

Abstract
To mimic the sandfly pool feeding process and characterize the cellular and biochemical events that occur during the early stages of promastigote–host interaction, we developed an ex vivo model of human blood infection with Leishmania promastigotes. Within 30 s of blood contact, Leishmania promastigotes bind natural anti–Leishmania antibodies, which then activate the classical complement pathway and opsonization by the third component of complement. The opsonized promastigotes undergo an immune adherence reaction and bind quantitatively to erythrocyte CR1 receptors; opsonized Leishmania amastigotes also bind to erythrocytes. Progression of infection implies promastigote transfer from erythrocytes to acceptor blood leukocytes. After 10 min of ex vivo infection, 25% of all leukocytes contain intracellular parasites, indicating that blood cells are the early targets for the invading promastigotes. We propose that adaptation to the immune adherence mechanism aids Leishmania survival, promoting rapid promastigote phagocytosis by leukocytes. This facilitates host colonization and may represent the parasite's earliest survival strategy. In light of this mechanism, it is unlikely that infection-blocking vaccines can be developed.
Keywords: Leishmania, infection, immune adherence, natural antibodies, complement
The Leishmanioses are a group of insect-transmitted parasitic diseases caused by trypanosomatid protozoans of the genus Leishmania. These protozoans have a heteroxenous life cycle, living first as sessile intracellular amastigotes in the vertebrate host, then as motile flagellated promastigotes in the gut lumen of the sandfly vector. Phlebotomine sandflies are telmophage diptera that feed from hematomas originated by laceration of superficial venules in the host dermis, and transmit the disease by inoculating promastigotes into the hematoma blood pool (1).
To understand the Leishmania infective process, it is critical to delineate the early sequence of events from promastigote inoculation to its final entry into the macrophage (2). After inoculation, the promastigote interacts with opsonic serum factors and activates the complement system (3). The third component of complement (C3)1–coated parasite adheres to mononuclear phagocytes (MN) through CR1 and CR3 complement receptors (CRs), and phagocytosis ensues without triggering the cells' respiratory burst (4). The promastigote then differentiates to the amastigote form within the phagolysosome, where it replicates. The infection is thought to spread after extensive parasite proliferation, leading to macrophage death, amastigote release into surrounding tissue, and entry into neighboring macrophages, all of which are aided by lymphatic and hematogenous dissemination of amastigotes and infected cells (5).
In vertebrate hosts, Leishmania parasites reside in MN; therefore promastigote–host cell interaction has been analyzed extensively in vitro with MN (6–9), PMN (10), and skin Langerhans cells (11). Promastigotes of several Leishmania species can also infect dendritic cells (12, 13), canine sarcoma cells (14), and human fibroblasts (15), although the route of entrance into sarcoma cells and fibroblasts does not appear to reflect the natural course of infection. Promastigote inoculation into the skin of laboratory animals has permitted study of the inflammatory reaction to Leishmania and has shown that the invading parasite interacts with myelomonocytic phagocytes before entering the macrophage (16–19). Therefore, in vitro analysis of promastigote–MN binding does not appear to reflect the first cellular interaction that follows promastigote invasion of a susceptible host.
In this study, we have analyzed the early stages of promastigote–host interaction in an ex vivo model of human blood infection with Leishmania promastigotes. Within seconds of blood contact, we observed that promastigotes are opsonized by host natural antibodies that activate the classical complement pathway and deposit C3 on the parasite surface. The C3-coated promastigotes undergo an immune adherence (IA) reaction and bind to erythrocytes, thereby enhancing parasite phagocytosis by blood leukocytes and facilitates host colonization.
Materials and Methods
Parasites and Cultures.
Leishmania donovani Khartoum 1246 (MHOM/SD/43/124) and Leishmania amazonensis Maria (MHOM/Br/79/Maria) isolates were provided by Dr. J.O. Hill (Trudeau Institute, Saranac Lake, NY). Promastigotes were cultured at 27°C in complete medium (RPMI 1640 supplemented with 10% heat-inactivated FCS [Imperial Labs.], 2 mM l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin) until they reached the stationary phase. Parasites were harvested by centrifugation (1,500 g, 15 min, 20°C), washed twice in PBS, pH 7.2, and adjusted to the desired concentration.
Amastigote Isolation.
L. amazonensis amastigotes were isolated from the footpads of infected BALB/c mice. Lesions were removed from the mice, the tissue disrupted, and amastigotes isolated by Percoll gradient centrifugation (20).
Promastigote Labeling with [111In]-oxine.
In brief, promastigotes (1–2 × 108 cells/ml) were incubated for 10 min at 20°C in serum-free RPMI 1640 medium with 0.1–0.2 mCi [111In]-oxine/ 108 cells (340 MBq/mg In, 1 mCi/ml; Amersham Int.). Promastigotes were then sedimented (11,000 g, 1 min), the pellet washed with PBS, adjusted to 107 cells/ml in complete medium, and incubated for 2 h at 27°C to allow release of poorly bound [111In]- oxine. Promastigotes were then resedimented and the concentration adjusted. The specific activity of the labeled population ranged from 0.7 to 0.9 cpm/cell.
Blood Collection and Cell Isolation.
Blood was drawn from healthy donors into preservative-free heparin (10 IU/ml). A 50% PBS-diluted blood sample was layered onto an equal volume of 72% Percoll (d = 1.094; Pharmacia Biotech AB, Uppsala, Sweden) and centrifuged (800 g, 30 min, 20°C), and the erythrocyte pellet was washed twice by centrifugation (500 g, 5 min, 20°C) and resuspended in PBS. Leukocytes were fractionated into MN and PMN populations (21), washed, and resuspended in PBS until use.
Antibodies and Sera.
Serum was prepared from healthy humans (NHS), rabbits (NRS), and mice (NMS) and stored in liquid nitrogen. NHS was adsorbed (Ads-NHS) for 30 min on ice with L. donovani promastigotes at a ratio of 1 ml of 50% diluted NHS:109 promastigotes. After incubation, the sample was centrifuged (11,000 g, 3 min) and the supernatant used for functional assays. IgM, obtained as euglobulin after exhaustive dialysis against distilled water, was freed of potentially contaminating IgG by filtration through a HiTrap Protein G column (Pharmacia Biotech AB). IgG was purified from the dialysate on a HiTrap Protein G column. SDS-PAGE analysis confirmed that both preparations were free of major contaminants. Mouse anti– human erythrocyte antibody was obtained from mice hyperimmunized with human erythrocytes. Rabbit anti–human CR1 receptor (R137) was donated by Prof. V. Nussenzweig and co-workers (New York University Medical Center, New York) and Dr. S. Rodríguez de Córdoba (Consejo Superior de Investigaciones Científicas, Centro de Investigaciones Biólogicas, Madrid, Spain). Goat anti–human immunoglobulin μ chain (goat anti-μ) was from Southern Biotechnology Associates. The anti–human CR1 mAb 543 (22) was from the American Type Culture Collection (HB 8592) and mAbs 3D9 (23) and J3D3 (24) were provided by Prof. J. Schifferli (Kantonsspital, Basel, Switzerland). The anti–human C3 α chain mAb SIM 27-49 (IgG2b) was developed in our laboratory; both 3D9 and SIM 27-49 were purified from mouse ascites fluid by protein A chromatography. Purified human C3 was the gift of Dr. F. Vivanco (Fundación Jiménez Díaz, Madrid, Spain).
Antibody Labeling with Sodium 125I.
Goat anti-μ (50 μg) or SIM 27-49 (25 μg) was labeled with 5 μl of sodium 125I (carrier-free, 105.36 mCi/ml; Du Pont/NEN) in Iodogen (Pierce Chemical Co., Rockford, IL)-coated tubes for 5 min at 20°C. The 125I-labeled antibody was isolated on a Sephadex G-25 column (Pharmacia Biotech AB) equilibrated in PFS buffer (PBS containing 2.5% FCS and 0.05% NaN3). Labeled antibody–containing fractions were pooled and specific activity was determined in a gamma counter.
Immune Adherence Assay in Normal Human Blood.
