

Abstract
Three novel streptococcal superantigen genes (spe-g, spe-h, and spe-j) were identified from the Streptococcus pyogenes M1 genomic database at the University of Oklahoma. A fourth novel gene (smez-2) was isolated from the S. pyogenes strain 2035, based on sequence homology to the streptococcal mitogenic exotoxin z (smez) gene. SMEZ-2, SPE-G, and SPE-J are most closely related to SMEZ and streptococcal pyrogenic exotoxin (SPE)-C, whereas SPE-H is most similar to the staphylococcal toxins than to any other streptococcal toxin. Recombinant (r)SMEZ, rSMEZ-2, rSPE-G, and rSPE-H were mitogenic for human peripheral blood lymphocytes with half-maximal responses between 0.02 and 50 pg/ml (rSMEZ-2 and rSPE-H, respectively). SMEZ-2 is the most potent superantigen (SAg) discovered thus far. All toxins, except rSPE-G, were active on murine T cells, but with reduced potency. Binding to a human B-lymphoblastoid line was shown to be zinc dependent with high binding affinity of 15–65 nM. Evidence from modeled protein structures and competitive binding experiments suggest that high affinity binding of each toxin is to the major histocompatibility complex class II β chain. Competition for binding between toxins was varied and revealed overlapping but discrete binding to subsets of class II molecules in the hierarchical order (SMEZ, SPE-C) > SMEZ-2 > SPE-H > SPE-G. The most common targets for the novel SAgs were human Vβ2.1- and Vβ4-expressing T cells. This might reflect a specific role for this subset of Vβs in the immune defense of gram-positive bacteria.
Keywords: superantigen, major histocompatibility complex class II, T cell receptor, streptococcal exotoxin, zinc dependency
S treptococcus pyogenes is a gram-positive pathogenic bacterium that produces a number of distinct but partially related streptococcal pyrogenic exotoxins (SPEs).1 These toxins belong to a bigger family of T cell mitogens or superantigens (SAgs), also found in viruses and other bacteria, but primarily in Staphylococcus aureus (1–6). SAgs simultaneously bind to MHC class II antigens and TCR molecules and stimulate a large number of T cells, which leads to high systemic levels of the cytokines TNF-α and IL-1β and of T cell mediators like IL-2 and IFN-γ (1, 7–9). The resulting pathological conditions include fever and shock (10–11).
SPE-A (12), SPE-C (13), and streptococcal SAg (SSA; reference 14) are significantly related to each other and to the S. aureus enterotoxins (SEs), building a homologous protein family with sequence identities from 20 to >90% (15). Bacterial SAgs are monomeric proteins, expressed as precursor molecules with typical bacterial signal peptides. The secreted mature toxins have molecular masses of 24–28 kD (4). SAgs bind to MHC class II outside the peptide binding groove and to all TCRs bearing a particular Vβ region (16–19). Crystal structures of SEA (20), SEB (21), SEC2 (22), SED (23), toxic shock syndrome toxin (TSST; reference 24), and SPE-C (25) show a conserved folding pattern, comprising a NH2-terminal β-barrel globular domain and a COOH-terminal globular domain based on a β-grasp motif.
All SAgs examined thus far, except SPE-C, have a generic binding site for the invariant α chain of MHC class II located in the NH2-terminal domain (26–28). There are two structural models for the α chain binding. TSST binds to the invariant α1 domain but lies across the top of the molecule, contacting peptide and preventing continued TCR contact (26–27). SEB binds out to one side of the α1 domain α chain and does not contact peptide residues (28). TCR contact is not excluded and indeed, SEB appears to rely on continued TCR/MHC class II contact for activity. The unique exception to α chain binding is SPE-C, which has surrendered its MHC class II α chain binding site in favor of a protein dimer interface (25, 29). Some toxins, like SEA, have in addition to their α chain binding a second high affinity binding site for the polymorphic MHC class II β chain, mediated by a tetrameric zinc coordination complex between four SEA residues and the highly conserved His81 of the HLA-DR1 β chain (20, 30, 31). This bifunctional binding mode enables SEA to cross-link MHC class II on the surface of APCs (32). SPE-C lacks the generic MHC class II α chain binding site and binds MHC class II only via the polymorphic β chain (29). As shown from the crystal structure, SPE-C also has a dimerization interface that might be stabilized by a second zinc binding site (25). Binding of SPE-C dimers to MHC class II represents an alternative mechanism for class II cross-linking. A third putative mode for class II cross-linking was suggested for SED based on the recently published SED crystal structure (23). Apparently, SED forms zinc-dependent dimers that bind to the MHC class II α chain in a SEB-like mechanism, using the NH2-terminal low affinity binding site.
SAgs can be identified by their particular Vβ profile. SPE-A preferably stimulates T cells that carry Vβ12.5 and Vβ14 TCRs (33), but stimulation of Vβ2 and Vβ15 has also been reported (34). SPE-C almost exclusively stimulates Vβ2 TCR (29).
SPE-B, a powerful streptococcal protease, has been shown to have mitogenic activity for Vβ8-bearing human T cells. However, rSPE-B has no SAg activity, suggesting that the purified SPE-B was contaminated with traces of other co-purified SAgs. In addition, several reports have also shown other purified streptococcal proteins to have Vβ8-like stimulation activity, later to be confirmed as a result of a potent contaminating SAg called SPE-X (35). All attempts to purify SPE-X or to identify the spe-x gene have so far been unsuccessful. Toyosaki et al. (36) described a SAg called mitogenic factor (MF) that activates Vβ2, Vβ7, Vβ8, and Vβ18 TCR, but its potency to stimulate T cells was 100–1,000 times weaker than that of SPE-A and SPE-C. It is therefore very unlikely that MF is the contaminating agent of other toxin preparations.
Another recently discovered toxin, the streptococcal mitogenic exotoxin Z (SMEZ), that displayed homology to other SAgs, was mitogenic for rabbit T cells (37). The potency and Vβ specificity for human and mouse T cells was not determined.
To identify other SAgs, the Oklahoma University S. pyogenes genome database (http://www.genome.ou.edu/strep.html) was screened using small regions of highly conserved protein sequences on all the streptococcal and staphylococcal SAgs. This screen yielded three complete and one incomplete open reading frames (ORFs) encoding potential SAgs. Each has been expressed as recombinant protein and examined for the potency to stimulate human and murine T cells. Here we describe the immunological and biochemical characterization of recombinant forms of SMEZ, SMEZ-2, SPE-G, and SPE-H. Despite high sequence homology, striking variation in function was observed. Both SMEZ and SMEZ-2 are probably responsible for the Vβ8-stimulating activity referred to as SPE-X.
Materials and Methods
Identification of Novel SAgs.
The novel SAgs were identified by searching the S. pyogenes M1 genome database at the University of Oklahoma with highly conserved β5 and α4 regions of streptococcal and staphylococcal SAgs, using a TBlastN search program.
The ORFs were defined by translating the DNA sequences around the matching regions and aligning the protein sequences to known SAgs using the computer program Gap. Multiple alignments and dendrograms were performed with Lineup and Pileup. We used the FASTA program for searching the SwissProt (Amos Bairoch, Switzerland) and PIR (Protein Identification Resource) protein databases.
The leader sequences of SPE-G and SPE-H were predicted using the SP Scan program. All computer programs are part of the Genetics Computer Group package (version 8).
Cloning of smez, smez-2, spe-g, and spe-h.
50 ng of S. pyogenes M1 (ATCC 700294) or S. pyogenes 2035 genomic DNA was used as a template to amplify the smez DNA fragment and the smez-2 DNA fragment, respectively, by PCR using the primers smez-fw (TGGGATCCTTAGAAGTAGATAATA) and smez-rev (AAGAATTCTTAGGAGTCAATTTC) and Taq Polymerase (Promega Corp.). The primers contain a terminal tag with the restriction enzyme recognition sequences BamHI and EcoRI, respectively. The amplified DNA fragment, encoding the mature protein without the leader sequence (37) was cloned into a T-tailed pBlueScript SKII vector (Stratagene).
Spe-g and spe-h were cloned by a similar approach, using the primers spe-g-fw (CTGGATCCGATGAAAATTTAAAAGATTTAA) and spe-g-rev (AAGAATTCGGGGGGAGAATAG), and spe-h-fw (TTGGATCCAATTCTTATAATACAACC) and spe-h-rev (AAAAGCTTTTAGCTGATTGACAC), respectively.
The DNA sequences of the subcloned toxin genes were confirmed by the dideoxy chain termination method using a Licor automated DNA sequencer (model 4200). As the DNA sequences from the genomic database are all unedited raw data, three subclones of every cloning experiment were analyzed to insure that no Taq polymerase–related mutations were introduced. The DNA sequence of the smez-2 gene has been annotated in EMBL/ Genbank/DDBJ under accession number AF086626.
