

Abstract
Autoantibodies and immune complexes are major pathogenic factors in autoimmune injury, responsible for initiation of the inflammatory cascade and its resulting tissue damage. This activation results from the interaction of immunoglobulin (Ig)G Fc receptors containing an activation motif (ITAM) with immune complexes (ICs) and cytotoxic autoantibodies which initiates and propagates an inflammatory response. In vitro, this pathway can be interrupted by coligation to FcγRIIB, an IgG Fc receptor containing an inhibitory motif (ITIM). In this report, we describe the in vivo consequences of FcγRII deficiency in the inflammatory response using a mouse model of IC alveolitis. At subthreshold concentrations of ICs that fail to elicit inflammatory responses in wild-type mice, FcγRII-deficient mice developed robust inflammatory responses characterized by increased hemorrhage, edema, and neutrophil infiltration. Bronchoalveolar fluids from FcγRII−/− stimulated mice contain higher levels of tumor necrosis factor and chemotactic activity, suggesting that FcγRII deficiency lowers the threshold of IC stimulation of resident cells such as the alveolar macrophage. In contrast, complement- and complement receptor–deficient mice develop normal inflammatory responses to suprathreshold levels of ICs, while FcRγ−/− mice are completely protected from inflammatory injury. An inhibitory role for FcγRII on macrophages is demonstrated by analysis of FcγRII−/− macrophages which show greater phagocytic and calcium flux responses upon FcγRIII engagement. These data reveal contrasting roles for the cellular receptors for IgG on inflammatory cells, providing a regulatory mechanism for setting thresholds for IC sensitivity based on the ratio of ITIM to ITAM FcγR expression. Exploiting the FcγRII inhibitory pathway could thus provide a new therapeutic approach for modulating antibody-triggered inflammation.
Keywords: Fc receptor, complement, immune complex, chemokine, cytokine
Immune complex (IC)1-induced inflammatory responses are significant contributors to the pathogenic mechanisms responsible for tissue destruction in a number of autoimmune conditions. The initiation of this inflammatory response by IgG-containing ICs may be triggered by either of two pathways: the soluble proteins of the complement system, or the cellular receptors for IgG, the Fc receptors (1). The binding of the complement component C1q to the IC leads to a cascade of proteolytic activation steps including the subsequent binding and proteolytic activation of C4 and C2 to form the C4bC2b complex or C3 convertase. The critical step of proteolytic conversion of C3 to C3b may lead subsequently to the activation of the late complement components C5–9 and the generation of several proinflammatory molecules, including the lytic membrane attack complex, the chemoattractants C5a and C3a, and independently lead to the interaction with the cellular receptors for C3 (2). Since these highly inflammatory molecules are found activated in inflammatory states, a direct causal relationship between complement activation and IC injury has often been assumed. However, the study of IC injury in mouse strains deficient in complement components and in the cellular receptors for IgG has allowed a systematic evaluation of these individual inflammatory pathways. Studies in FcR-deficient or complement-deficient mice have demonstrated the dominant role of the FcR pathway in initiating autoimmune injury. Thus, IC-induced inflammation in the skin, as modeled by the Arthus reaction, is dependent on FcR and not complement activation (3, 4). Similarly, the activation of FcRs is required for IC-triggered glomerulonephritis in a murine model of systemic lupus (5). Taken together, these data argue for a prominent role for FcR engagement rather than complement in IgG-triggered systemic autoimmunity in a wide variety of tissue sites.
In the mouse, IgG-containing ICs may interact with one of three cellular IgG receptors, two of which, FcγRI and FcγRIII, lead to cellular activation and are multimeric receptors containing both a ligand-binding subunit and the associated signaling subunit, the immunoreceptor tyrosine-based activation motif (ITAM)-containing γ chain (6–11). The third receptor for IgG, FcγRII, is a single subunit receptor, and its immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing cytoplasmic domain (12–14) inhibits ITAM-mediated cellular activation signals through the recruitment of the inositol phosphatase SHIP (15). Coligation of FcγRII and an ITAM-containing receptor leads to aborted FcεRI and FcγRIII-triggered activation in mast cells (16, 17) and B cell receptor–triggered activation in B cells (18–20). FcγRII is widely expressed on all hematopoietic lineage cells, including monocytes and granulocytes, raising the possibility that this receptor may function to modulate IC-triggered inflammatory responses in vivo by determining thresholds for activation of inflammatory cells. The present study was undertaken to investigate this possibility in an in vivo murine model of IC-triggered alveolitis. In this passive model of acute IC disease, the antigen OVA is injected intravenously followed by the delivery of specific anti-OVA IgG to the airways by intratracheal administration. ICs are formed in situ in the lung and incite an acute inflammatory response within 4 h. The resident alveolar macrophage is the dominant cell type present in the alveolar spaces and is therefore a likely contributor to the initiation of the IC-triggered inflammatory cascade, elaborating a number of cytokines, including IL-1 (21–23), TNF-α (24, 25), IL-6 (26), and platelet-activating factor (PAF [23, 27]), that act in both a paracrine and autocrine manner. The production of chemokines, including macrophage-inflammatory protein (MIP)-1 (28), MIP-2, cytokine-induced neutrophil chemoattractant 1 (CINC-1 [29]), IL-8 (30), and C5a (31–33), result in the recruitment of other inflammatory cell types, predominantly the neutrophil, to the lung (for a review, see reference 34). Production of vasoactive substances and vascular injury (35) result in both hemorrhage and edema. This response is highlighted histologically by hemorrhage, neutrophil infiltration, and perivascular edema.