Promastigote binding to human cells was analyzed by adding 50 μl of 111In- labeled promastigotes (107 cells/ml) to 100 μl of heparinized whole blood and incubating at 37°C for 1 min (all IA assay incubations were carried out in a 37°C waterbath). The reaction was terminated by transferring tube contents onto a 2-ml discontinuous gradient of 1 ml 63% (d = 1.077) and 1 ml 72% Percoll solutions and centrifuging (500 g, 3 min, 20°C). Three gradient fractions were collected, one each from the buffer-Percoll interface (fraction 1), from the 63–72% Percoll interface (fraction 2), and from the erythrocyte pellet (fraction 3); they were filtered through glass fiber discs (GF/C; Whatman Int.) washed three times in cold PBS, and the retained [111In] cpm were determined.
Measurement of the Immune Adherence Reaction Kinetics.
111 In-labeled promastigotes (50 μl; 107 cells/ml) were mixed with 100 μl of heparinized blood and incubated at 37°C for varying time periods (0–240 s). After incubation, the samples were layered onto 1.5-ml 72% Percoll and centrifuged (500 g, 3 min, 20°C). The Percoll solution (free parasites) and the erythrocyte pellet (erythrocyte-bound parasites) were collected separately, and processed as above. The promastigote viability is calculated for each point by adding promastigote-retained [111In] cpm in the upper and lower gradient fractions, and the 100% viability value, as all promastigote [111In] cpm at time 0 of incubation. The erythrocyte-bound promastigote profile is calculated as promastigote-retained [111In] cpm bound to erythrocytes (lower fraction only) relative to total viable parasites at each time point. The analyses were performed in duplicate.
Immune Adherence On-Rate Constant (k+1) Measurement in Human Blood.
Duplicate 50-μl samples of 111In-labeled promastigotes (1–2 × 107 cells/ml) and 100 μl of heparinized blood were incubated at 37°C for varying time periods (0–240 s). Tube contents were transferred onto 1.5 ml of 72% Percoll, centrifuged, collected, filtered, and processed as before. A plot of log B max/ B max − B ti against incubation times (t i) gives a straight line of slope m; B max is the percentage of maximum promastigote binding, and B ti the percentage of promastigotes bound at times t i. Assuming 5 × 106 erythrocytes/μl of blood, the erythrocyte concentration [E] is 5.5 × 10−12 M; k +1 was calculated as 2.303 m/[E] mol−1 s−1 (25) and is given as the mean ± SEM of three experiments.
Binding Kinetics of NHS C3 and IgM Antibodies to Leishmania Promastigotes.
50 μl of a 2 × 108/ml promastigote suspension was mixed with 50 μl of 50% untreated NHS or 50% NHS adjusted to 10 mM EGTA, 7 mM MgCl2 in PBS to block classical complement pathway activation (26), and incubated at 37°C for varying time periods. The reaction was terminated by addition of 1 ml of cold (4°C) PFS and promastigotes were washed twice by centrifugation (11,000 g, 1 min, 20°C). The cell pellet was resuspended in 0.2 ml of PFS containing 2 × 105 cpm of [125I]-SIM 27-49 (specific activity 107 cpm/μg) and incubated on ice for 1 h. Promastigotes were then washed twice by centrifugation (11,000 g, 1 min) in cold PFS and the [125I]-SIM 27-49 cpm bound was determined.
Promastigote C3b deposition dependence on both IgM and IgG natural antibodies was analyzed by incubating 50 μl of a 2 × 108/ml promastigote suspension with 50 μl of 50% NHS, 50% Ads-NHS, or 50% Ads-NHS supplemented with either 100 μg of purified IgM or IgG at 37°C for 3 min. Samples were then processed as above and promastigote-bound [125I]-SIM 27-49 cpm was determined.
To analyze the kinetics of IgM binding to promastigotes, 50-μl aliquots of 50% NHS were mixed with 50 μl of promastigotes (2 × 108 cells/ml) and incubated at 37°C for varying time periods. The reaction was terminated and promastigotes were washed three times (11,000 g, 1 min) with cold PFS. The pellet was resuspended in 0.2 ml of PFS containing 2 × 105 cpm of 125I-goat anti-μ (specific activity 107 cpm/μg), incubated 1 h on ice, washed twice as above, and promastigote-bound antibody was determined. For these analyses, duplicate samples were processed.
Equilibrium Constant (Kd) Measurement of the Immune Adherence Reaction.
50 μl of 33% NHS and 50 μl of 111In-labeled promastigotes (107 cells/ml) were mixed with 50 μl of each of nine erythrocyte suspensions at concentrations from 1% (0.76 × 107 cells) to 30% (22.8 × 107 cells). Tubes were incubated at 37°C for 3 min. After incubation, tube contents were layered onto 1.5 ml of 72% Percoll and centrifuged (500 g, 3 min, 20°C). The Percoll solution and the erythrocyte pellet were collected separately, filtered, and erythrocyte-bound 111In-labeled promastigote cpm were determined. K d and V max values were obtained by direct linear plot (27), representing the number of erythrocyte-bound promastigotes at each erythrocyte concentration [Ei] tested (y-axis), against erythrocyte concentration; in this case, each [Ei] point is assigned its negative value on the x-axis.
Characterization of the Immune Adherence Reaction.
To simplify analysis of serum components and the nature of the erythrocyte IA receptor, blood was replaced by NHS and washed erythrocytes. 50 μl each of 111In-labeled promastigotes (107 cells/ml), 50% or 33% NHS, and a 10% (vol/vol) washed erythrocyte suspension were incubated at 37°C for 3 min, gradient centrifuged, collected, filtered, and processed as above. The analyses were performed in duplicate.
To analyze IA dependence on divalent ions, 50-μl samples of 33% NHS were pretreated by incubating for 10 min at 20°C with varying concentrations (0–10 mM) of disodium EDTA. To test the thermostability of serum factors involved in the reaction, serum was incubated at 56°C for 45 min. To investigate IA reaction dependence on functional C3, NHS C3 and C4 thiolester bonds were blocked with methylamine hydrochloride (MA) (28), and then serum was dialyzed against PBS for 1 h at 4°C. Control or MA-treated samples were supplemented with purified C3 at a final concentration of 0.25, 0.5, or 1.5 mg/ml, then assayed for IA. IA reaction dependence on natural anti–Leishmania antibody was analyzed by replacing 33% NHS with Ads-NHS or Ads-NHS supplemented with IgM or IgG at a final concentration of 0.66 mg/ml.
To disrupt CR1 structure and function, erythrocytes were treated with 2-aminoethyl isothiouronium (AET) (29). Erythrocytes were trypsin-digested as described (30); after incubation, cells were washed twice in PBS, adjusted to 10%, and used in the IA assay.
To block the erythrocyte IA receptor, aliquots of a 10% erythrocyte suspension were incubated for 1 h on ice with pretitrated dilutions of R137, mAbs 3D9, 543, J3D3, or mouse anti–human erythrocyte antiserum. Heat-inactivated NRS, NHS, NMS, and normal mouse IgG controls were used at the same dilutions as the corresponding antibodies. After incubation, antibody-treated erythrocytes were washed twice in PBS, adjusted to 10%, and used in the IA assay.
Erythrocyte–promastigote IA rosettes were prepared by incubating 50 μl of promastigotes (107 cells/ml) with 100 μl of human blood at 37°C for 30 s (method A). A 5-μl aliquot was gently dispersed into 1 ml of PBS and the IA rosettes examined under a bright-field inverted microscope (×20 phase-contrast objective). Alternatively, 50 μl each of promastigotes, 33% NHS, and a 10% washed erythrocyte suspension were incubated at 37°C for 1 min (method B). After incubation, parasites were stained with 1% acridine orange (AO) diluted 1:50,000 (31). A 10-μl aliquot was gently dispersed into 200 μl of PBS, and the IA rosettes were examined in an UV-fluorescence microscope and photographed.
Amastigote IA Assay.