Expression and Purification of rSMEZ, rSMEZ-2, rSPE-G, and rSPE-H.
Subcloned smez, smez-2, and spe-g fragments were cut from pBlueScript SKII vectors, using restriction enzymes BamHI and EcoRI (GIBCO BRL), and cloned into pGEX-2T expression vectors (Pharmacia Biotech). Due to an internal EcoRI restriction site within the spe-h gene, the pBlueScript:spe-h subclone was digested with BamHI and HindIII and the spe-h fragment was cloned into a modified pGEX-2T vector that contains a HindIII 3′ cloning site.
rSMEZ, rSMEZ-2, and rSPE-H were expressed in Escherichia coli DH5α cells as glutathione-S-transferase (GST) fusion proteins. Cultures were grown at 37°C and induced for 3–4 h after adding 0.2 mM isopropyl-β-d-thiogalactopyranoside. GST– SPE-G fusion protein was expressed in cells grown at 28°C.
The GST fusion proteins were purified on glutathione (GSH) agarose as previously described (29) and the mature toxins were cleaved off from GST by trypsin digestion. All recombinant toxins, except rSMEZ, were further purified by two rounds of cation exchange chromatography using carboxy methyl sepharose (Pharmacia Biotech). The GST-SMEZ fusion protein was trypsin digested on the GSH-column and the flow-through containing the SMEZ was collected.
Gel Electrophoresis.
All purified recombinant toxins were tested on a 12.5% SDS-polyacrylamide gel according to Laemmli's procedure. The isoelectric point of the recombinant toxins was determined by isoelectric focusing on a 5.5% polyacrylamide gel using ampholine, pH 5–8 (Pharmacia Biotech). The gel was run for 90 min at 1 Watt constant power.
Toxin Proliferation Assay.
Human PBLs were purified from blood of a healthy donor by Histopaque Ficoll (Sigma Chemical Co.) fractionation. The PBLs were incubated in 96-well round-bottomed microtiter plates at 105 cells per well with RPMI-10 (RPMI with 10% FCS) containing varying dilutions of recombinant toxins. The dilution series was performed in 1:5 steps from a starting concentration of 10 ng/ml of toxin. Pipette tips were changed after each dilution step. After 3 d, 0.1 μCi [3H]thymidine was added to each well and cells were incubated for another 24 h. Cells were harvested and counted on a scintillation counter.
Mouse leukocytes were obtained from spleens of five different mouse strains (SJL, B10.M, B10/J, C3H, and BALB/c). Splenocytes were washed in DMEM-10, counted in 5% acetic acid, and incubated on microtiter plates at 105 cells per well with DMEM-10 and toxins as described for human PBLs.
TCR Vβ Analysis.
Vβ enrichment analysis was performed by anchored multiprimer amplification (38). Human PBLs were incubated with 20 pg/ml of recombinant toxin at 106 cells/ml for 3 d. A twofold volume expansion of the culture followed with medium containing 20 ng/ml IL-2. After another 24 h, stimulated and resting cells were harvested and RNA was prepared using Trizol reagent (GIBCO BRL). A 500 bp β chain DNA probe was obtained by anchored multiprimer PCR as previously described (38), radiolabeled, and hybridized to individual Vβs and a Cβ DNA region dot-blotted on a Nylon membrane. The membrane was analyzed on a Storm PhosphorImager using ImageQuant software (Molecular Dynamics). Individual Vβs were expressed as a percentage of all the Vβs determined by hybridization to the Cβ probe.
Jurkat Cell Assay.
Jurkat cells (a human T cell line) and LG-2 cells (a human B lymphoblastoid cell line, homozygous for HLA-DR1) were harvested in log phase and resuspended in RPMI-10. 100 μl of the cell suspension, containing 105 Jurkat cells and 2 × 104 LG-2 cells were mixed with 100 μl of varying dilutions of recombinant toxins on 96-well plates. After incubating overnight at 37°C, 100-μl aliquots were transferred onto a fresh plate and 100 μl (104) of SeI cells (IL-2–dependent murine T cell line) per well were added. After incubating for 24 h, 0.1 μCi [3H]thymidine was added to each well and cells were incubated for another 24 h. Cells were harvested and counted on a scintillation counter. As a control, a dilution series of IL-2 was incubated with SeI cells.
Computer-aided Modelling of Protein Structures.
Protein structures of SMEZ2, SPE-G and SPE-H were created on a Silicon Graphics computer using InsightII/Homology software (Biosym Technologies). The SAgs SEA, SEB, and SPE-C were used as reference proteins to determine structurally conserved regions (SCRs). Coordinate files for SEA (1ESF), SEB (1SEB), and SPE-C (1AN8) were downloaded from the Brookhaven Protein Database. The primary amino acid sequences of the reference proteins and SMEZ-2, SPE-G, and SPE-H, respectively, were aligned, and coordinates from superimposed SCRs were assigned to the model proteins. The loop regions between the SCRs were generated by random choice. MolScript software (39) was used for displaying the computer-generated images.
Radiolabeling and LG-2 Binding Experiments.
Recombinant toxin was radioiodinated by the chloramine T method as previously described (29). Labeled toxin was separated from free iodine by size exclusion chromatography using Sephadex G25 (Pharmacia Biotech). LG2 cells were used for cell binding experiments, as previously described (29). In brief, cells were harvested, resuspended in RPMI-10, mixed at 106 cells/ml with 125I-tracer toxin (1 ng) and 0.0001–10 μg of unlabeled toxin, and incubated at 37°C for 1 h. After washing with ice cold RPMI-1 the pelleted cells were analyzed in a gamma counter.
For zinc binding assays, the toxins were incubated in RPMI-10 alone, RPMI-10 with 1 mM EDTA, or RPMI-10 with 1 mM EDTA and 2 mM ZnCl2. Scatchard analysis was performed as described elsewhere (40).
For competitive binding studies, 1 ng of 125I-tracer toxin (rSMEZ, rSMES-2, rSPE-G, rSPE-H, rSEA, rSPE-C, or rTSST) was incubated with 0.0001–10 μg of unlabeled toxin (rSMEZ, rSMES-2, rSPE-G, rSPE-H, rSEA, rSEB, rSPE-C, and rTSST) for 1 h. For SEB inhibition studies, 20 ng of 125I-rSEB was used as tracer and samples were incubated for 4 h.
Results
Identification and Sequence Analysis of Novel SAgs.
The Oklahoma University S. pyogenes M1 genome database is accessible via the internet and contains a collection of more than 300 DNA sequence contigs derived from a shot gun plasmid library of the complete S. pyogenes M1 genome. The currently available DNA sequences cover ∼95% of the total genome. We searched this database with a highly conserved SAg peptide sequence, using a search program that screens the DNA database for peptide sequences in all six possible reading frames. We found 8 significant matches and predicted the ORFs by aligning translated DNA sequences to complete protein sequences of known SAgs.
Five matches gave complete ORFs with significant homology to streptococcal and staphylococcal SAgs. Three of these ORFs correlate to SPE-C, SSA, and the recently described SMEZ (37), respectively. The remaining two ORFs could not be correlated to any known protein in the SwissProt and PIR databases. We named these novel putative sag genes spe-g and spe-h.
One ORF could not be generated completely due to its location close to the end of a contig. The DNA sequence of the missing 5′-end is located on another contig, and individual contigs have yet to be assembled in the database. However, the available sequence shows an ORF for the 137 COOH-terminal amino acid residues of a putative novel SAg that could not be found in the existing protein databases. We named this thus far incomplete gene spe-j. In two cases, a complete ORF could not be defined due to several out-of-frame mutations. Although DNA sequencing errors on the unedited DNA sequences cannot be ruled out completely, the high frequency of inserts and deletions probably represent natural mutation events on pseudogenes, which are no longer used.
To produce recombinant proteins of SMEZ, SPE-G, and SPE-H, individual genes (coding for the mature toxins without leader sequence) were amplified by PCR and subcloned for DNA sequencing. Both S. pyogenes M1 and S. pyogenes 2035 genomic DNA were used and individual toxin gene sequences were compared between the two strains. The spe-h gene was isolated from the M1 strain but could not be amplified from strain 2035 genomic DNA, suggesting a restricted strain specificity for this toxin. The spe-g gene was cloned from both M1 and 2035, and DNA sequence analysis of both genes showed no differences. The full-length smez gene was isolated from both strains, but DNA sequence comparison revealed some striking differences. The smez gene of strain 2035 showed nucleotide changes in 36 positions (or 5%) compared with smez from strain M1 (Fig. 1). The deduced protein sequences differed in 17 amino acid residues (or 8.1%). This difference was sufficient to indicate a new gene. We named this gene smez-2, because it is 95% homologous to smez.