FcγRII-deficient mice develop augmented inflammatory responses in IC alveolitis with increased hemorrhage, edema, and neutrophil infiltration, whereas γ chain–deficient mice are protected. C3-deficient and C5aR–deficient mice reveal wild-type responses to IC-induced inflammation. The cell type likely to play a major role in the development of alveolitis, the alveolar macrophage, was shown to have enhanced phagocytic capacity and enhanced calcium influx in response to ICs when lacking FcγRII. These results demonstrate the roles of the two opposing FcγR systems in the inflammatory response and suggest alternative immunotherapeutic approaches in the treatment of IC disease.
Materials and Methods
Mice.
Wild-type, γ−/−, and FcRII−/− mice were on a BALB/c background. The C3−/− mice were a gift of Mike Carroll (Harvard University School of Medicine, Boston, MA), and the C5aR−/− mice were a gift of Craig Gerard (Children's Hospital, Boston, MA). All of the mice were bred and housed at Taconic Farms and used at the age of 6–12 wk.
Lung Reverse Passive Arthus Reaction.
The reverse passive Arthus reaction was initiated by the intravenous injection of the tail vein with OVA (Sigma Chemical Co.) 20 μg/g in 200 μl PBS solution. The mice were anesthetized by inhalation of Metofane and subsequently injected intratracheally with 60 μl of PBS containing either 30 or 300 μg of rabbit anti–chicken egg albumin IgG (ICN Pharmaceuticals, Inc.) or PBS alone. After 4 h the mice were killed, and either a bronchoalveolar lavage (BAL) was performed with 1 ml of PBS three times per mouse, or the lungs were removed for formalin fixation or myeloperoxidase (MPO) assay.
Quantitation of Edema and Hemorrhage.
Edema was assessed by a vasopermeability index in mice that received 200 μl of 2% Evan's blue dye (Sigma Chemical Co.) in PBS intravenously. The vasopermeability index is the ratio of the OD630 of the pooled 3 ml BAL to a 1:20 dilution of serum, and represents the extravasation of protein-bound Evan's blue dye into the bronchoalveolar space.
The amount of alveolar hemorrhage was calculated by multiplying the total cell count by the percentage of RBCs determined by manual differentials of Wright-Giemsa–stained cytospin BAL samples.
Quantitation of Neutrophil Infiltration.
Total lung neutrophil activity was assessed by MPO assay. After three freeze–thaw cycles, lungs were homogenized (Polytron homogenizer; Tekmar Co.) four times for 10 s at a setting of 4, in 10 vol of ice-cold buffer (50 mM potassium phosphate, pH 7.4). After centrifugation at 25,000 g for 20 min, the pellet was resuspended in 10 vol of lysis buffer (50 mM potassium phosphate, pH 6.0, 0.5% HTAB, 10 mM EDTA), homogenized, freeze–thawed, and sonicated for 15 s. 50 μl lysate samples were added to 450 μl of reaction buffer (50 mM potassium phosphate, pH 6.0, containing 0.167 mg/ml O-dianisidine dihydrochloride and 0.005% H2O2). MPO activity in supernatants was assayed by measuring the change (per minute) in absorbance at 460 nm resulting from decomposition of H2O2.
TNF Assay.
Levels of soluble murine TNF-α were measured, in duplicate, in appropriately diluted samples of BAL. The TNF-α concentration was determined with a mouse TNF-α ELISA kit as described in the manufacturer's instructions (Genzyme Corp.) and compared with a standard curve.
Cell Preparation and Chemotaxis Assay.
BALB/c wild-type mouse bone marrow 0.75 × 106 cells in 100 μl RPMI 1640 medium, 0.25% human serum albumin (HSA) were added to the top chamber of a 6.5-mm diameter, 3-μm pore polycarbonate Transwell culture insert (Costar Corp.) and incubated for 1.5 h with either RPMI/0.25% HSA or BAL diluted 1:2 in RPMI/ 0.25% HSA. The neutrophils that transmigrated into the lower chamber were vigorously suspended and counted with a FACScan® (Becton Dickinson) for 20 s at 60 μl/min with gating on forward and side scatter.
Phagocytosis Assays.
Adherent peritoneal macrophages were obtained from mice after flushing the abdominal cavity with ice-cold PBS/1% BSA. IgG2b-opsonized SRBCs were prepared as described previously (11). In brief, TNP-derivitized SRBCs were extensively washed and incubated with subagglutinating quantities of culture supernatants of the IgG2b producing anti-DNP hybridoma U12.5 (a gift of Jay Unkeless, Mount Sinai Medical Center, New York). After washing away free antibody, IgG2b-opsonized RBCs were added to adherent macrophages for 1 h at 37°C. Unphagocytosed RBCs were removed by osmotic lysis, and phagocytosis plates were fixed with PBS/0.25% glutaraldehyde before microscopic examination.
Calcium Flux Measurements.
Peritoneal macrophages were suspended at a concentration of 2 × 106 cells/ml in DMEM with 10% HI-FCS, 2 μM fura-2/AM (Molecular Probes, Inc.) for 30 min at 35°C. Fura-2/AM–loaded cells were incubated with mAb 2.4G2 (5 μg/ml) for 30 min at 4°C. Cells were stimulated with goat anti–rat IgG mAb (Cappel) at 25 ng/ml. Relative fluorescence was monitored in a Perkin-Elmer fluorimeter.