IA rosettes formed by human erythrocytes and opsonized amastigotes were prepared in duplicate with 50 μl each of a 10% washed erythrocyte suspension, amastigotes (2 × 107 cells/ml), and 33% NHS, with or without 10 mM disodium EDTA. In control samples, PBS replaced NHS. Samples were incubated at 37°C for 3 min; aliquots were then AO-stained as before and amastigotes associated with three or more erythrocytes were counted as rosettes.
Leukocyte-dependent IA Rosette Dissociation.
Blood samples were divided into two aliquots; one was used as a control and the other was centrifuged through 72% Percoll to isolate erythrocytes. Erythrocytes were washed twice in PBS and resuspended in autologous plasma to their initial concentration in blood. Duplicate aliquots of 50 μl of the platelet- and leukocyte-depleted sample or the untreated control were mixed with 50 μl of 111In-labeled L. donovani promastigotes (107 cells/ml) and 50 μl of PBS, and incubated at 37°C for 0 to 30 min. At the end of each incubation period, mixtures were centrifuged through 1.5 ml of 72% Percoll (500 g, 3 min). Fractions containing the erythrocyte pellet and the buffer-Percoll interface were collected separately, filtered, and processed as before.
Promastigote Acceptor Capacity of Blood Leukocyte Populations.
Leukocyte acceptor capacity was analyzed in a two-step assay. First, IA rosettes were preformed by method B; then 0.2 ml (5 × 107 cells/ml) of each of the leukocyte populations under study (unfractionated leukocytes, PMN, and MN) and a PBS control were added to four series of tubes containing preformed IA rosettes at a 20:1 leukocyte/promastigote ratio. Duplicate samples were incubated at 37°C for 0 to 30 min; after incubation, tube contents were centrifuged through 1.5 ml of 72% Percoll (800 g, 3 min). Cells at the buffer-Percoll interface and the erythrocyte pellet were collected separately, filtered, and processed as before.
Ex Vivo Infection Assay of Human Blood with Leishmania Promastigotes.
1 vol of fresh heparinized blood was mixed with 1 vol of a L. donovani promastigote suspension (107 cells/ml) and incubated at 37°C for varying time periods. Infection was terminated by adding 1 vol of 0.4% paraformaldehyde in PBS, and tube contents were then loaded onto 2 vol of 72% Percoll. After centrifugation (850 g, 10 min), the leukocyte population at the plasma-Percoll interface was collected, diluted with 20 vol of PBS, and sedimented (500 g for 10 min). To reduce the number of contaminating free promastigotes, the leukocyte pellet was resuspended in RPMI 1640 (5 ml medium/ml blood) and recentrifuged (80 g, 5 min). Cell monolayers were prepared in a Cytospin 2 centrifuge (Shandon Southern Products) or as manually prepared cell smears. The percentage of leukocytes with intracellular parasites was determined by microscopic examination of cell monolayers stained with modified Giemsa reagent and photographed.
Results
Interaction of Leishmania spp Promastigotes with Normal Human Blood.
To study promastigote fate during the period immediately after their inoculation into humans and to determine the host cell populations parasitized in hematoma, we developed an ex vivo model of Leishmania spp infection. Data shown are for L. donovani and, when indicated, for L. amazonensis promastigotes, although similar results were obtained with Leishmania infantum and Leishmania major (data not shown); they may thus be considered applicable to Leishmania spp.
To mimic natural infection conditions, fresh human blood from different donors was infected with 111In-labeled L. donovani promastigotes and the blood–parasite mixture centrifuged through a discontinuous Percoll density gradient to separate cell populations and determine 111In-labeled promastigote distribution (Fig. 1). 1 min after promastigote addition, nearly 100% of the parasites cosedimented with the erythrocyte pellet. In contrast, promastigotes added to EDTA-treated blood or mixed with PBS alone were located at the Percoll-buffer interface after centrifugation. Promastigote cosedimentation with erythrocytes was specific, unrelated to the erythrocyte blood group, and dependent on the presence of serum factors and divalent ions in the reaction mixture. These data show that the parasites bind by IA to erythrocytes immediately after contact with host blood.
Figure 1.

Promastigote binding to human blood cell populations. 111In-labeled L. donovani promastigotes (5 × 105 cells) were incubated at 37°C for 1 min with 100 μl of (A) heparinized blood; (B) blood supplemented with 10 mM EDTA; and (C) PBS. After incubation, the samples were centrifuged (500 g, 3 min, 20°C) through a discontinuous Percoll gradient. Three fractions (1–3), one each from the buffer-Percoll interface (1); the 63–72% Percoll interface (2); and from the erythrocyte pellet (3) were collected, processed as described in Materials and Methods, and the percentage of [111In] cpm in each fraction determined. Data are the mean values of duplicate samples from a representative experiment of seven performed.
Kinetic and Promastigote Viability Profiles of the IA Reaction.
The kinetics are shown of erythrocyte–promastigote (E-P) interaction and the promastigote viability profile over a 4-min ex vivo infection period (Fig. 2). Within the first 30 s, the IA reaction has reached completion, and the promastigote inoculum binds completely to erythrocytes. To calculate the on-rate constant (k +1) of this reaction, it is assumed that the E-P interaction occurs between cell particles (1,000:1 to 500:1 ratio) rather than between erythrocyte CR1 receptors and promastigote-bound C3b ligands. In the incubation mixture [E] >> [P], and the interaction is treated as a pseudo-first order process, giving a k +1 value of 3 × 1010 mol−1 s−1. Provided that promastigote-associated [111In] cpm parallels parasite viability, >90% of all viable promastigotes remain erythrocyte-bound during the incubation period. Promastigote viability was high during the first 60 s of incubation; between 60 and 120 s, a sharp decrease in the initial erythrocyte-bound [111In] cpm was observed due to complement lysis. A similar promastigote lysis course is obtained by incubating 111In-labeled promastigotes in the presence of 25% NHS; under these conditions, the 111In-labeled promastigote half-life measured was ∼90 s, by which time promastigote flagellar mobility was already impaired (data not shown).
Figure 2.
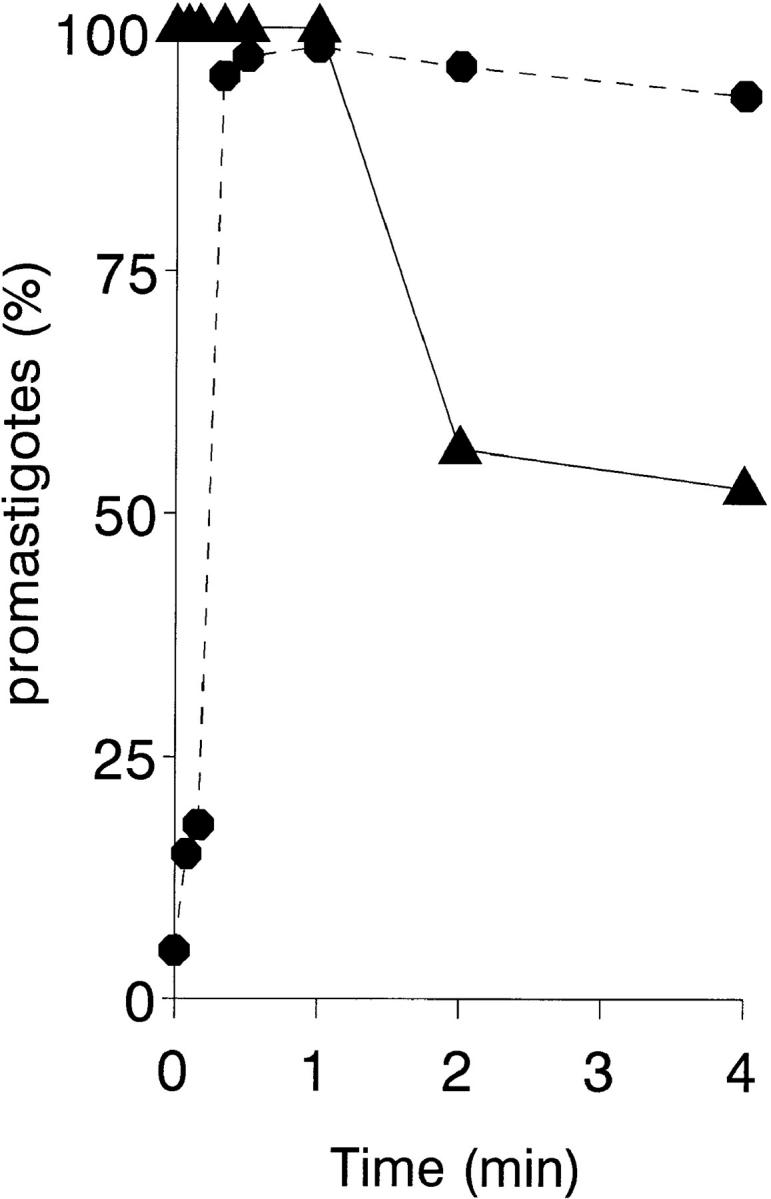
Kinetic and promastigote viability course of the IA reaction. 111In-labeled L. donovani promastigotes (5 × 105 cells) were mixed with 100 μl of heparinized blood and incubated at 37°C for 0, 10, 20, 30, 60, 120, and 240 s. After incubation, the samples were centrifuged (500 g, 3 min, 20°C) through 72% Percoll. The Percoll solution (free parasites) and the erythrocyte pellet (erythrocyte-bound parasites) were collected separately, filtered, and the percentage of [111In] cpm retained was determined. Promastigote viability (▴) and erythrocyte-bound promastigote (•) profiles were calculated as described in Materials and Methods. Data are the mean value of duplicate samples from a representative experiment of three similar experiments performed.