Figure 1.
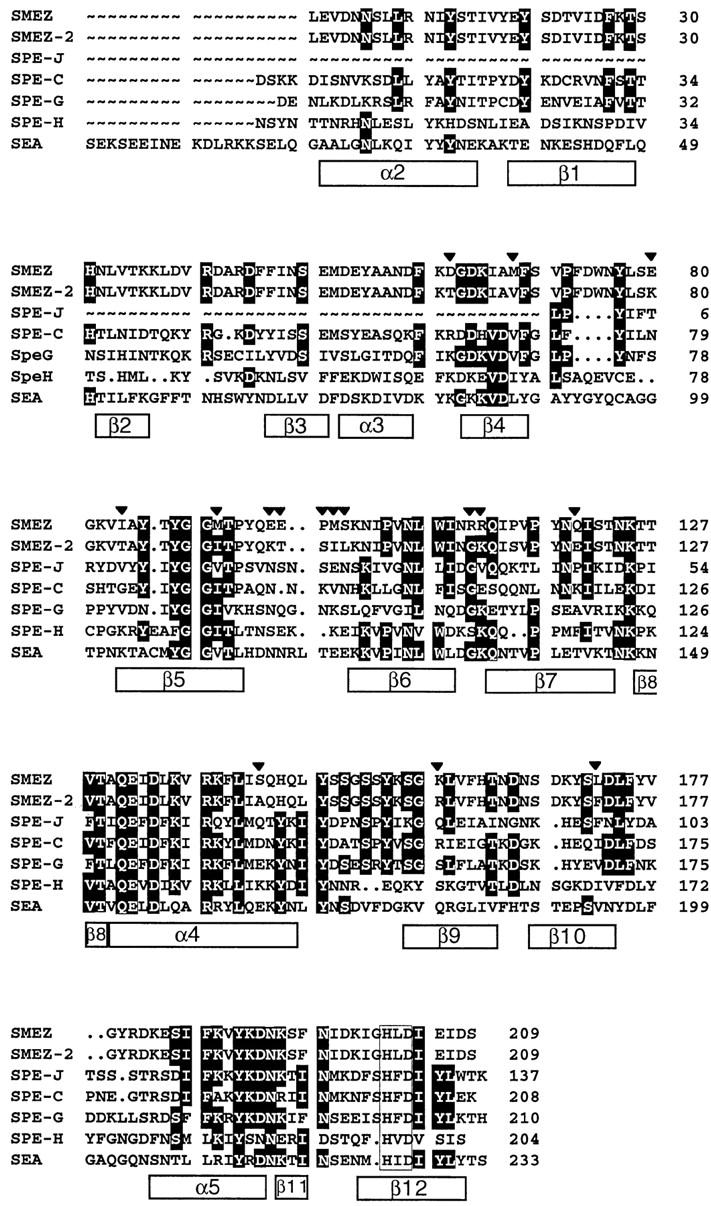
Multiple alignment of SAg protein sequences. The protein sequences of mature toxins were aligned using the PileUp program on the GCG package. Regions of high sequence identity are in black boxes. The boxes below the sequences indicate the structural elements of SPE-C, as determined from the crystal structure (25). Regions with highest homology correspond to the β4, β5, α4, and α5 regions in SPE-C. The clear box near the COOH terminus represents a primary zinc binding motif, a common feature of all toxins shown here. The arrows on top of the sequence alignment show the regions of sequence diversity between SMEZ and SMEZ-2. The NH2-terminal part of SPE-J is missing, as the corresponding DNA sequence is not yet available in the database.
The most significant difference between SMEZ and SMEZ-2 is an exchanged pentapeptide sequence at position 96–100, where the EEPMS sequence of SMEZ is converted to KTSIL in SMEZ-2 (Fig. 1). A second cluster is at position 111–112, where an RR dipeptide is exchanged for GK in SMEZ-2. The remaining 10 different residues are spread over almost the entire primary sequence.
A revised SAg family tree, based on primary amino acid sequence homology (Fig. 2) now shows three general subfamilies: group A comprises SPE-C, SPE-J, SPE-G, SMEZ, and SMEZ-2; group B comprises SEC1-3, SEB, SSA, SPE-A, and SEG; and group C comprises SEA, SEE, SED, SEH, and SEI. Two SAgs, TSST and SPE-H, do not belong to any one of those subfamilies.
Figure 2.
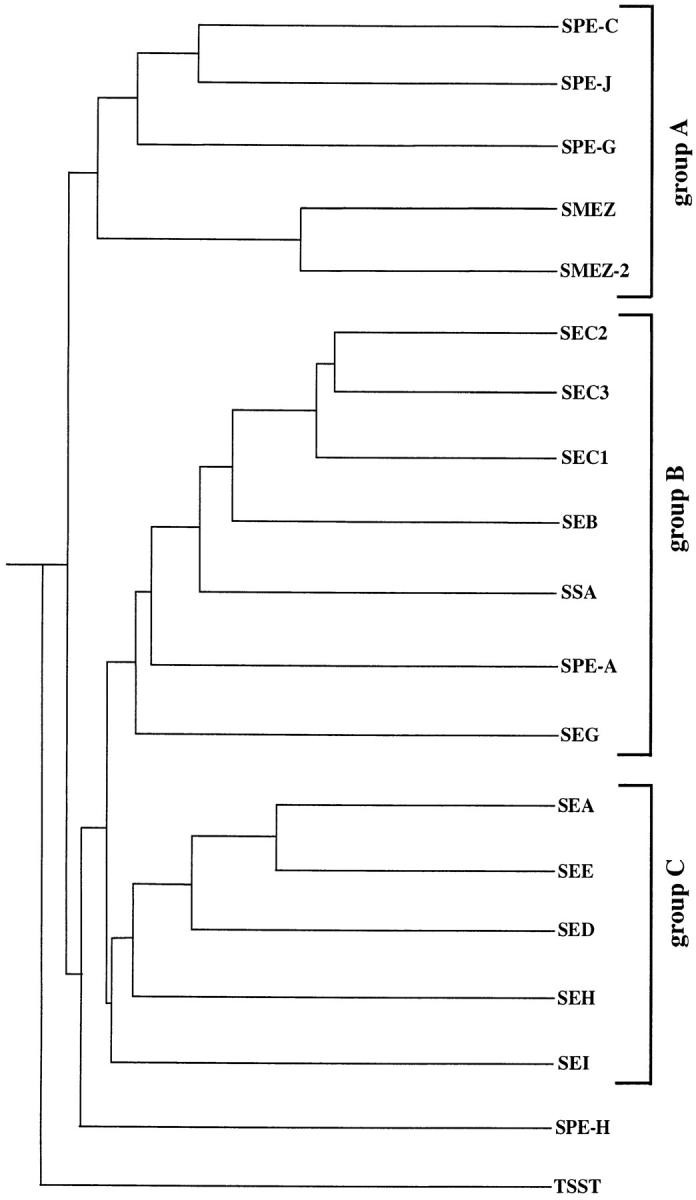
SAg family tree. The family tree was created using the PileUp program on the GCG package and is based on primary amino acid sequence homology. The SAg family can be divided into three groups (or subfamilies) and two individual branches (SPE-H and TSST). SMEZ, SMEZ-2, SPE-G, and SPE-J build a subfamily together with SPE-C. The complete DNA sequence of the spe-j gene is not yet available, so only the 135 COOH-terminal amino acid residues of SPE-J could be used for the alignment.
SMEZ, SMEZ-2, SPE-G, and SPE-J are most closely related to SPE-C, increasing the number of this subfamily from two to five members. SPE-G shows the highest protein sequence homology with SPE-C (38.4% identity and 46.6% similarity). The homology of SPE-J to SPE-C is even more significant (56% identity and 62% similarity), but this comparison is only preliminary due to the missing NH2-terminal sequence. SMEZ shows 30.9/40.7% homology to SPE-C, and SMEZ-2 is 92/93% homologous to SMEZ. SPE-H builds a new branch in the family tree and is most closely related to SED, showing 25% identity and 37.3% similarity.
Multiple alignment of SAg protein sequences (Fig. 1) shows that similarities are clustered within structure-determining regions, represented by α4, α5, β4, and β5 regions. This applies to all toxins of the SAg family (data not shown) and explains why SAgs like SPE-C and SEA have very similar overall structures despite their rather low sequence identity of 24.4%.
Although SPE-H is less related to SPE-C, it shows two common features with the “SPE-C subfamily”: (i) a truncated NH2-terminus, lacking the α1 region and (ii) a primary zinc binding motif (H-X-D) at the COOH terminus (Fig. 1). It has been shown for several SAgs that this motif is involved in a zinc-coordinated binding to the β chain of HLA-DR1 (18, 23).