Results
Previous studies on the FcR dependence and complement independence of IC-triggered inflammation were based on studies using models involving the skin and kidney (3–5, 36, 37). It has been suggested that IC-induced inflammation in the lung may display a greater dependence on complement activation pathways than other anatomic sites (32, 38). To delineate the relative contributions of complement- and FcR-triggered activation pathways in IC alveolitis, the inflammatory reactions of wild-type, C3- deficient, and C5aR-deficient mice were compared. Mice were killed 4 h after challenge, and lungs were processed for either histological or quantitative assessment of inflammation. Quantitative analysis included RBC counts of BAL fluid for an index of hemorrhage; measurement of Evan's blue dye extravasation into bronchoalveolar fluid for edema evaluation; and MPO assay of whole lung tissue as an indicator of neutrophil infiltration. Wild-type, C3−/−, C5aR−/−, and FcR γ−/− mice challenged with 300 μg rabbit anti-OVA IgG alone had little or no inflammatory reaction observable histologically or by quantitative assessment of hemorrhage, edema, or neutrophil infiltration (see Fig. 2, and data not shown). Wild-type mice treated with both intravenous OVA and 300 μg of anti-OVA to induce in situ formation of ICs developed robust inflammatory responses characterized by alveolar hemorrhage, neutrophil infiltration, and perivascular edema (Figs. 1 and 2). Contrary to the conclusion of a prior report that C5aR deficiency significantly attenuated inflammation in this model (32, 38), we observed minimally reduced inflammatory responses in IC-treated complement-deficient animals both qualitatively and quantitatively (Figs. 1 and 2). Histological analysis revealed that complement-deficient mice exhibited minimally attenuated hemorrhage with somewhat fewer RBCs and PMNs seen in BAL specimens (Fig. 1). Quantitative assessment revealed lowered edematous responses, but no statistically significant differences in BAL RBC counts or total lung MPO activity were detectable between wild-type and complement-deficient animals (Fig. 2). In contrast to the lack of protection in C3- and C5aR-deficient mice, protection in FcR γ−/− mice was complete. Alveolar hemorrhage is easily seen in wild-type, C3−/−, and C5aR−/− mice but not in γ−/− mice (Fig. 1 A, top panels). BAL fluids (Fig. 1, bottom panels) reveal that the airways of wild-type, C3−/−, and C5aR−/− mice are filled with RBCs and recruited neutrophils, but that the airways of γ−/− mice show little signs of inflammation and BAL specimens contain resident alveolar macrophages and few RBCs and few neutrophils. These data demonstrate that IC-triggered inflammation in the lung is FcR dependent, as has been observed in the skin and kidney, and establish the general importance of the FcR γ activation pathway, as opposed to complement, in IC injury regardless of tissue site.
Figure 2.
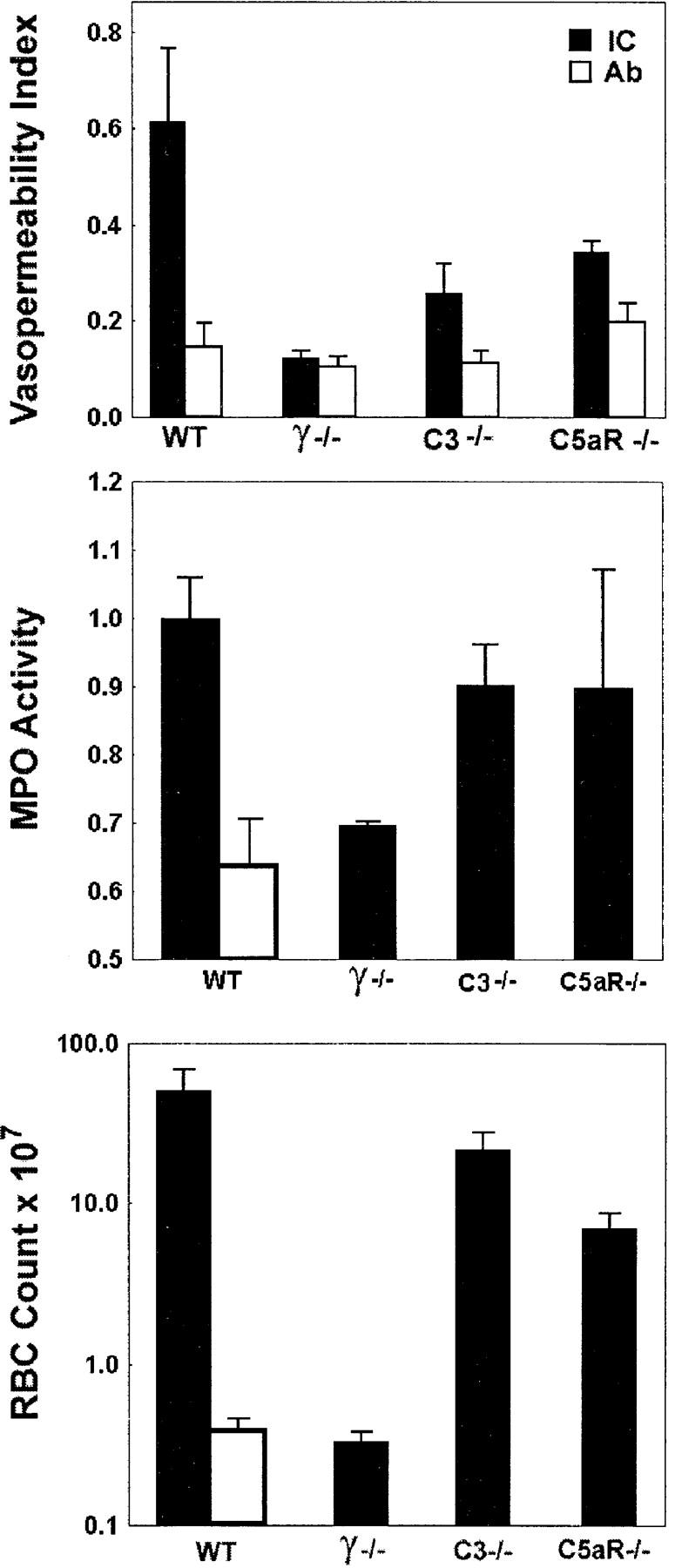