Promastigote Opsonization in the Presence of Nonimmune Serum.
Complement activation by Leishmania promastigotes is believed to occur mainly through the alternative pathway (3). The rapid kinetics observed for the IA reaction suggested that this is not the case, for which the activation pathway of the complement cascade by L. amazonensis and L. donovani promastigotes was studied. Classical pathway activation requires both Ca2+ and Mg2+, whereas the alternative pathway requires only Mg2+; differential cation chelation thus permits identification of the activation pathway. In NHS adjusted to 10 mM EGTA and 7 mM MgCl2, Ca2+ is effectively chelated but free Mg2+ is available for alternative pathway activation. The course of C3 deposition on the promastigote surface is shown in the presence of 25% NHS or Mg-EGTA–treated 25% NHS (Fig. 3 A). After 2–3 min of incubation, C3 binding reaches a maximum and remains unchanged for the remainder of the time course. In contrast, C3 deposition in Mg-EGTA–treated serum follows a slower course that varies depending on the Leishmania species studied.
Figure 3.


Promastigote opsonization kinetics in NHS. (A) Promastigote complement activation pathway in NHS (closed symbols) and Mg-EGTA–treated NHS (open symbols). Duplicate aliquots (107 cells) of L. donovani (•, ○) and L. amazonensis (▴, ▵) promastigotes were incubated at 37°C in 25% NHS or 10 mM EGTA/7 mM Mg2Cl-treated 25% NHS, for 0, 0.25, 0.5, 1, 2, 3, 4, 5, 7.5, 10, and 15 min. Promastigotes were then washed twice by centrifugation (11,000 g, 1 min) in cold PFS and incubated 1 h on ice with mAb [125I]SIM 27-49. After incubation, promastigotes were washed twice (11,000 g, 1 min) and [125I]SIM 27-49 cpm determined. C3 deposition is expressed as a percentage of the point of maximum [125I]SIM 27-49 binding. Data are the mean value of duplicate samples taken from one representative experiment of three performed. (B) IgM binding kinetics to L. donovani and L. amazonensis promastigotes. Duplicate aliquots (107 cells) of L. donovani (•) and L. amazonensis (▴) promastigotes were incubated at 37°C in 25% NHS for 0, 0.25, 0.5, 1, 2, 3, 4, 5, 7, and 10 min. After reaction, promastigotes were washed three times (11,000 g, 1 min) with cold PFS and incubated for 1 h on ice with 125I-goat anti-μ antibody. Results are expressed as the percentage of IgM binding relative to the point of maximum binding. Each point value represents the mean of duplicate samples taken from a representative experiment of five performed.
The finding that Leishmania promastigotes activate complement by the classical pathway prompted a search for anti-Leishmania antibodies in NHS. In 25% NHS, promastigotes are rapidly coated with IgG and IgM antibodies. The kinetic profile of IgM binding to L. amazonensis and L. donovani is shown (Fig. 3 B). IgM binding to both species follows the same pattern, presenting a very rapid forward reaction. After 30–60 s of incubation, IgM binding reaches a maximum, followed by a rapid, marked release of the bound antibody, amounting to ∼50% of that initially fixed. IgG binding to the parasite follows a similar course (data not shown). Promastigote C3 deposition dependence on IgM or IgG binding was analyzed in the presence of NHS, Ads-NHS with L. donovani promastigotes, and Ads-NHS supplemented with IgM or IgG purified from the same serum pool. After one adsorption cycle, Ads-NHS C3 deposition capacity was reduced to 15% of that of NHS. Ads-NHS supplemented with purified IgM (100 μg) recovered 79% of its initial activity. In contrast, IgG addition (100 μg) only showed 24% activity recovery. Therefore, IgM binding to promastigotes is required for promastigote opsonization.
Characterization of the IA Reaction.
As the IA process is strictly dependent on serum factor activity and the presence of divalent ions in the incubation mixture, the role of these factors and the nature of the erythrocyte IA receptor were then studied. Serum and erythrocytes were subjected to several treatments, and their capacity to sustain the IA reaction tested (Table I). IA-promoting activity was lost from serum after heat inactivation, in the presence of ≥1 mM EDTA, or after blockage of C3/C4 thiolester bonds by MA treatment. Purified C3 at physiological concentrations cannot alone restore the serum activity; however, when purified C3 was added to the mixture containing MA-treated serum, the IA reaction was restored to 42% of positive control levels. Under these conditions, the partial IA recovery must be due largely to alternative pathway C3b deposition. In addition, Ads-NHS supplemented with IgM, but not with IgG, recovers most of the IA capacity lost through adsorption, indicating that IgM binding to promastigotes is a prerequisite for the IA reaction.
Table I.
Characterization of the IA Reaction
| Concentration* | IA reaction (%)‡ | |||
|---|---|---|---|---|
| Serum treatment | ||||
| Nil | 93.2 ± 3.0 | |||
| Heat 56°C, 45 min | 33% | 6.4 ± 0.3 | ||
| EDTA | 10 mM | 9.7 ± 1.6 | ||
| MA | 100 mM | 9.0 ± 1.1 | ||
| PBS + C3 | 1.5 mg/ml | 12.8 ± 1.0 | ||
| MA + C3 | 100 mM + 1.5 mg/ml | 41.9 ± 1.0 | ||
| Ads-NHS | 16% | 8.4 ± 1.0 | ||
| Ads-NHS + IgG | 16% + 0.66 mg/ml | 16.2 ± 1.3 | ||
| Ads-NHS + IgM | 16% + 0.66 mg/ml | 87.3 ± 1.0 | ||
| Erythrocyte treatment | ||||
| Nil | 88.0 ± 2.0 | |||
| Trypsin | 153 IU/ml | 6.0 ± 0.4 | ||
| AET | 6.4% wt/vol | 10.0 ± 2.0 | ||
| R137 | 1/10 | 8.4 ± 1.0 | ||
| NRS | 1/10 | 89.0 ± 3.0 | ||
| Mouse anti-HRBC | 1/2,000 | 89.0 ± 7.0 | ||
| NMS | 1/2,000 | 89.0 ± 1.8 | ||
| mAb 3D9 | 10 μg/ml | 6.4 ± 0.8 | ||
| mAb 543 | 10 μg/ml | 92.0 ± 0.6 | ||
| mAb J3D3 | 1/20 | 90.0 ± 0.3 | ||
| NMIgG§ | 10 μg/ml | 92.0 ± 0.7 |
Final concentration in the assay.
IA reaction (%) was calculated as [111In] cpm in the erythrocyte pellet/ total [111In] cpm × 100.
Normal mouse IgG.