Fusion proteins of GST-SMEZ, GST-SMEZ-2, and GST-SPE-H were completely soluble and gave yields of ∼30 mg/liter. The GST-SPE-G fusion was insoluble when grown at 37°C, but mostly soluble when expressed in cells growing at 28°C. Although soluble GST-SPE-G yields were 20–30 mg/per liter, solubility decreased after cleavage of the fusion protein with trypsin. Soluble rSPE-G was achieved by diluting the GST-SPE-G to <0.2 mg/ml before cleavage. After cation exchange chromatography, purified rSPE-G could be stored at ∼0.4 mg/ml.
rSMEZ could not be separated from GST by ion exchange chromatography. Isoelectric focusing revealed that the isoelectric points of the two proteins are too similar to allow separation (data not shown). Therefore, rSMEZ was released from GST by cleaving with trypsin while still bound to the GSH agarose column. rSMEZ was collected with the flow through.
The purified recombinant toxins were applied to SDS-PAGE and isoelectric focusing (Fig. 3). Each toxin ran as a single band on the SDS PAA gel, confirming their purity and their calculated molecular weights of 24.33 (SMEZ), 24.15 (SMEZ-2), 24.63 (SPE-G), and 23.63 (SPE-H) (Fig. 3 A). The isoelectric focusing gel (Fig. 3 B) shows a significant difference between rSMEZ and rSMEZ-2. Like most other staphylococcal and streptococcal toxins, rSMEZ-2 possesses a slightly basic isoelectric point at pH 7–8, but rSMEZ is acidic with an isoelectric point at pH 6–6.5.
Figure 3.


Gel electrophoresis of the purified recombinant toxins. (A) Two micrograms of purified recombinant toxin were run on a 12.5% SDS-polyacrylamide gel to show the purity of the preparations. (B) 2 μg of purified recombinant toxin were run on an isoelectric focusing gel (5.5% PAA, pH 5–8). The isoelectric point of rSMEZ-2, rSPE-G, and rSPE-H is similar and was estimated at pH 7–8. The IEP of rSMEZ was estimated at pH 6–6.5.
T Cell Proliferation and Vβ Specificity.
To ensure the native conformation of the purified recombinant toxins, a standard [3H]thymidine incorporation assay was performed to test for their potency to stimulate PBLs. All toxins were active on human T cells (Fig. 4). rSEA, rSEB, rSPE-C, and rTSST were included as reference proteins. The mitogenic potency of these toxins was lower than previously described, but is regarded as a more accurate figure. In previous studies performed by us (29), a higher starting concentration of toxin (100 ng/ml) was used and tips were not changed in between dilutions. This led to significant carry-over across the whole dilution range. In this study, the starting concentration was 10 ng/ml and tips were changed in between dilutions, thereby preventing any carry-over.
Figure 4.

Stimulation of human T cells with recombinant toxins. PBLs were isolated from human blood samples and incubated with varying concentrations of recombinant toxin. After 3 d, 0.1 μCi [3H]thymidine was added and cells were incubated for another 24 h, before being harvested and counted on a gamma counter. ○, unstimulated; ▴, rSMEZ; □, rSMEZ-2; ♦, rSPE-G; ┘, rSPE-H.
The half-maximal responses (P50 values) for rSPE-G and rSPE-H were 2 and 50 pg/ml, respectively. No activity was detected at <0.02 pg/ml and 0.1 pg/ml, respectively. Both toxins are therefore less potent than rSPE-C. rSMEZ was similar in potency to rSPE-C, with a P50 value of 0.08 pg/ml and no detectable proliferation at <0.5 fg/ml. rSMEZ-2 showed the strongest mitogenic potency of all toxins tested or, as far as we are aware, described elsewhere. The P50 value was determined at 0.02 pg/ml and rSMEZ-2 was still active at <0.1 fg/ml. All P50 values are summarized in Table I.
Table I.
Potency of Recombinant Toxins on Human and Mouse T Cells
| Proliferation potential P50 [pg/ml] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Toxin | Human | SJL | B10.M | B10/J | C3H | BALB/c | ||||||
| SEA | 0.1 | 20 | 12 | 1.8 | 19 | 1,000 | ||||||
| SEE | 0.2 | 10 | 12 | 1.5 | 50 | 15 | ||||||
| SEB | 0.8 | 7,000 | 80,000 | 5,000 | 10,000 | 1,000 | ||||||
| TSST | 0.2 | 20 | 1,000 | 1.2 | 100 | 10 | ||||||
| SPE-C | 0.1 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | ||||||
| SMEZ | 0.08 | 80 | 80 | 100 | 9,000 | 200 | ||||||
| SMEZ-2 | 0.02 | 100 | 15 | 10 | 800 | 18 | ||||||
| SPE-G | 2 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | ||||||
| SPE-H | 50 | 15 | 800 | 5,000 | 100 | 1,000 | ||||||
Human PBLs and mouse T cells were stimulated with varying amounts of recombinant toxin. The P50 value reflects the concentration of recombinant toxin required to induce 50% maximal cell proliferaton. No proliferation was detected for rSPE-C and rSPE-G at any concentration tested on murine T cells.
Murine T cells from five different mouse strains were tested for their mitogenic response to rSMEZ, rSMEZ-2, rSPE-G, and rSPE-H (Table I). rSPE-G showed no activity against any of the mouse strains tested. rSPE-H, rSMEZ, and rSMEZ-2 showed varied potency depending on the individual mouse strain. For example, rSMEZ-2 was 500-fold more potent than rSPE-H in the B10/J strain, whereas rSPE-H was 7.5-fold more active than rSMEZ-2 in the SJL strain.
The most consistently potent toxin on murine T cells was rSMEZ-2, with P50 values of 10 pg/ml in B10/J and 800 pg/ml in C3H. rSMEZ varied between 80 pg/ml in SJL and B10.M and 9,000 pg/ml in C3H. The P50 value for rSPE-H was between 15 pg/ml in SJL and 5,000 pg/ml in B10/J.
The human TCR Vβ specificity of the recombinant toxins was determined by multiprimer anchored PCR and dot-blot analysis using a panel of 17 human Vβ DNA regions. The Vβ enrichment after stimulation with toxin was compared with the Vβ profile of unstimulated PBLs (Table II). The sum total of all Vβs stimulated by rSMEZ, rSMEZ-2, and rSPE-G was close to 100%, suggesting that the Vβs used in the panel represent all the targeted Vβs. On the other hand, the total of the Vβs stimulated by rSPE-H was only 75%. It is therefore likely that rSPE-H also stimulated some less common Vβs, which are not represented in the panel. The most dramatic response was seen with all toxins, except rSMEZ-2, on Vβ2.1 bearing T cells (21-fold for rSMEZ, 45-fold for rSPE-G, and 22-fold for rSPE-H). In contrast, rSMEZ-2 gave only a 2.5-fold increase of Vβ2.1 T cells. SPE-G also targeted Vβ4.1, Vβ6.9, Vβ9.1, and Vβ12.3 (three- to fourfold). A moderate enrichment of Vβ12.6, Vβ9.1, and Vβ23.1 (four- to eightfold) was observed with rSPE-H. Both rSMEZ and rSMEZ-2 targeted Vβ4.1 and Vβ8.1 with similiar efficiency (three- to sevenfold). This finding is of particular interest, because Vβ8.1 activity had been found in some, but not all, S. pyogenes culture supernatants and in crude preparations of SPE-A and SPE-C. Moreover, SPE-B has often been claimed to have Vβ8 specific activity, but has since been shown to be a contaminant previously called SPE-X (35). The ability of rSMEZ and rSMEZ-2 to stimulate the Vβ8.1 Jurkat cell line was tested (Fig. 5). rSMEZ was less potent than the control toxin (rSEE), showing a half-maximal response of 0.2 ng/ml, compared with 0.08 ng/ml with rSEE, but rSMEZ-2 was more potent than rSEE (0.02 ng/ml). No proliferation activity was observed with the negative control toxin rSEA.
Table II.