IC alveolitis: quantitative assessment of edema, neutrophilic infiltration, and hemorrhage in wild-type (WT), γ−/−, C3−/−, and C5aR−/− mice. (Top) Vasopermeability index, as assessed by Evan's blue dye extravasation into the bronchoalveolar space. (Middle) Neutrophilic infiltration, as assessed by neutrophil-specific MPO assay. MPO activity in lung lysates was measured at OD450. (Bottom) Alveolar hemorrhage, as measured by total RBC counts of BAL. Mice received OVA (IC, black bars) or PBS (Ab, white bars) intravenously, followed by 300 μg anti-OVA intratracheally. Experimental groups included three to seven mice per group. PBS controls were not performed for all genotypes. Data are presented as mean ± SEM. The P values for edematous, neutrophilic, and hemorrhagic responses were as follows: 0.02, 0.01, and 0.03, γ−/− vs. wild-type; 0.03, 0.08, and 0.05, C3−/− vs. wild-type; and 0.08, 0.45, and 0.09, C5aR−/− vs. wild-type, respectively. Statistical analysis was performed with an unpaired two-tailed Student's t test.
Figure 1.

IC alveolitis in wild-type (WT), γ−/−, C3−/−, and C5aR−/− mice. (Top panels) Hematoxylin and eosin– stained formalin-fixed lung sections (original magnification: ×200). (Bottom panels) Wright-Giemsa stains of cytospin preparations of bronchoalveolar fluid samples. Mice received OVA intravenously, followed by 300 μg anti-OVA intratracheally.
Since FcγRII is able to inhibit activation responses generated by the γ chain–associated receptors, FcγRI and III in vitro, we next set out to determine the in vivo relevance of this inhibitory pathway in IC-triggered inflammation. To evaluate the role of FcγRII in the acute inflammatory response in this FcR-dependent model, wild-type and FcγRII−/− mice were challenged with a low dose of polyclonal anti-OVA rabbit IgG (30 μg) either in the presence or absence of intravenously injected OVA. Both wild-type and FcγRII−/− mice challenged with rabbit anti-OVA IgG alone had little or no inflammatory reaction observable histologically (data not shown) or by quantitative assessment of hemorrhage, edema, or neutrophil infiltration. Similarly, at these low doses of antibody, wild-type mice challenged with both OVA and rabbit anti-OVA IgG had little observable inflammatory responses (Fig. 3, left). However, the inflammatory response of FcγRII−/− mice was enhanced, with increased responses of all three parameters of inflammation (Fig. 3, right; and Fig. 4). Histological changes noted in FcγRII−/− mice included enhanced alveolar hemorrhage, increased interstitial neutrophil infiltration, and perivascular edema. These results indicate that FcγRII functions to set the threshold level for IC-triggered inflammation, which is relieved upon deletion of this gene.
Figure 3.
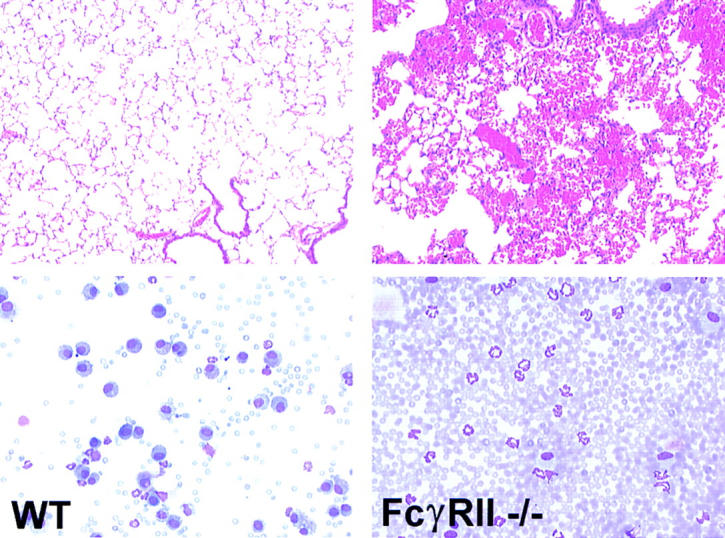
IC alveolitis in wild-type (WT) and FcγRII−/− mice. (Top panels) Hematoxylin and eosin–stained formalin-fixed lung sections (original magnification: ×200). (Bottom panels) Wright-Giemsa stains of cytospin preparations of bronchoalveolar fluid samples. Mice received OVA intravenously, followed by 30 μg anti-OVA intratracheally.
Figure 4.