The involvement of C3 suggested that erythrocyte CR1 participates in the IA reaction. To test this, erythrocytes were treated with AET (29) or trypsin (30), which destroy or inactivate CR1. In previous studies we confirmed (data not shown), that erythrocyte CR1 recognition by rabbit anti-CR1–specific antiserum (R137) decays in parallel to erythrocyte incubation time with AET, and that AET-treated erythrocytes no longer bind opsonized promastigotes; again, after AET or trypsin treatment, no IA reaction was observed (Table I). Anti-CR1–specific polyclonal antibody R137 and mAb 3D9 blocked the E-P interaction in a dose-dependent manner. In contrast, the reaction was not inhibited either by the mAb 543 or the mAb J3D3, which recognize CR1 epitopes not directly related to the C3b binding site (22, 24, 32), or by mouse anti–human erythrocyte serum. As a control, preincubation of erythrocytes with NRS, NMS, or normal mouse IgG had no effect on E-P interaction.
The dissociation constant (K d) for the E-P interaction was obtained from Langmuir saturation isoterm data (Fig. 4 A) plotted by the direct linear method (Fig. 4 A, inset); the K d value was either 2 × 107 (the erythrocyte number binding 50% of the promastigote inoculum) or, in terms of erythrocyte molarity, 2 × 10−13 M.
Figure 4.

Analysis of promastigote–erythrocyte interaction. (A) Dissociation (K d) constant measurement. 50 μl each of 33% NHS and 111In-labeled promastigotes (107 cells/ml) were mixed with 50 μl each of nine erythrocyte suspensions at concentrations ranging from 1 to 30% and incubated at 37°C for 3 min. After incubation, erythrocyte-bound promastigotes (•) were isolated by centrifugation (500 g, 3 min, 20°C) through 72% Percoll, and [111In] cpm determined. K d and V max values were obtained by direct linear plot as described in Materials and Methods. The inset shows the first quadrant of the plot, where the intersection points that allow K d and V max calculation are clustered. Data are shown from one of two similar experiments. (B) Microscopic analysis of the IA rosettes formed in normal human blood. Promastigote rosettes were formed by method A (Materials and Methods). A 5-μl aliquot was dispersed into 1 ml of PBS and IA rosettes examined under a bright-field inverted microscope (×280). (C) Detail of an AO-stained promastigote rosette. Rosettes were formed by method B (Materials and Methods), and stained with 1% AO diluted 1:50,000. A 10-μl aliquot was gently dispersed into 200 μl of PBS and the IA rosettes examined in an UV-fluorescence microscope (×1,350).
Within 30 s of parasite addition to blood, promastigotes and erythrocytes formed visible IA rosettes consisting of aggregates of erythrocytes centered on a promastigote (Fig. 4 B). IA rosettes were also formed by method B, followed by AO staining to permit visualization of the parasite surrounded by erythrocytes in the complexes (Fig. 4 C).
Amastigote IA.
To determine whether amastigotes also undergo IA, they were isolated from L. amazonensis–infected BALB/c mice and incubated with washed erythrocytes and NHS as described in method B. NHS was replaced by either PBS or EDTA-treated NHS in control samples. After incubation, amastigotes were stained with AO and those with three or more bound erythrocytes were counted as IA rosettes (Fig. 5 A). More than 90% of amastigotes were attached to erythrocytes after 3 min of incubation (Fig. 5 B), whereas no reaction was observed in controls.
Figure 5.


Amastigote IA. (A) AO-stained amastigote rosettes with human erythrocytes. Rosettes were formed as described in Materials and Methods. After incubation, samples were stained and photographed (×790). (B) Amastigote IA reaction. 50-μl aliquots each of a 10% washed erythrocyte suspension, L. amazonensis amastigotes (2 × 107 cells/ml), and 33% NHS with or without 10 mM EDTA, or PBS as control, were incubated at 37°C for 3 min. After incubation, samples were AO-stained and amastigotes associated with two or more erythrocytes counted as IA rosettes. A, amastigotes; E, erythrocytes. Data are expressed as the mean values of duplicate samples. Similar results were obtained in two individual experiments.
Leukocyte-dependent IA Rosette Dissociation.
Progress of infection must imply transfer of promastigotes from IA rosettes to leukocyte acceptor cells. Promastigote transfer was studied with a cellular immune complex uptake assay (33) used to test whether leukocytes affect IA rosette dissociation during an ex vivo infection. 111In-labeled promastigote IA rosettes formed in normal and leukocyte-depleted blood were incubated for varying periods of time and the parasite fraction cosedimenting with erythrocytes was determined. After 15 min of infection, ∼6% of the original IA rosettes had dissociated in the leukocyte-depleted sample, compared with 36% in the untreated sample (Fig. 6 A). Thus, the promastigote dissociation rate in the presence of leukocytes is sixfold more rapid than in their absence. The promastigote acceptor capacity of unfractionated blood leukocytes, PMN, and MN was then tested by incubating 111In-labeled promastigote IA rosettes with the isolated leukocyte populations for varying periods of time. After each incubation period, leukocytes and erythrocyte-bound promastigotes were separated by Percoll centrifugation, and the [111In] cpm in each cell fraction determined. The kinetics and percentages of promastigotes released from the IA rosettes are shown (Fig. 6 B). In the presence of PMN, which have the highest acceptor capacity, IA rosette dissociation is very rapid, showing 79 and 93% promastigote release after 4 and 10 min of incubation, respectively, whereas IA rosette promastigote dissociation in the absence of leukocytes did not exceed 10%. After 4 min of incubation, isolated PMN showed 1.5-fold higher acceptor capacity on a per cell basis than did unfractionated leukocytes, and approximately twice that of MN.
Figure 6.


Promastigote transfer from IA rosettes to leukocyte acceptor cells. (A) Leukocyte-dependent IA rosette dissociation. For each time point, duplicate aliquots (50 μl) of normal and platelet- and leukocyte-depleted blood samples were incubated with 50 μl of 111In-labeled L. donovani promastigotes (5 × 105 cells) and 50 μl of PBS, then incubated at 37°C for 1, 2, 5, 7.5, 15, and 30 min. After reaction, tube contents were loaded onto 72% Percoll and centrifuged (500 g, 5 min). Fractions containing the buffer-Percoll interface and the erythrocyte pellet were collected separately, filtered, and [111In] cpm determined. Percentage of IA rosettes dissociated in normal blood (•) and in the platelet- and leukocyte-depleted sample (▴). Background IA rosette dissociation after 1 min of incubation equals 4.5% of total [111In] cpm released. Data are expressed as the mean ± SEM of three experiments. (B) Promastigote acceptor capacity of blood cell populations. 111In-labeled promastigote transfer was analyzed in a two-step assay. Duplicate samples of IA rosettes, prepared by method B, were incubated at a 20:1 leukocyte/promastigote ratio with unfractionated leukocytes, PMN, MN, and PBS at 37°C for 0, 4, 10, and 30 min. After incubation, tube contents were loaded onto 72% Percoll and centrifuged (800 g, 3 min). Cells at the buffer-Percoll interface and the erythrocyte pellet were collected separately, filtered, and [111In] cpm determined. Acceptor cell populations: PMN (▪), unfractionated leukocytes (•), MN (▴), and PBS control (✚). Data are the mean value of duplicate samples from one representative experiment of four performed.
After incubation and centrifugation, the parasites band to the top of the Percoll solution, where they form large, low-density IA complexes with leukocytes. If IA rosette dissociation is physiological, leukocytes should be engaged in active parasite phagocytosis. To test this prediction, human blood was infected ex vivo with promastigotes at a 1.5:1 parasite/ leukocyte ratio. The infection was terminated at different time intervals, the leukocytes isolated, and cell smears were Giemsa stained. After 5 min of incubation, numerous infected leukocytes could be observed, and after 10 min, ∼25% of the cells had intracellular parasites. At 60 min after infection nearly all blood phagocytes were parasitized, and their phagocytic efficiency was in the order PMN > MN > eosinophils. Few apparently intact leishmanias were observed in PMN and eosinophils, suggesting that these cells are engaged in parasite lysis. In contrast, leishmanias within monocytes showed a more clearly delimited morphology. Infected lymphoid cells were not found.