Vβ Specificity of Recombinant Toxins on Human PBLs
| Percentage of Vβ enrichment | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vβ | Resting | SMEZ | SMEZ-2 | SPE-G | SPE-H | |||||
| 1.1 | 0.2 | 0.3 | 0.4 | 1.2 | 1 | |||||
| 2.1 | 0.4 | 8.4 | 1 | 17.9 | 8.6 | |||||
| 3.2 | 4.8 | 3.1 | 2.5 | 3 | 2.4 | |||||
| 4.1 | 3.5 | 24.8 | 14.4 | 11.2 | 5.2 | |||||
| 5.1 | 6.2 | 1.4 | 2.5 | 5.7 | 2.2 | |||||
| 5.3 | 5.6 | 2.2 | 4.1 | 4.7 | 4.1 | |||||
| 6.3 | 3 | 0.8 | 2.3 | 4.7 | 3.5 | |||||
| 6.4 | 5.4 | 2.1 | 5.9 | 9.6 | 5.6 | |||||
| 6.9 | 6.9 | 3.5 | 9.3 | 19.1 | 12.2 | |||||
| 7.3 | 3.5 | 15.3 | 7.3 | 3.2 | 12.6 | |||||
| 7.4 | 9 | 13.5 | 11.7 | 2.9 | 6.3 | |||||
| 8.1 | 8.7 | 20.7 | 36 | 4.5 | 2.4 | |||||
| 9.1 | 0.3 | 0.05 | 0 | 1.2 | 2.3 | |||||
| 12.3 | 0.8 | 1.6 | 2 | 3.2 | 2.6 | |||||
| 12.5 | 3 | 1.2 | 2 | 3 | 2.3 | |||||
| 15.1 | 0.6 | 0.5 | 0.7 | 1.2 | 0.8 | |||||
| 23.1 | 0.2 | 0.1 | 0.3 | 0.8 | 1 | |||||
| Total % | 62.1 | 99.7 | 102.8 | 97.1 | 75.2 | |||||
Human PBLs were incubated with 20 pg/ml of recombinant toxin for 4 d. Relative enrichment of Vβ cDNAs was analyzed from RNA of stimulated and resting PBLs by anchored primer PCR and reverse dot-blot to a panel of 17 different Vβ cDNAs (38). The figures represent the percentage of each Vβ with respect to total Cβ.
Figure 5.

Jurkat cell assay. Jurkat cells (bearing a Vβ8 TCR) and LG-2 cells were mixed with varying concentrations of recombinant toxin and incubated for 24 h, before SeI cells were added. After 1 d, 0.1 μCi [3H]thymidine was added and cells were counted after another 24 h. The Vβ8 targeting SEE was used as a positive control. The negative control was SEA. Both SMEZ and SMEZ-2 were potent stimulators of Jurkat cells, indicating their ability to specifically target Vβ8-bearing T cells. ○, unstimulated; ▴, rSEA; □, rSEE; ♦, rSMEZ; ▪, rSMEZ-2.
MHC Class II Binding.
To determine if there were significant structural differences, the protein structures of SMEZ-2, SPE-G, and SPE-H were modelled onto the superimposed structurally conserved regions of SEA, SEB, and SPE-C (Fig. 6). The models showed that in all three proteins, the two amino acid side chains of the COOH-terminal primary zinc binding motif are in close proximity to a third potential zinc ligand to build a zinc binding site, similar to the zinc binding site observed in SEA and SPE-C.
Figure 6.


Computer generated models of protein structures. The models were created on a Silicon Graphic computer using InsightII/Homology software. SEA, SEB, and SPE-C were used as reference proteins to determine structurally conserved regions. The loop regions were generated by random choice. MolScript software (39) was used for displaying the computer generated images. (a) All models show a potential zinc binding site within the β-grasp motif of the toxin structure. Two zinc binding residues are provided by a primary zinc motif (H-X-D) and the third ligand (H or D) comes from either the β9 or β10 strand. The amino acid residues of the β1-β2 loop that corresponds to the HLA-DRI α chain binding site in SEA and SEB are shown on the right side of the models. In all three protein models, this loop region is less hydrophobic than in SEA and SEB, suggesting the lack of the α chain binding site. A, Crystal structures of SPE-C (25); B, SMEZ-2; C, SPE-G; D, SPE-H. (b) SMEZ-2 model, showing the predicted location of the 17 residues that are different between SMEZ and SMEZ-2. The first residue is from SMEZ-2, followed by the position on the primary protein sequence and the corresponding residue on SMEZ.
The zinc binding residues in SPE-C are H167, H201, and D203, and it is thought that H81 from the HLA-DR1 β chain binds to the same zinc cation to form a regular tetrahedral complex. The two ligands of the primary zinc binding motif, H201 and D203, are located on the β12 strand, which is part of the β-grasp motif, a common structural domain of SAgs. The third ligand, H167, comes from the β10 strand (25).
In the model of SPE-G (Fig. 6 a, B) three potential zinc binding ligands (H167, H202, and D204) are located at corresponding positions. In the SMEZ-2 and the SPE-H models (Fig. 6 a, A and C), the two corresponding β12 residues are H202, D204 and H198, D200, respectively. The third ligand in SPE-H (D160) and SMEZ-2 (H162) comes from the β9 strand and is most similar to H187 in SEA. It has been shown from crystal structures that H167 of SPE-C and H187 of SEA are spatially and geometrically equivalent sites (20, 25).
All SAgs examined so far, except SPE-C, bind to a conserved motif in the MHC class II α1 domain. In SEB and TSST, hydrophobic residues on the loop between the β1 and β2 strands project into a hydrophobic depression in the MHC II α1 domain. This loop region has changed its character in SPE-C, where the hydrophobic residues (F44, L45, Y46, and F47 in SEB) are substituted by the less hydrophobic residues T33, T34, and H35. A comparison of this region on the computer-generated models revealed that the generic HLA-DR1 α chain binding site might also be missing. As the loop regions are generated by random choice, no conclusions can be made from their conformation in the models. However, in none of the three models does the β1-β2 loop have the required hydrophobic features observed in SEB and TSST (21, 24). The residues are I25, D26, F27, K28, T279 and S30 in SMEZ-2; T31, T32, N33, and S34 in SPE-G; and K28, N29, S30, P31, D32, I33, V34, and T35 in SPE-H.
SMEZ-2 differs from SMEZ in only 17 amino acids. Fig. 6 b shows the computer-generated model of SMEZ-2 with the position of those 17 residues. Most of the exchanges are located on loop regions, most significantly on the β5-β6 loop with five consecutive residues replaced. The potential zinc binding site and the β1-β2 loop are not affected by the replaced amino acids.
The TCR Vβ specificity differs between SMEZ and SMEZ-2 by one Vβ. SMEZ strongly stimulates Vβ2 T cells, but SMEZ-2 does not (Table II). One or more of the 17 exchanged residues in SMEZ/SMEZ-2 might therefore be directly involved in TCR binding. The exact position of the TCR binding site can not be predicted from the model as several regions have been implicated in TCR binding for different toxins. Crystal structures of SEC2 and SEC3, complexed with a TCR-β chain, indicated the direct role of several residues located on α2, the β2-β3 loop, the β4-β5 loop, and α4 (41). On the other hand, binding of TSST to the TCR involves residues from α4, the β7-β8 loop, and the α4-β9 loop (24). The SMEZ-2 model shows three residues, which might contribute to TCR binding. In SMEZ, Lys is exchanged for Glu at position 80 and Thr is exchanged for Ile at position 84, both on the β4-β5 loop. On the COOH-terminal end of the α4 helix, Ala is replaced by Ser at position 143.
The results from the computer-modeled protein structures suggest that all four toxins, SMEZ, SMEZ-2, SPE-G, and SPE-H, might bind to the HLA-DR1 β chain in a zinc dependent fashion, similar to SEA and SPE-C, but might not be able to interact with the HLA-DR1 α site, a situation that has previously only been observed with SPE-C (25, 29).
To find out whether or not zinc is required for binding of the toxins to MHC class II, a binding assay was performed using human LG-2 cells (which are MHC class II– expressing cells homozygous for HLA-DR1). Direct binding of 125I-labeled toxins was completely abolished in the presence of 1 mM EDTA (Fig. 7, Table III). When 2 mM ZnCl2 was added, binding to the LG-2 cells could be restored completely. These results show that the toxins bind in a zinc dependent mode, most likely to the HLA-DR1 β chain similarly to SEA and SPE-C. However, it does yet not exclude the possibility of an additional binding to the HLA-DR1 α chain.
Figure 7.

Zinc dependent binding of SMEZ-2 to LG-2 cells. LG-2 cells were incubated in duplicates with 1 ng of 125I-labeled rSMEZ-2 and increasing amounts of unlabeled toxin at 37°C for 1 h, and then the cells were washed and counted. ○, incubation in media; ▴, incubation in media plus 1 mM EDTA; □, incubation in media plus 1 mM EDTA, 2 mM ZnCl2.
Table III.
Binding Affinities and Zinc Dependencies for SAgs to Human Class II
| Toxin | MHC class II binding kd [nM] | zinc dependency | ||
|---|---|---|---|---|
| SEA | 36/1,000 | ++ | ||
| SEB | 340 | − | ||
| TSST | 130 | − | ||
| SPE-C | 70 | ++ | ||
| SMEZ | 65/1,000 | ++ | ||
| SMEZ-2 | 25/1,000 | ++ | ||
| SPE-G | 16/1,000 | ++ | ||
| SPE-H | 37/2,000 | ++ |
The binding affinities of the toxins to MHC class II were determined by Scatchard analysis using LG-2 cells. Zinc dependency was determined by binding of recombinant toxins to LG-2 cells in the presence and absence of EDTA, as described in the Materials and Methods section.