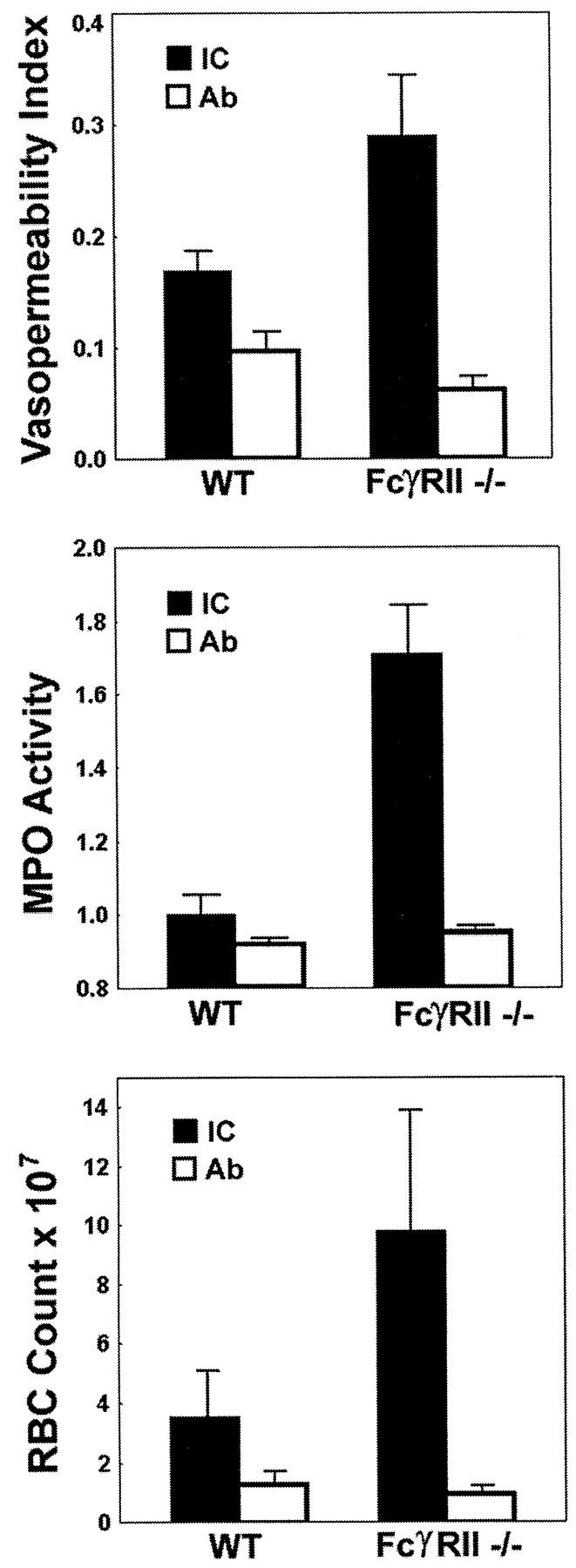
IC alveolitis: quantitative assessment of edema, neutrophilic infiltration, and hemorrhage in wild-type (WT) and FcγRII−/− mice. (Top) Vasopermeability index, as assessed by dye extravasation into the bronchoalveolar space. (Middle) Neutrophilic infiltration, as assessed by neutrophil-specific MPO assay. MPO activity in lung lysates was measured at OD450. (Bottom) Alveolar hemorrhage, as measured by total RBC counts of BAL. Mice received OVA (IC, black bars) or PBS (Ab, white bars) intravenously, followed by 30 μg anti-OVA intratracheally. Experimental groups included four to seven mice per group. Data are presented as mean ± SEM. The P values for IC-stimulated edematous, neutrophilic, and hemorrhagic responses were 0.03, 0.05, and 0.002, FcγRII−/− vs. wild-type, respectively. Statistical analysis was performed with an unpaired two-tailed Student's t test.
The enhanced infiltration of neutrophils seen in IC-stimulated lungs of FcγRII−/− mice suggested that the local production of recruitment factors that include inflammatory cytokines and chemokines might be enhanced in the lungs of FcγRII−/− mice. Indeed, the level of TNF-α production in BAL fluid is enhanced twofold in FcγRII−/− mice (Fig. 5 A). Furthermore, FcγRII−/− BAL fluid contains more than twofold increased neutrophil chemotactic activity compared with control BAL fluid from wild-type stimulated lungs (Fig. 5 B). On the other hand, FcγRII−/−, γ−/−, and wild-type neutrophils show equivalent migratory responses when exposed to chemokine-containing BAL fluid obtained from IC-stimulated wild-type mice (not shown). These data are consistent with the hypothesis that the enhanced neutrophil infiltrative responses seen in FcγRII−/− mice is the result of a lower threshold of IC-triggered activation of FcγR-bearing resident cells, including the alveolar macrophage. This activation of resident cells leads to the increased local production of secondary mediators of inflammation, including TNF-α and chemokines, leading to enhanced recruitment of circulating neutrophils.
Figure 5.

Enhanced cytokine and chemokine production in IC-stimulated FcγRII−/− mice. (A) TNF-α production in wild-type (WT) and FcγRII−/− mice. ELISA measurements of BAL aliquots. (B) Chemotactic activity of wild-type and FcγRII−/− BAL. Migration of wild-type neutrophils elicited by dilutions of BAL fluids. Chemotactic responses are expressed as the percentage of the total neutrophils loaded into the upper chamber which migrated into the lower BAL fluid–containing chamber. Mice received OVA (black bars) or PBS (white bars) intravenously, followed by 30 μg anti-OVA intratracheally. Experimental groups included four to seven mice per group. Data are presented as mean ± SEM. The P values for IC-stimulated TNF and chemokine production responses were 0.02 and 0.005, FcγRII−/− vs. wild-type, respectively. Statistical analysis was performed with an unpaired two-tailed Student's t test.