Discussion
In natural leishmaniosis transmission, promastigotes undergo a brief but critical encounter with host blood during the sandfly blood meal. As macrophages are considered direct targets of the invading parasites, it was of interest to ascertain whether promastigotes interact with other host cell lineages before taking final residence in the macrophage. Therefore, we developed an ex vivo model of blood infection and found that upon contact with human blood, promastigotes are immediately coated by natural IgM and IgG anti–Leishmania antibodies. Promastigote–IgM association kinetics is very rapid, and parasite epitopes are saturated within 1 min of incubation (Fig. 3 B). We have determined that anti–Leishmania antibodies comprise 15–30% of total NHS IgM (∼1.6 × 10−7 M) (manuscript in preparation). As antibody activity depends on both its avidity and its local concentration (34, 35), the velocity of the opsonizing reaction is not unexpected.
Fresh sera from most vertebrates contain natural antibodies that react with Leishmania promastigotes (36, 37); in nonimmune human sera, IgM and IgG antibodies have been reported that bind and agglutinate Leishmania promastigotes (38, 39), as well as IgG that recognize Leishmania Gal α 1→ 3 Gal epitopes (40). In spite of data indicating that IgM anti–Leishmania antibodies activate the classical complement pathway and facilitate parasite phagocytosis (41), there appears to be a consensus that, with the reported exceptions of L. donovani (38, 39) and metacyclic peanut lectin nonagglutinable L. major forms (42), promastigotes of the genus Leishmania activate complement by the alternative pathway (43, 44). The results reported here show that in humans natural IgM binding to Leishmania is the mechanism that triggers activation of the classical complement pathway. In 25% NHS, C3 binding to L. donovani and L. amazonensis promastigotes reaches its maximum level after 2–3 min of incubation. Only when the classical pathway is blocked with Mg-EGTA does C3 binding follow alternative pathway kinetics (Fig. 3 A). These results can safely be extended to Leishmania spp, as similar complement activation patterns are also observed with L. major and L. infantum promastigotes and with Crithidia choanomastigotes (data not shown). Furthermore, the role of antibody in promastigote opsonization contradicts the view that Leishmania invade host cells by nonopsonic phagocytosis (45).
After incubation with human blood, the opsonized promastigotes bind to erythrocyte CR1, forming IA rosettes (Fig. 4, B and C). The erythrocyte–promastigote–C3b interaction is extremely rapid (k +1 = 3 × 1010 mol−1 s−1) and is complete within 30 s of initiation. The close parallel between Leishmania IA and the binding of opsonized immune complexes to erythrocytes (46, 47) indicate that these reactions share the same mechanism. For Leishmania, IgM binding is a prerequisite for opsonization, and promastigote-bound C3b is the only parasite ligand involved in erythrocyte IA (Table I). Besides promastigotes, opsonized Leishmania amastigotes also bind to human erythrocytes (Fig. 5, A and B), suggesting that IA may contribute to amastigote dissemination in the infected host. In addition to Leishmania, erythrocyte CR1 participates in Plasmodium cytoadherence to primate erythrocytes (48), and probably also plays a part in red cell adhesion of serum-opsonized trypanosomes (49). We suggest that erythrocyte CR1 receptor–mediated IA may thus have a paramount role in parasite invasion of primates.
After the IA reaction, continuity of infection requires that erythrocyte-bound promastigotes be shuttled to acceptor leukocytes through a cooperative mechanism involving Leishmania-bound C3b and iC3b fragments and cell complement CR1 and CR3 receptors (50, 51). Although it has been proposed that promastigote Zn-proteinase (52) and leukocyte CR1 receptors (53) participate in promastigote-surface iC3b ligand generation, the erythrocyte CR1 receptor is the most logical physiological cofactor candidate for the enzymatic degradation of promastigote-bound C3b to iC3b by factor I. In addition to processing antigen-bound C3b to iC3b and C3dg fragments, erythrocytes efficiently enhance antigen-C3dg uptake by CR2 B lymphocyte receptors (54); this mechanism may well be exploited by Leishmania parasites to divert the host protective immune response.
In the ex vivo assay, at 10 min after infection, ∼25% of blood phagocytes were parasitized in the same proportions at which they are found in human blood (PMN > monocytes > eosinophils), implying that there is no preferential leukocyte target in early Leishmania infection. However, on a per cell basis isolated PMN do engulf approximately twice as many promastigotes as MN. Similar phagocytic capacity has been observed, although after much longer infection periods, in isolated human blood leukocytes infected in vitro with L. donovani amastigotes (55), and in tissue samples analyzed shortly after promastigote inoculation into hamsters (17) and mice (18, 19). These ex vivo and in vivo experimental data, although seemingly identical, derive from different mechanisms. Based on the leukocyte composition of murine blood (56), the expected percentage of Leishmania-containing PMN after ex vivo infection should be ∼16%, rather than the 90% observed in the in vivo experiments. The ex vivo infection model therefore provides information on the type and proportion of leukocytes infected in situ, while in vivo data reflect the cellular infiltrate in the parasite-triggered inflammatory reaction. Nonprimate in vivo systems would thus appear to be unsuitable for recreating the very different early Leishmania infection conditions in primates.
IA plays a central role in the clearance of opsonized microorganisms and foreign particles from the circulation via their removal by phagocytes (57). Accordingly, we propose that Leishmania adaptation to the host IA mechanism aids parasite survival by promoting rapid promastigote phagocytosis and leukocyte colonization. During Leishmania opsonization in human blood, promastigotes undergo three continuous reactions: natural antibody binding, followed by C3 deposition, followed by subsequent adherence to erythrocytes. Each reaction proceeds at a different velocity (Fig. 7). Promastigote–IgM binding and erythrocyte–promastigote IA are very rapid, whereas promastigote C3 deposition by the classical pathway is a slower process. It is believed that IA proceeds faster than C3b deposition because erythrocyte CR1 receptors cluster on the cell surface and establish multipoint interactions with C3b ligands (58). This cooperative effect increases in such extent the overall CR1–C3b interaction avidity, that only very few C3b molecules (between 60 and 3,000 molecules/cell) are required to produce a positive IA reaction (59). A similar phenomenon appears to occur in Leishmania IA. Within 30 s of ex vivo infection, >80% of Leishmania epitopes recognized by IgM are occupied, the IA reaction is completed, and promastigotes are being shuttled to blood phagocytes. Complement lysis of parasites is still negligible at this time, as only ∼5% (10,000 molecules) of the total C3 promastigote acceptor capacity has been deposited. Since in the presence of 25% NHS, 50% [111In] cpm release from labeled promastigotes is registered by 90 s, the IA mechanism could substantially aid Leishmania survival through promotion of promastigote phagocytosis by leukocytes, which provide a safe haven for the parasite.
Figure 7.

Kinetic profile of Leishmania IA reaction. The time course of the opsonizing reactions involved in the Leishmania IA mechanism. IgM binding (▪), C3 deposition (▴), and the promastigote–erythrocyte interaction (•) are taken from Fig. 3, B and A, and Fig. 2, respectively. Data are expressed as a percentage of the point of maximum reaction.
The results of this study modify the current view of human Leishmania infection by emphasizing the fundamental role of natural antibodies in triggering parasite opsonization and confirming that, under physiological conditions, promastigotes activate complement only through the classical pathway. In addition, these data show that, under physiological conditions, Leishmania uses the IA mechanism to infect the host, and suggest that this adaptation may be an extremely early and very important parasite strategy for host invasion.
For some, immunoprophylaxis is the only hope for leishmaniosis control (60). The participation of natural antibodies in the mechanism used by Leishmania to infect the host renders improbable the development of infection-blocking vaccines. Emphasis should be placed on therapies aimed at protecting the host against high parasite load, which should limit disease transmission.
Acknowledgments
The authors thank Manuela Díez and Inmaculada Moreno for excellent technical assistance; Prof. Victor Nussenzweig and Dr. Santiago Rodríguez de Córdoba for providing the R137 anti–human CR1 antiserum; Prof. Jurg Schifferli for mAbs 3D9 and J3D3; Dr. Fernando Vivanco for purified human C3; Dr. Joseph O. Hill for Leishmania strains; and Catherine Mark for editorial assistance and sound advice on the manuscript.