The biphasic binding of SEA to HLA-DR1 can be deduced from Scatchard analysis. It shows that SEA possesses a high affinity binding site of 36 nM (which is the zinc dependent β chain binding site) and a low affinity binding site of 1 μM (α chain binding site). On the other hand, only one binding site for HLA-DR1 was deduced from Scatchard analysis with SEB, TSST, and SPE-C, respectively (Table III). Therefore, Scatchard analysis was performed with radiolabeled rSMEZ, rSMEZ-2, rSPE-G, and rSPE-H using LG-2 cells. All four toxins showed multiphasic curves with at least two binding sites on LG-2 cells, a high affinity site of 15–65 nM, and a low affinity site of 1–2 μM (Fig. 8, Table III).
Figure 8.

Scatchard analysis of SMEZ-2 binding to LG-2 cells. 1 ng 125I-labeled rSMEZ-2 was incubated in duplicates with LG-2 cells and a twofold dilution series of cold toxin (10 μg to 10 pg). After 1 h, cells were washed and counted. Scatchard plots were performed as described elsewhere (40). ⋄, low affinity binding of SMEZ-2 to LG-2 cells; □, high affinity binding of SMEZ-2 to LG-2 cells.
In a further attempt to determine the orientation of the toxins on MHC class II, competition binding experiments were performed. The recombinant toxins and reference toxins (rSEA, rSEB, rSPE-C, and rTSST) were radiolabeled and tested with excess of unlabeled toxin for binding to LG-2 cells. The results are summarized in Fig. 10. Both rSEA and rSPE-C inhibited binding of labeled rSMEZ, rSMEZ-2, rSPE-G, and rSPE-H. However, rSPE-C only partially inhibited binding (50%) of the labeled rSMEZ-2 (Fig. 9). rSEB did not compete with any other toxin, even at the highest concentration tested. rTSST was only slightly competitive against 125I-labeled rSMEZ, rSMEZ-2 and rSPE-G, respectively, and did not inhibit rSPE-H binding at all.
Figure 10.

Summary of competitive binding experiments. Efficiency of each labeled toxin to compete with a 10,000-fold molar excess of any other unlabeled toxin for binding to LG-2 cells. White boxes, no competition; lightest gray boxes, 25% competition; medium gray boxes, 50% competition; dark gray boxes, 75% competition; black boxes, 100% competition. The results within the boxed area at the bottom right have been published elsewhere (29).
Figure 9.

Competition binding study with SMEZ-2. LG-2 cells were incubated in duplicates with 1 ng of 125I-labeled rSMEZ-2 and increasing amounts of unlabeled rSMEZ-2, rSEA, rSEB, rTSST, or rSPE-C. After 1 h cells were washed and counted. ○, rSMEZ-2; ▴, rSEA; □, rSEB; ┌, rTSST; ♦, rSPE-C.
Reciprocal competition experiments were performed. rSMEZ, rSMEZ-2, and rSPE-H prevented 125I-rSEA from binding to LG-2 cells. However, only partial competition (50%) was observed even at the highest toxin concentrations (10,000-fold molar excess). rSPE-G did not prevent binding of 125I-rSEA, and 125I-rTSST binding was only partially inhibited by rSMEZ, rSMEZ-2, and rSPE-H, but not by rSPE-G. Significantly, none of the toxins inhibited 125I-rSEB binding, even at the highest concentration tested.
In a further set of competition binding experiments, rSMEZ, rSMEZ-2, rSPE-G, and rSPE-H were tested for competition against each other. Both rSMEZ and rSMEZ-2 competed equally with each other and also prevented binding of labeled rSPE-G and rSPE-H. In contrast, rSPE-G and rSPE-H did not inhibit any other toxin binding, suggesting that these toxins had the most restricted subset of MHC class II molecules, which represent specific receptors.
Discussion
This paper reports the identification of four novel SAgs from S. pyogenes: SPE-G, SPE-H, SPE-J, and SMEZ-2. The spe-g, spe-h, and spe-j genes were found by screening the S. pyogenes M1 genomic database at the University of Oklahoma using short peptide sequences of highly conserved regions. An attempt to clone smez from strain 2035 led to the identification of another novel sag gene (smez-2) based on high sequence homology (95% DNA sequence identity). These SAgs represent the only streptococcal toxins thus far identified on the basis of their nucleotide sequence and not by analysis of purified proteins.
SMEZ-2, SPE-G, and SPE-J are most similar to SPE-C and the recently identified SMEZ (37). SPE-H is more closely related to the staphylococcal SAgs than to any streptococcal toxin. SMEZ-2 is the most potent SAg so far discovered with a half-maximal response to stimulate T cells approximately five times more active than that of SPE-C.
The function of these new SAgs provides an interesting insight into the nature of SAg activation and the variation that exists within this family of potent T cell mitogens. Perhaps the most striking feature is the high frequency with which Vβ2 and Vβ4 T cells are targeted. Out of the seven SAgs examined thus far, five target Vβ2 and three target Vβ4 as their principal T cell subgroup. The reason for this remains an intriguing mystery but may suggest that there is a specific role for Vβ2-bearing T cells in streptococcal induced immune defense. On the other hand, it might simply reflect the fact that the Vβ2 domain has features more amenable to ligation by this group of toxins. Only a careful analysis of the Vβ2–toxin interaction will resolve this issue.
The data represented in this paper suggest that SMEZ, SMEZ-2, SPE-G, and SPE-H all bind to the MHC class II β chain, but not to the α chain. Evidence that supports binding exclusively to the β chain are as follows. (a) Protein structure models predict a putative zinc binding site similar to SEA and SPE-C, but suggest that the β1-β2 hydrophobic loop in SEA and SEB is significantly less hydrophobic in all four new streptococcal toxins. (b) All toxins bind to LG-2 cells in a completely zinc dependent fashion. (c) Scatchard analysis revealed a high affinity binding site between 15 and 65 nM, similar to the HLA-DR1 β chain affinities of SEA and SPE-C. (d) All toxins compete with SEA and SPE-C for binding to HLA-DR1, but inhibit neither SEB nor TSST.
A putative zinc binding region was predicted for all four toxins, comprising the COOH-terminal primary zinc binding motif (H-X-D) on the β12 strand and a third ligand from the β9 strand (in SMEZ, SMEZ-2, and SPE-G) or the β10 strand (in SPE-G). The role for zinc in binding to MHC class II was confirmed by complete inhibition of cell surface binding by 1 mM EDTA. Moreover, calculated binding affinities were well in the range of the high affinity binding determined for SEA and SPE-C to the MHC class II β chain. Interestingly, the binding affinity for SPE-G (15 nM) is the highest so far determined for any toxin, yet rSPE-G is a weaker stimulator of human T cells than SEA, SPE-C, SMEZ, or SMEZ-2, indicating that affinity for MHC class II does not equate with potency towards T cells. A possible explanation is that although SPE-G binds tightly to MHC class II, it might have a much lower affinity for the TCR, or alternatively might assume an orientation on class II that is not optimal for TCR ligation. This possibility might indicate that the orientation of a toxin bound to MHC class II might be an important consideration in how well TCRs are ligated and signals are triggered.
Competitive binding studies provided evidence for the exclusive binding to the MHC class II β chain. Neither SEB nor TSST binding was inhibited by any of the new toxins. The ability to partially compete with TSST might best be explained by steric hindrance between the toxins across the top of the MHC class II molecule, rather than by direct competition for the α chain site. Exclusive MHC class II β chain binding would therefore predict strong inhibition of SPE-C, but this was not the case. Only SMEZ effectively inhibited SPE-C. The interpretation of this data is that SPE-C targets a broader range of MHC class II molecules compared with any of the new toxins, whereas SPE-G, which did not inhibit any other toxin, has a much more restricted repertoire of MHC class II molecules. From these data, a clear hierarchical order of binding can be shown: (SPE-C, SMEZ) > SMEZ-2 > SPE-H > SPE-G. The most likely explanation for the nature of these MHC class II subsets is that they are defined by the bound antigenic peptide. Indeed, peptide restriction has been reported for several other SAgs (42). For example, SEB and TSST both bind to the HLA-DR1 α chain, but do not cross-compete at all, because peptides restrict their binding to different HLA-DR1 subsets. Moreover, Thibodeau et al. (43) reported that only a fraction of available MHC class II molecules are actually able to bind a given SAg with high affinity. This might also explain the multiphasic Scatchard plots of SMEZ, SMEZ-2, SPE-G, and SPE-H binding when toxins have a variety of affinities ranging from nanomolar to micromolar defined by different peptides. Other possible explanations might be (a) binding of the toxins to a second, as yet undetermined class II site; (b) binding to different class II isotypes; and (c) binding to a non–class II receptor.
