Chem. 1990;265:11788–11795. [PubMed] [Google Scholar]
- 36.Springer BA, Sligar SG, Olson JS, Phillips GN. Chem Rev. 1994;94:699–714. [Google Scholar]
- 37.Aranda R, Levin EJ, Schotte F, Anfinrud PA, Phillips GN., Jr Acta Crystallogr D. 2006;62:776–783. doi: 10.1107/S0907444906017318. [DOI] [PubMed] [Google Scholar]
- 38.Seno Y, Go N. J Mol Biol. 1990;216:95–109. doi: 10.1016/S0022-2836(05)80063-4. [DOI] [PubMed] [Google Scholar]
- 39.Springer BA, Egeberg KD, Sligar SG, Rohlfs RJ, Mathews AJ, Olson JS. J Biol Chem. 1989;264:3057–3060. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}