In the unstimulated state, the normal cellular population of the alveolar air spaces is dominated by the alveolar macrophage. FACS® analysis of isolated alveolar macrophages stained with the RII/RIII-specific mAb, 2.4G2, from wild-type, RII-deficient, and γ chain–deficient mice demonstrated high levels of staining, indicating that both FcγRII and III are expressed on these cells (data not shown). To further investigate the function of macrophage-expressed FcγRII in vitro, resting peritoneal macrophages were isolated from wild-type and FcγRII−/− mice and subjected to FcγR cross-linking assays. To assess the early membrane events associated with combined FcγRII and III cross-linking, resident macrophages were treated with the rat mAb 2.4G2 and a secondary goat anti–rat IgG F(ab′)2. It has been previously shown that γ chain–deficient macrophages have abrogated calcium fluxes upon FcγRII/III cross-linking, indicating that cross-linking of FcγRII alone does not result in calcium influx (39). An inhibitory role for FcγRII is revealed when wild-type and FcγRII−/− macrophages are compared for calcium influx responses (Fig. 6 A). Cross-linking of both FcγRII and III receptors on wild-type macrophages generates a rapid [Ca2+] influx of 60 nM, whereas in the absence of coexpressed FcγRII, 2.4G2 cross-linking of FcγRIII alone generates a larger [Ca2+] influx of 110 nM. This enhanced calcium influx is likely the result of the stimulated γ chain activation pathway unbridled from the inhibitory effect of FcγRII-mediated SHIP recruitment. To further assess the functional consequences of FcγRII deficiency, wild-type and FcγRII−/− macrophages were compared for phagocytic activity (Fig. 6 B). It has been previously demonstrated that antibody- mediated macrophage phagocytosis requires γ chain expression both in vivo and in vitro (11, 36). In the current study, the lack of FcγRII expression is associated with enhanced phagocytosis of IgG2b-coated RBCs. These in vitro studies are consistent with FcγRII functioning as a general regulator of ITAM-mediated activation in inflammatory cells and is likely to account for the enhanced inflammation observed in the FcγRII−/− mice challenged with ICs.
Figure 6.
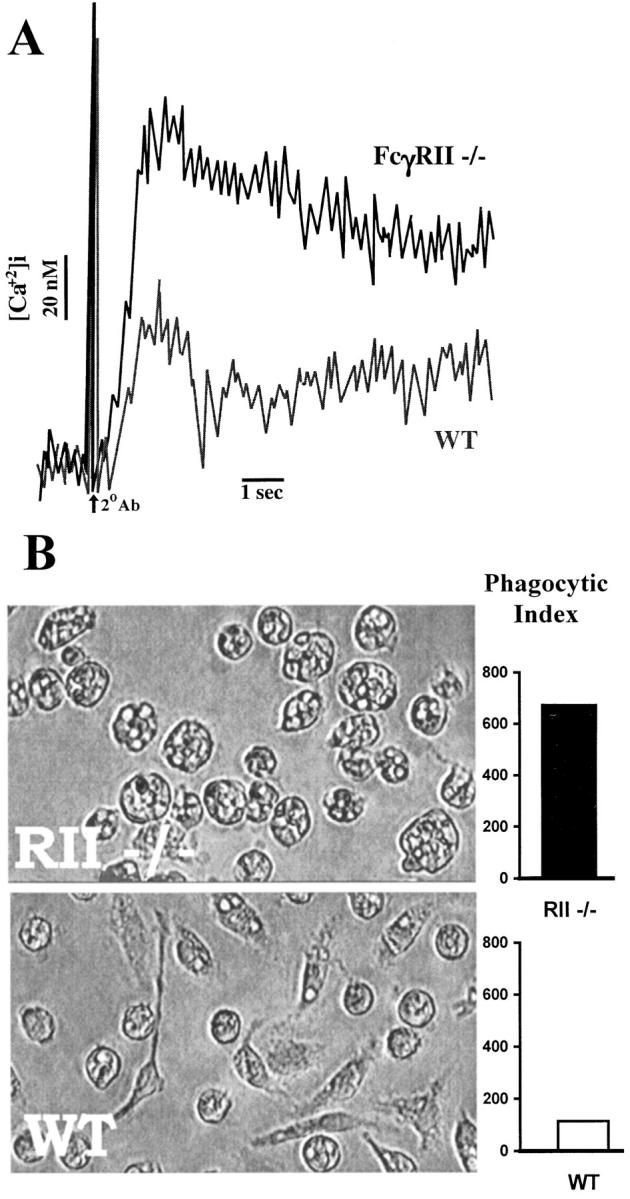
Enhanced FcγR-triggered responses of FcγRII−/− macrophages. (A) Phagocytosis of IgG2b-coated RBCs is enhanced in FcγRII−/−: phagocytic index is expressed as the number of RBCs ingested by 500 representative macrophages. (B) Enhanced calcium flux responses upon FcR ligation in FcγRII−/− mice. Peritoneal macrophages were preincubated with mAb 2.4G2 before stimulation with the secondary antibody, goat anti–rat IgG mAb. Relative fluorescence indicative of intracellular Ca2+ is shown for wild-type (WT) and FcγRII−/− mice.