This work was supported by grant FIS 94/215 from the Fondo de Investigaciones Sanitarias (Spain).
Footnotes
Abbreviations used in this paper: Ads-NHS, adsorbed NHS; AET, 2-aminoethyl isothiouronium; AO, acridine orange; C3, the third component of complement; C3dg, degradation fragment of C3; CR, complement receptor; goat anti-μ, goat anti–human immunoglobulin μ chain; IA, immune adherence; iC3b, inactivated C3b; MA, methylamine hydrochloride; MN, mononuclear phagocyte; NHS, normal human serum; NMS, normal mouse serum; NRS, normal rabbit serum; PFS, PBS containing 2.5% FCS and 0.05% NaN3.
References
- 1.Ribeiro JMC. Blood-feeding arthropods: live syringes or invertebrate pharmacologists? . Infect Agents Dis. 1995;4:143–152. [PubMed] [Google Scholar]
- 2.Mosser DM, Rosenthal LA. Leishmania–macrophage interactions: multiple receptors, multiple ligands and diverse cellular responses. Semin Cell Biol. 1993;4:315–322. doi: 10.1006/scel.1993.1038. [DOI] [PubMed] [Google Scholar]
- 3.Brittingham A, Mosser DM. Exploitation of the complement system by Leishmaniapromastigotes. Parasitol Today. 1996;12:444–447. doi: 10.1016/0169-4758(96)10067-3. [DOI] [PubMed] [Google Scholar]
- 4.Wright SD, Silverstein SC. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J Exp Med. 1983;158:2016–2023. doi: 10.1084/jem.158.6.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill JO. Pathophysiology of experimental leishmaniasis: pattern of development of metastatic disease in the susceptible host. Infect Immun. 1986;52:364–369. doi: 10.1128/iai.52.2.364-369.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang K-P. Leishmania donovani: promastigote–macrophage surface interactions in vitro. Exp Parasitol. 1979;48:175–189. doi: 10.1016/0014-4894(79)90097-3. [DOI] [PubMed] [Google Scholar]
- 7.Pearson RD, Romito R, Symes PH, Harcus JL. Interaction of Leishmania donovanipromastigotes with human monocyte–derived macrophages: parasite entry, intracellular survival, and multiplication. Infect Immun. 1981;32:1249–1253. doi: 10.1128/iai.32.3.1249-1253.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klempner MS, Cendron M, Wyler DJ. Attachment of plasma membrane vesicles of human macrophages to Leishmania tropicapromastigotes. J Infect Dis. 1983;148:377–384. doi: 10.1093/infdis/148.3.377. [DOI] [PubMed] [Google Scholar]
- 9.Locksley RM, Heinzel FP, Frankhauser JE, Nelson CS, Sadick MD. Cutaneous host defense in leishmaniasis: interaction of isolated dermal macrophages and epidermal Langerhans cells with the insect-stage promastigote. Infect Immun. 1988;56:336–342. doi: 10.1128/iai.56.2.336-342.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearson RD, Steigbigel RT. Phagocytosis and killing of the protozoan Leishmania donovaniby human polymorphonuclear leukocytes. J Immunol. 1981;127:1438–1443. [PubMed] [Google Scholar]
- 11.Axelrod O, Klaus S, Frankenburg S. Antigen presentation by epidermal Langerhans cells in experimental cutaneous leishmaniasis. Parasite Immunol. 1994;16:593–598. doi: 10.1111/j.1365-3024.1994.tb00315.x. [DOI] [PubMed] [Google Scholar]
- 12.Ridley MJ, Wells CW. Macrophage–parasite interaction in the lesions of cutaneous leishmaniasis. An ultrastructural study. Am J Pathol. 1986;123:79–85. [PMC free article] [PubMed] [Google Scholar]
- 13.Williams RO. Invasion of murine dendritic cells by Leishmania major and . Leishmania mexicana J Parasitol. 1988;74:186–187. [PubMed] [Google Scholar]
- 14.Lewis DH. Infection of tissue culture cells of low phagocytic ability by . Leishmania mexicana mexicana Ann Trop Med Parasitol. 1974;68:327–336. doi: 10.1080/00034983.1974.11686955. [DOI] [PubMed] [Google Scholar]
- 15.Schwartzman JD, Pearson RD. The interaction of Leishmania donovanipromastigotes and human fibroblasts in vitro. Am J Trop Med Hyg. 1985;34:850–855. doi: 10.4269/ajtmh.1985.34.850. [DOI] [PubMed] [Google Scholar]
- 16.Rey H. Cellular reactions in the dermal connective tissue of the hamster to . Leishmania brasiliensis J Infect Dis. 1943;72:117–124. [Google Scholar]
- 17.Wilson ME, Innes DJ, Sousa A, Pearson RD. Early histopathology of experimental infection with Leishmania donovaniin hamsters. J Parasitol. 1987;73:55–63. [PubMed] [Google Scholar]
- 18.Pompeu ML, Freitas LA, Santos MLV, Khouri M, Barral-Netto M. Granulocytes in the inflammatory process of BALB/c mice infected by Leishmania amazonensis.A quantitative approach. Acta Trop. 1991;48:185–193. doi: 10.1016/0001-706x(91)90046-m. [DOI] [PubMed] [Google Scholar]
- 19.Beil WJ, Meinardus-Hager G, Neugebauer D-C, Sorg C. Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J Leukoc Biol. 1992;52:135–142. doi: 10.1002/jlb.52.2.135. [DOI] [PubMed] [Google Scholar]
- 20.Chang, K.-P., and L.D. Hendricks. 1985. Laboratory cultivation and maintenance of Leishmania. In Leishmaniasis. K.-P. Chang and R.S. Bray, editors. Elsevier, Amsterdam. 1–30.
- 21.Metcalf, J.A., J.I. Gallin, W.M. Nauseef, and R.K. Root. 1986. Neutrophil purification. In Laboratory Manual of Neutrophil Function. J.A. Metcalf, J.I. Gallin, W.M. Nauseef, and R.K. Root, editors. Raven Press, New York. 2–6.