Another interesting finding is the complete lack of proliferation response when SPE-G and SPE-C were tested on murine T cells, although it has been shown that SPE-C binds to murine MHC class II (I-E) (29). Two possible mechanisms can be evoked to explain this feature. One possibility is that there is no equivalent to human Vβ2 TCR in mice. This seems unlikely, as all other Vβ2 specific toxins are potent stimulators of mouse T cells. The second possibility is that the orientation of SPE-C and SPE-G on mouse class II does not allow ligation of murine TCR molecules.
Both SMEZ and SMEZ-2 specifically stimulated human Vβ8 T cells. A Vβ8-specific activity had previously been detected in contaminated preparations of SPE-A and SPE-B and in crude extracts of certain S. pyogenes strains. The agent responsible for this specific activity remained elusive, but was believed to be another SAg, named SPE-X (35). SMEZ and SMEZ-2 are both potent stimulators of Vβ8 and thus are good candidates for the elusive SPE-X. Interestingly, Abe et al. (44) reported a selective expansion of T cells expressing Vβ2 and Vβ8 from patients with Kawasaki disease (KD), an acute multisystem vasculitis of young children most prevalent in Japan (45). Epidemiologic evidence strongly supports an infectious etiology for this disease. The causative agent for KD is unknown, but SAgs have been implicated in triggering the disease (44, 46). The Vβ2 and Vβ8 specificity of SMEZ correlates well with the selective T cell expansion in KD patients reported by Abe and coworkers (44). Further studies are underway to examine the frequency of these new SAgs and whether expression can be related to diseases such as KD.
The identification of the novel toxins increases the total number of known SAgs to eight and the confirmed bacterial SAg family to nineteen. However, individual streptococcal strains seem to carry only a limited number of SAg genes. A recent study by Cockerill et al. (47) examined the frequency of toxin genes in strains isolated from patients with invasive streptococcal disease. Only one strain carried three different toxins genes, spe-a, spe-c, and ssa. In this study, we show that strain 2035 lacks the smez and spe-h genes, whereas the M1 strain lacks the smez-2 gene. Variable sag gene frequencies among different strains is most likely due to horizontal gene transfer by mobile genetic elements such as phage or insertion element (12, 48). It is clear from this limited analysis of the four new sag genes that they too are subject to variable existence throughout the S. pyogenes strains. A larger screening of clinical isolates for these new sag genes is currently underway to determine which are the most common SAgs.
By expanding the family of SAgs, much stronger predictions about the general mechanism of SAg activation can be made. Despite the variation of SAgs, it appears that there is a significant preference for binding of certain Vβs, such as Vβ2 and Vβ4 and for exclusive binding of the MHC class II β chain. However, the actual binding to class II appears to be more complex and variable, with a clear hierarchy of binding, most likely due to peptide restriction. This complex binding pattern to MHC class II might in fact be very finely tuned and might have an impact on the fate of SAg-activated T cells in terms of apoptosis, anergy, and the development of autoimmune diseases. It may also represent the difference between host susceptibility and resistance to streptococcus mediated disease.
Acknowledgments
We thank Nick Fleming for technical assistance with DNA sequence analysis and Dr. Vic Arcus for his help with the MolScript computer software. We gratefully acknowledge the Streptococcal Genome Sequencing Project (funded by USPHS/NIH grant AI38406) and Bruce A. Roe, S.P. Linn, L. Song, X. Yuan, S. Clifton, M. McShan and J. Ferretti.
This study was supported by the Health Research Council of New Zealand.
Abbreviations used in this paper
- GST
glutathione-S-transferase
- KD
Kawasaki disease
- ORF
open reading frame
- SAg
superantigen
- SCR
structurally conserved region
- SE
staphylococcal enterotoxin
- SMEZ
streptococcal mitogenic exotoxin Z
- SPE
streptococcal pyrogenic exotoxin
- SSA
streptococcal SAg
- TSST
toxic shock syndrome toxin
References
- 1.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. 1990;248:705–711. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- 2.Huber BT, Hsu PN, Sutkowski N. Virus-encoded superantigens. Microbiol Rev. 1996;60:473–482. doi: 10.1128/mr.60.3.473-482.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alouf, J.E., H. Knoell, and W. Koehler. 1991. The family of mitogenic, shock-inducing and superantigenic toxins from staphylococci and streptococci. In Sourcebook of Bacterial Protein Toxins. J.E. Alouf and J.H. Freer, editors. Academic Press, San Diego. 367–414.
- 4.Betley MJ, Borst DW, Regassa LB. Staphylococcal enterotoxins, toxic shock syndrome toxin and streptococcal exotoxins: a comparative study of their molecular biology. Chem Immunol. 1992;55:1–35. [PubMed] [Google Scholar]
- 5.Ren K, Bannan JD, Pancholi V, Cheung AL, Robbins JC, Fischetti VA, Zabriskie JB. Characterization and biological properties of a new staphylococcal exotoxin. J Exp Med. 1994;180:1675–1683. doi: 10.1084/jem.180.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munson SH, Tremaine MT, Betley MJ, Welch RA. Identification and characterization of staphylococcal enterotoxin types G and I from Staphylococcus aureus. . Infect Immun. 1998;66:3337–3348. doi: 10.1128/iai.66.7.3337-3348.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herman A, Kappler JW, Marrack P, Pullen AM. Superantigens: mechanism of T-cell stimulation and role in immune responses. Annu Rev Immunol. 1991;9:745–772. doi: 10.1146/annurev.iy.09.040191.003525. [DOI] [PubMed] [Google Scholar]
- 8.Janeway CA, Jr, Yagi J, Conrad PJ, Katz ME, Jones B, Vroegop S, Buxser S. T-cell responses to Mls and to bacterial proteins that mimic its behavior. Immunol Rev. 1989;107:61–68. doi: 10.1111/j.1600-065x.1989.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 9.Fast DJ, Schlievert PM, Nelson RD. Toxic shock syndrome-associated staphylococcal and streptococcal pyrogenic toxins are potent inducers of tumor necrosis factor production. Infect Immun. 1989;57:291–294. doi: 10.1128/iai.57.1.291-294.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotzin BL, Leung DY, Kappler J, Marrack P. Superantigens and their potential role in human disease. Adv Immunol. 1993;54:99–166. doi: 10.1016/s0065-2776(08)60534-9. [DOI] [PubMed] [Google Scholar]
- 11.Bohach GA, Fast DJ, Nelson RD, Schlievert PM. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit Rev Microbiol. 1990;17:251–272. doi: 10.3109/10408419009105728. [DOI] [PubMed] [Google Scholar]
- 12.Weeks CR, Ferretti JJ. Nucleotide sequence of the type A streptococcal exotoxin (erythrogenic toxin) gene from Streptococcus pyogenesbacteriophage T12. Infect Immun. 1986;52:144–150. doi: 10.1128/iai.52.1.144-150.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goshorn SC, Bohach GA, Schlievert PM. Cloning and characterization of the gene, speC, for pyrogenic exotoxin type C from . Streptococcus pyogenes Mol Gen Genet. 1988;212:66–70. doi: 10.1007/BF00322445. [DOI] [PubMed] [Google Scholar]
- 14.Mollick JA, Miller GG, Musser JM, Cook RG, Grossman D, Rich RR. A novel superantigen isolated from pathogenic strains of Streptococcus pyogeneswith aminoterminal homology to staphylococcal enterotoxins B and C. J Clin Invest. 1993;92:710–719. doi: 10.1172/JCI116641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Den Busche RA, Lyon JD, Bohach GA. Molecular evolution of the staphylococcal and streptococcal pyrogenic toxin gene family. Mol Phylogenet Evol. 1993;2:281–292. doi: 10.1006/mpev.1993.1027. [DOI] [PubMed] [Google Scholar]
- 16.Dellabona P, Peccoud J, Kappler J, Marrack P, Benoist C, Mathis D. Superantigens interact with MHC class II molecules outside of the antigen groove. Cell. 1990;62:1115–1121. doi: 10.1016/0092-8674(90)90388-u. [DOI] [PubMed] [Google Scholar]