Discussion
Although the inflammatory cascade induced by ICs ultimately involves a complex interplay of cells and the molecular mediators of inflammation, the initiation of the IgG-triggered cascade has been found to be singularly dependent on engagement and activation of the ITAM-containing γ chain–associated Fc receptors (1). Gene-targeted mice that lack these receptors are protected from the pathological consequences of cytotoxic IgG and IgG-containing soluble ICs (3–5, 36, 37). The present study adds to prior studies in highlighting the requirement for γ chain activation in IC-triggered inflammatory responses in the lung, kidney, and skin. These data argue for a general model of IC-mediated injury, regardless of tissue site, in which the regulation of the FcγR γ activation pathway(s) rather than complement activation is critical to the initiation of the inflammatory cascade.
IC injury may result from activation of resident tissue inflammatory cells and/or the recruitment of circulating monocytes or neutrophils to sites of deposition. In the skin, resident FcγR-bearing mast cells are necessary for initiation of the IC-triggered Arthus reaction (40). The FcγR-bearing resident cell in the lung responsible for initiating the inflammatory response is likely to be the alveolar macrophage. These cells elaborate several inflammatory mediators critical to IC-induced alveolitis, including the cytokines IL-1, TNF-α, IL-6 (21–26), and several chemokines, including MIP-1 and C-X-C cytokines (28–33; for a review, see reference 34). The production of these inflammatory mediators during the IC alveolitis response is enhanced in FcγRII−/− mice, including the cytokine TNF-α as well as chemokines. This is consistent with the hypothesis that IC stimulation of FcγR-bearing tissue resident cells, like the alveolar macrophage, regulates the initiation of the inflammatory response. The balance of FcγR γ stimulatory and FcγRII inhibitory inputs likely determines the cellular threshold of IC-stimulated activation of these resident cells. The absence of SHIP-mediated inhibitory effects on early γ chain signaling events in FcγRII-deficient macrophages is manifested as increased calcium influxes upon cross-linking of FcγRIII. This unbridling of FcγRIII results in enhanced phagocytosis of IgG2b-opsonized RBCs by FcγRII−/− macrophages.
These results demonstrate the regulatory role of the activating and inhibitory Fcγ receptors in initiating IC alveolitis, challenging the viewpoint that the relative contributions of complement and FcγR pathways is dependent on tissue site (38). These data suggest instead that the shared mechanisms of IC-induced injury in the skin, lung, and kidney are likely indicative of a common pathway of autoantibody pathogenesis at other tissue sites. Two potential therapeutic strategies are suggested by these data. Upregulation of the FcγRII inhibitory pathway or downregulation of the FcγR γ pathway would reduce IC-triggered inflammatory responses. The ratio of expression of the activating FcγRIII and inhibitory FcγRII receptors is likely to determine the IC concentration threshold required to trigger effector cell activation. The identification of cytokines or agents that raise the ratio of surface expression of FcγRII/ FcγRIII would likely raise the IC threshold required for cellular activation, potentially providing a novel class of antiinflammatory therapeutics.
Acknowledgments
The authors would like to thank Devon White and Fred Vital for technical assistance, and Cynthia Ritter for administrative assistance. We would like to express our respect and appreciation for the memory of Dr. Nathan Seriff.
This work was supported in part by grants from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (to R. Clynes) and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (to J.V. Ravetch).
Abbreviations used in this paper
- BAL
bronchoalveolar lavage
- HSA
human serum albumin
- IC
immune complex
- ITAM
immunoreceptor tyrosine-based activation motif
- ITIM
immunoreceptor tyrosine-based inhibition motif
- MIP
macrophage-inflammatory protein
- MPO
myeloperoxidase
References
- 1.Ravetch JV, Clynes RA. Divergent roles for Fc receptors and complement in vivo. Annu Rev Immunol. 1998;16:421–432. doi: 10.1146/annurev.immunol.16.1.421. [DOI] [PubMed] [Google Scholar]
- 2.Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP. Control of the complement system. Adv Immunol. 1996;61:201–283. doi: 10.1016/s0065-2776(08)60868-8. [DOI] [PubMed] [Google Scholar]
- 3.Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 4.Sylvestre D, Clynes R, Ma M, Warren H, Carroll MC, Ravetch JV. Immunoglobulin G–mediated inflammatory responses develop normally in complement-deficient mice. J Exp Med. 1996;184:2385–2392. doi: 10.1084/jem.184.6.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 6.Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. [PubMed] [Google Scholar]
- 7.Cambier JC, Daeron M, Fridman W, Gergely J, Kinet JP. New nomenclature for the Reth motif (or ARH1/TAM/ARAM/YXXL) Immunol Today. 1994;16:110. doi: 10.1016/0167-5699(95)80105-7. [DOI] [PubMed] [Google Scholar]
- 8.Ernst LK, van der Winkle JG, Chiu IM, Anderson CL. Association of the high affinity receptor for IgG (FcγRI) with the γ subunit of the IgE receptor. Proc Natl Acad Sci USA. 1993;90:6023–6027. doi: 10.1073/pnas.90.13.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanier LL, Yu G, Phillips JH. Co-association of CD3γ with a receptor (CD16) for IgG on human natural killer cells. Nature. 1989;341:803–806. doi: 10.1038/342803a0. [DOI] [PubMed] [Google Scholar]