- 22.Esparza I, Fox RI, Schreiber RD. Interferon-γ–dependent modulation of C3b receptors (CR1) on human peripheral blood monocytes. J Immunol. 1986;136:1360–1365. [PubMed] [Google Scholar]
- 23.O'Shea JJ, Brown EJ, Seligmann BE, Metcalf JA, Frank MM, Gallin JI. Evidence for distinct intracellular pools of receptors for C3b and C3bi in human neutrophils. J Immunol. 1985;134:2580–2587. [PubMed] [Google Scholar]
- 24.Cook J, Fischer E, Boucheix C, Mirsrahi M, Jouvin M-H, Weiss L, Jack MR, Kazatchkine MD. Mouse monoclonal antibodies to the human C3b receptor. Mol Immunol. 1985;22:531–539. doi: 10.1016/0161-5890(85)90176-2. [DOI] [PubMed] [Google Scholar]
- 25.Mason DW, Williams AF. The kinetics of antibody binding to membrane antigens in solution and at the cell surface. Biochem J. 1980;187:1–20. doi: 10.1042/bj1870001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fine DP, Marney SR, Jr, Colley DG, Sergent JS, Des RM, Prez C3 shunt activation in human serum chelated with EGTA. J Immunol. 1972;109:807–809. [PubMed] [Google Scholar]
- 27.Eisenthal R, Cornish-Bowden A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem J. 1974;139:715–720. doi: 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hostetter MK, Krueger RA, Schmeling DJ. The biochemistry of opsonization: central role of the reactive thiolester of the third component of complement. J Infect Dis. 1984;150:653–661. doi: 10.1093/infdis/150.5.653. [DOI] [PubMed] [Google Scholar]
- 29.Ezzell JL, Wilcox LA, Bernshaw NJ, Parker CJ. Induction of the paroxysmal nocturnal hemoglobinuria phenotype in normal human erythrocytes: effects of 2-aminoethylisothiouronium bromide on membrane proteins that regulate complement. Blood. 1991;77:2764–2773. [PubMed] [Google Scholar]
- 30.Dorval BL, Cosio FG, Birmingham DJ, Hebert LA. Human erythrocytes inhibit complement-mediated solubilization of immune complexes. J Immunol. 1989;142:2721–2727. [PubMed] [Google Scholar]
- 31.Mosser DM, Edelson PJ. The third component of complement (C3) is responsible for the intracellular survival of Leishmania major. . Nature. 1987;327:329–331. doi: 10.1038/327329b0. [DOI] [PubMed] [Google Scholar]
- 32.Nickells M, Hauhart R, Krych M, Bala V, Subramanian, Geoghegan-Barek K, Marsh HC, Jr, Atkinson JP. Mapping epitopes for 20 monoclonal antibodies to CR1. Clin Exp Immunol. 1998;112:27–33. doi: 10.1046/j.1365-2249.1998.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emlen W, Burdick G, Carl V, Lachmann PJ. Binding of model immune complexes to erythrocyte CR1 facilitates immune complex uptake by U937 cells. J Immunol. 1989;142:4366–4371. [PubMed] [Google Scholar]
- 34.Avrameas S, Ternynck T. Natural autoantibodies: the other side of the immune system. Res Immunol. 1995;146:235–248. doi: 10.1016/0923-2494(96)80259-8. [DOI] [PubMed] [Google Scholar]
- 35.Bachmann MF, Kalinke U, Althage A, Freer G, Burkhart C, Roost H-P, Aguet M, Hengartner H, Zinkernagel RM. The role of antibody concentration and avidity in antiviral protection. Science. 1997;276:2024–2027. doi: 10.1126/science.276.5321.2024. [DOI] [PubMed] [Google Scholar]
- 36.Ulrich M, Trujillo D, Ortiz, Convit J. The effect of fresh serum on the leptomonads of Leishmania.I. Preliminary report. Trans R Soc Trop Med Hyg. 1968;62:825–830. doi: 10.1016/0035-9203(68)90011-4. [DOI] [PubMed] [Google Scholar]
- 37.Schmunis GA, Herman R. Characteristics of so-called natural antibodies in various normal sera against culture forms of Leishmania. . J Parasitol. 1970;56:889–896. [PubMed] [Google Scholar]
- 38.Pearson RD, Steigbigel RT. Mechanism of lethal effect of human serum upon . Leishmania donovani J Immunol. 1980;125:2195–2201. [PubMed] [Google Scholar]
- 39.Mosser DM, Burke SK, Coutavas EE, Wedgwood JF, Edelson PJ. Leishmaniaspecies: mechanisms of complement activation by five strains of promastigotes. Exp Parasitol. 1986;62:394–404. doi: 10.1016/0014-4894(86)90048-2. [DOI] [PubMed] [Google Scholar]
- 40.Avila JL, Rojas M, Galili U. Immunogenic Galα1→ 3Gal carbohydrate epitopes are present on pathogenic American Trypanosoma and Leishmania. . J Immunol. 1989;142:2828–2834. [PubMed] [Google Scholar]
- 41.Navin TR, Krug EC, Pearson RD. Effect of immunoglobulin M from normal human serum on Leishmania donovanipromastigote agglutination, complement-mediated killing, and phagocytosis by human monocytes. Infect Immun. 1989;57:1343–1346. doi: 10.1128/iai.57.4.1343-1346.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puentes SM, Sacks DL, Da Silva RP, Joiner KA. Complement binding by two developmental stages of Leishmania majorpromastigotes varying in expression of a surface lipophosphoglycan. J Exp Med. 1988;167:887–902. doi: 10.1084/jem.167.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mosser DM, Edelson PJ. Activation of the alternative complement pathway by Leishmaniapromastigotes: parasite lysis and attachment to macrophages. J Immunol. 1984;132:1501–1505. [PubMed] [Google Scholar]
- 44.Puentes SM, Dwyer DM, Bates PA, Joiner KA. Binding and release of C3 from Leishmania donovanipromastigotes during incubation in normal human serum. J Immunol. 1989;143:3743–3749. [PubMed] [Google Scholar]
- 45.Ofek I, Goldhar J, Keisari Y, Sharon N. Nonopsonic phagocytosis of microorganisms. Ann Rev Microbiol. 1995;49:239–276. doi: 10.1146/annurev.mi.49.100195.001323. [DOI] [PubMed] [Google Scholar]
- 46.Medof ME, Prince GM, Oger JJ-F. Kinetics of interaction of immune complexes with complement receptors on human blood cells: modification of complexes during interaction with red cells. Clin Exp Immunol. 1982;48:715–725. [PMC free article] [PubMed] [Google Scholar]
- 47.Edberg JC, Wright E, Taylor RP. Quantitative analyses of the binding of soluble complement–fixing antibody/dsDNA immune complexes to CR1 on human red blood cells. J Immunol. 1987;139:3739–3747. [PubMed] [Google Scholar]
- 48.Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparumrosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388:292–295. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- 49.Wallace JM, Wormall A. Red-cell adhesion in trypanosomiasis of man and other animals. II. Some experiments on the mechanism of the reaction. Parasitology. 1931;23:346–359. [Google Scholar]
- 50.Da Silva RP, Fenton B, Hall, Joiner KA, Sacks DL. CR1, the C3b receptor, mediates binding of infective Leishmania majormetacyclic promastigotes to human macrophages. J Immunol. 1989;143:617–622. [PubMed] [Google Scholar]
- 51.Rosenthal LA, Sutterwala FS, Kehrli ME, Mosser DM. Leishmania major–human macrophage interactions: cooperation between Mac-1 (CD11b/CD18) and complement receptor type 1 (CD35) in promastigote adhesion. Infect Immun. 1996;64:2206–2215. doi: 10.1128/iai.64.6.2206-2215.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang K-P, Mosser DM. Role of the Leishmaniasurface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol. 1995;155:3102–3111. [PubMed] [Google Scholar]
- 53.Sutterwala FS, Rosenthal LA, Mosser DM. Cooperation between CR1 (CD35) and CR3 (CD11b/ CD18) in the binding of complement-opsonized particles. J Leukoc Biol. 1996;59:883–890. doi: 10.1002/jlb.59.6.883. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen CH, Matthiesen SH, Lyng I, Leslie GQ. The role of complement receptor type 1 (CR1, CD35) in determining the cellular distribution of opsonized immune complexes between whole blood cells: kinetic analysis of the buffering capacity of erythrocytes. Immunology. 1997;90:129–137. doi: 10.1046/j.1365-2567.1997.00138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang K-P. Leishmanicidal mechanisms of human polymorphonuclear phagocytes. Am J Trop Med Hyg. 1981;30:322–333. doi: 10.4269/ajtmh.1981.30.322. [DOI] [PubMed] [Google Scholar]
- 56.Mirkovich AM, Galelli A, Allison AC, Modabber FZ. Increased myelopoiesis during Leishmania majorinfection in mice: generation of “safe targets”, a possible way to evade the effector immune mechanism. Clin Exp Immunol. 1986;64:1–7. [PMC free article] [PubMed] [Google Scholar]
- 57.Nelson DS. Immune adherence. Adv Immunol. 1963;3:131–180. [Google Scholar]
- 58.Arnaout MA, Dana N, Melamed J, Medicus R, Colten HR. Low ionic strength or chemical cross-linking of monomeric C3b increases its binding affinity to the human complement C3b receptor. Immunology. 1983;48:229–237. [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper NR. Immune adherence by the fourth component of complement. Science. 1969;165:396–398. doi: 10.1126/science.165.3891.396. [DOI] [PubMed] [Google Scholar]
- 60.Modabber, F. 1996. Vaccine: the only hope to control Leishmaniasis. In Molecular and Immune Mechanisms in the Pathogenesis of Cutaneous Leishmaniasis. F.J. Tapia, G. Cáceres-Dittmar, and M.A. Sánchez, editors. R.G. Landes Company, Austin, TX. 223–236.