- 17.Fraser JD. High-affinity binding of staphylococcal enterotoxins A and B to HLA-DR. Nature. 1989;339:221–223. doi: 10.1038/339221a0. [DOI] [PubMed] [Google Scholar]
- 18.Fleischer B, Schrezenmeier H. T cell stimulation by staphylococcal enterotoxins. Clonally variable response and requirement for major histocompatibility complex class II molecules on accessory or target cells. J Exp Med. 1988;167:1697–1707. doi: 10.1084/jem.167.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mollick JA, Cook RG, Rich RR. Class II MHC molecules are specific receptors for staphylococcus enterotoxin A. Science. 1989;244:817–820. doi: 10.1126/science.2658055. [DOI] [PubMed] [Google Scholar]
- 20.Schad EM, Zaitseva I, Zaitsev VN, Dohlsten M, Kalland T, Schlievert PM, Ohlendorf DH, Svensson LA. Crystal structure of the superantigen staphylococcal enterotoxin type A. EMBO (Eur Mol Biol Organ) J. 1995;14:3292–3301. doi: 10.1002/j.1460-2075.1995.tb07336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swaminathan S, Furey W, Pletcher J, Sax M. Crystal structure of staphylococcal enterotoxin B, a superantigen. Nature. 1992;359:801–806. doi: 10.1038/359801a0. [DOI] [PubMed] [Google Scholar]
- 22.Papageorgiou AC, Acharya KR, Shapiro R, Passalacqua EF, Brehm RD, Tranter HS. Crystal structure of the superantigen enterotoxin C2 from Staphylococcus aureusreveals a zinc-binding site. Structure. 1995;3:769–779. doi: 10.1016/s0969-2126(01)00212-x. [DOI] [PubMed] [Google Scholar]
- 23.Sundstrom M, Abrahmsen L, Antonsson P, Mehindate K, Mourad W, Dohlsten M. The crystal structure of staphylococcal enterotoxin type D reveals Zn2+-mediated homodimerization. EMBO (Eur Mol Biol Organ) J. 1996;15:6832–6840. [PMC free article] [PubMed] [Google Scholar]
- 24.Acharya KR, Passalacqua EF, Jones EY, Harlos K, Stuart DI, Brehm RD, Tranter HS. Structural basis of superantigen action inferred from crystal structure of toxic-shock syndrome toxin-1. Nature. 1994;367:94–97. doi: 10.1038/367094a0. [DOI] [PubMed] [Google Scholar]
- 25.Roussel A, Anderson BF, Baker HM, Fraser JD, Baker EN. Crystal structure of the streptococcal superantigen SPE-C: dimerization and zinc binding suggest a novel mode of interaction with MHC class II molecules. Nat Struct Biol. 1997;4:635–643. doi: 10.1038/nsb0897-635. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Urban RG, Strominger JL, Wiley DC. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science. 1994;266:1870–1874. doi: 10.1126/science.7997880. [DOI] [PubMed] [Google Scholar]
- 27.Hurley JM, Shimonkevitz R, Hanagan A, Enney K, Boen E, Malmstrom S, Kotzin BL, Matsumura M. Identification of class II major histocompatibility complex and T cell receptor binding sites in the superantigen toxic shock syndrome toxin 1. J Exp Med. 1995;181:2229–2235. doi: 10.1084/jem.181.6.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seth A, Stern LJ, Ottenhoff TH, Engel I, Owen MJ, Lamb JR, Klausner RD, Wiley DC. Binary and ternary complexes between T-cell receptor, class II MHC and superantigen in vitro. Nature. 1994;369:324–327. doi: 10.1038/369324a0. [DOI] [PubMed] [Google Scholar]
- 29.Li PL, Tiedemann RE, Moffat SL, Fraser JD. The superantigen streptococcal pyrogenic exotoxin C (SPE-C) exhibits a novel mode of action. J Exp Med. 1997;186:375–383. doi: 10.1084/jem.186.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson KR, Tiedemann RE, Urban RG, Lowe SC, Strominger JL, Fraser JD. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J Exp Med. 1995;182:711–720. doi: 10.1084/jem.182.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozono H, Parker D, White J, Marrack P, Kappler J. Multiple binding sites for bacterial superantigens on soluble class II MHC molecules. Immunity. 1995;3:187–196. doi: 10.1016/1074-7613(95)90088-8. [DOI] [PubMed] [Google Scholar]
- 32.Tiedemann RE, Fraser JD. Cross-linking of MHC class II molecules by staphylococcal enterotoxin A is essential for antigen-presenting cell and T cell activation. J Immunol. 1996;157:3958–3966. [PubMed] [Google Scholar]
- 33.Braun MA, Gerlach D, Hartwig UF, Ozegowski JH, Romagne F, Carrel S, Kohler W, Fleischer B. Stimulation of human T cells by streptococcal “superantigen” erythrogenic toxins (scarlet fever toxins) J Immunol. 1993;150:2457–2466. [PubMed] [Google Scholar]
- 34.Kline JB, Collins CM. Analysis of the interaction between the bacterial superantigen streptococcal pyrogenic exotoxin A (SpeA) and the human T-cell receptor. Mol Microbiol. 1997;24:191–202. doi: 10.1046/j.1365-2958.1997.3381696.x. [DOI] [PubMed] [Google Scholar]
- 35.Fleischer B, Necker A, Leget C, Malissen B, Romagne F. Reactivity of mouse T-cell hybridomas expressing human Vβ gene segments with staphylococcal and streptococcal superantigens. Infect Immun. 1996;64:987–994. doi: 10.1128/iai.64.3.987-994.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toyosaki T, Yoshioka T, Tsuruta Y, Yutsudo T, Iwasaki M, Suzuki R. Definition of the mitogenic factor (MF) as a novel streptococcal superantigen that is different from streptococcal pyrogenic exotoxins A, B, and C. Eur J Immunol. 1996;26:2693–2701. doi: 10.1002/eji.1830261122. [DOI] [PubMed] [Google Scholar]
- 37.Kamezawa Y, Nakahara T, Nakano S, Abe Y, Nozaki-Renard J, Isono T. Streptococcal mitogenic exotoxin Z, a novel acidic superantigenic toxin produced by a T1 strain of . Streptococcus pyogenes Infect Immun. 1997;65:3828–3833. doi: 10.1128/iai.65.9.3828-3833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudson KR, Robinson H, Fraser JD. Two adjacent residues in Staphylococcal enterotoxins A and E determine T cell receptor Vβ specificity. J Exp Med. 1993;177:175–185. doi: 10.1084/jem.177.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallography. 1991;24:946–950. [Google Scholar]
- 40.Cunningham BC, Jhurani P, Ng P, Wells JA. Receptor and antibody epitopes in human growth hormone identified by homologue scanning mutagenesis. Science. 1989;243:1330–1336. doi: 10.1126/science.2466339. [DOI] [PubMed] [Google Scholar]
- 41.Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Mariuzza RA. Crystal structure of a T-cell receptor beta-chain complexed with a superantigen. Nature. 1996;384:188–192. doi: 10.1038/384188a0. [DOI] [PubMed] [Google Scholar]
- 42.Wen R, Cole GA, Surman S, Blackman MA, Woodland DL. Major histocompatibility complex class II– associated peptides control the presentation of bacterial superantigens to T cells. J Exp Med. 1996;183:1083–1092. doi: 10.1084/jem.183.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thibodeau J, Cloutier I, Lavoie PM, Labrecque N, Mourad W, Jardetzky T, Sekaly RP. Subsets of HLA-DR1 molecules defined by SEB and TSST-1 binding. Science. 1994;266:1874–1878. doi: 10.1126/science.7997881. [DOI] [PubMed] [Google Scholar]
- 44.Abe J, Kotzin BL, Jujo K, Melish ME, Glode MP, Kohsaka T, Leung DY. Selective expansion of T cells expressing T-cell receptor variable regions V beta 2 and V beta 8 in Kawasaki disease. Proc Natl Acad Sci USA. 1992;89:4066–4070. doi: 10.1073/pnas.89.9.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Jpn J Allergol. 1967;16:178. [PubMed] [Google Scholar]
- 46.Leung DY, Giorno RC, Kazemi LV, Flynn PA, Busse JB. Evidence for superantigen involvement in cardiovascular injury due to Kawasaki syndrome. J Immunol. 1995;155:5018–5021. [PubMed] [Google Scholar]
- 47.Cockerill FR, Thompson RL, Musser JM, Schlievert PM, Talbot J, Holley KE, Harmsen WS, Ilstrup DM, Kohner PC, Kim MH, et al. Molecular, serological, and clinical features of 16 consecutive cases of invasive streptococcal disease. Clin Infect Dis. 1998;26:1448–1458. doi: 10.1086/516376. [DOI] [PubMed] [Google Scholar]
- 48.Kapur V, Reda KB, Li LL, Ho LJ, Rich RR, Musser JM. Characterization and distribution of insertion sequence IS1239 in . Streptococcus pyogenes Gene. 1994;150:135–140. doi: 10.1016/0378-1119(94)90872-9. [DOI] [PubMed] [Google Scholar]