- 10.Ra C, Jouvin MH, Blank U, Kinet JP. A macrophage Fcγ receptor and the mast cell receptor for immunoglobulin E share an identical subunit. Nature. 1989;341:752–754. doi: 10.1038/341752a0. [DOI] [PubMed] [Google Scholar]
- 11.Takai T, Li M, Sylvestre DL, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 12.Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, Webster P, Sautes C, Mellman I, Fridman WH. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 13.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 14.Daeron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, Fridman WH. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negative BCR-, TCR- and FcR- dependent cell activation. Immunity. 1995;3:635–646. doi: 10.1016/1074-7613(95)90134-5. [DOI] [PubMed] [Google Scholar]
- 15.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 16.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 17.Daeron M, Malbec O, Latour S, Arock M, Fridman WH. Regulation of high-affinity IgE receptor– mediated mast cell activation by murine low-affinity IgG receptors. J Clin Invest. 1995;95:577–585. doi: 10.1172/JCI117701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Rourke L, Tooze R, Fearon DT. Co-receptors of B lymphocytes. Curr Opin Immunol. 1997;9:324–329. doi: 10.1016/s0952-7915(97)80077-5. [DOI] [PubMed] [Google Scholar]
- 19.Phillips NE, Parker DC. Fc-dependent inhibition of mouse B cell activation by whole anti-mu antibodies. J Immunol. 1983;130:602–606. [PubMed] [Google Scholar]
- 20.Phillips NE, Parker DC. Cross-linking of B lymphocyte Fc gamma receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J Immunol. 1984;132:627–632. [PubMed] [Google Scholar]
- 21.Lentsch AB, Czermak BJ, Bless NM, Ward PA. NF-kappaB activation during IgG immune complex-induced lung injury: requirements for TNF-alpha and IL-1beta but not complement. Am J Pathol. 1998;152:1327–1336. [PMC free article] [PubMed] [Google Scholar]
- 22.Shanley TP, Peters JL, Jones ML, Chensue SW, Kunkel SL, Ward PA. Regulatory effects of endogenous interleukin-1 receptor antagonist protein in immunoglobulin G immune complex–induced lung injury. J Clin Invest. 1996;97:963–970. doi: 10.1172/JCI118520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warren JS. Relationship between interleukin-1 beta and platelet-activating factor in the pathogenesis of acute immune complex alveolitis in the rat. Am J Pathol. 1992;141:551–560. [PMC free article] [PubMed] [Google Scholar]
- 24.Warren JS, Barton PA, Mandel DM, Matrosic K. Intrapulmonary tumor necrosis factor triggers local platelet-activating factor production in rat immune complex alveolitis. Lab Invest. 1990;63:746–754. [PubMed] [Google Scholar]
- 25.Warren JS, Yabroff KR, Remick DG, Kunkel SL, Chensue SW, Kunkel RG, Johnson KJ, Ward PA. Tumor necrosis factor participates in the pathogenesis of acute immune complex alveolitis in the rat. J Clin Invest. 1989;84:1873–1882. doi: 10.1172/JCI114374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shanley TP, Foreback JL, Remick DG, Ulich TR, Kunkel SL, Ward PA. Regulatory effects of interleukin-6 in immunoglobulin G immune-complex-induced lung injury. Am J Pathol. 1997;151:193–203. [PMC free article] [PubMed] [Google Scholar]
- 27.Warren JS, Mandel DM, Johnson KJ, Ward PA. Evidence for the role of platelet-activating factor in immune complex vasculitis in the rat. J Clin Invest. 1989;83:669–678. doi: 10.1172/JCI113931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shanley TP, Schmal H, Friedl HP, Jones ML, Ward PA. Role of macrophage inflammatory protein-1 alpha (MIP-1 alpha) in acute lung injury in rats. J Immunol. 1995;154:4793–4802. [PubMed] [Google Scholar]
- 29.Shanley TP, Schmal H, Warner RL, Schmid E, Friedl HP, Ward PA. Requirement for C-X-C chemokines (macrophage inflammatory protein-2 and cytokine-induced neutrophil chemoattractant) in IgG immune complex-induced lung injury. J Immunol. 1997;158:3439–3448. [PubMed] [Google Scholar]
- 30.Mulligan MS, Jones ML, Bolanowski MA, Baganoff MP, Deppeler CL, Meyers DM, Ryan US, Ward PA. Inhibition of lung inflammatory reactions in rats by an anti-human IL-8 antibody. J Immunol. 1993;150:5585–5595. [PubMed] [Google Scholar]
- 31.Mulligan MS, Schmid E, Beck-Schimmer B, Till GO, Friedl HP, Brauer RB, Hugli TE, Miyasaka M, Warner RL, Johnson KJ, Ward PA. Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest. 1996;98:503–512. doi: 10.1172/JCI118818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bozic CR, Lu B, Hopken UE, Gerard C, Gerard NP. Neurogenic amplification of immune complex inflammation. Science. 1996;273:1722–1725. doi: 10.1126/science.273.5282.1722. [DOI] [PubMed] [Google Scholar]
- 33.Mulligan MS, Yeh CG, Rudolph AR, Ward PA. Protective effects of soluble CR1 in complement- and neutrophil-mediated tissue injury. J Immunol. 1992;148:1479–1485. [PubMed] [Google Scholar]
- 34.Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann NY Acad Sci. 1996;796:104–112. doi: 10.1111/j.1749-6632.1996.tb32572.x. [DOI] [PubMed] [Google Scholar]
- 35.Varani J, Ward PA. Mechanisms of neutrophil-dependent and neutrophil-independent endothelial cell injury. Biol Signals. 1994;3:1–14. doi: 10.1159/000109521. [DOI] [PubMed] [Google Scholar]
- 36.Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 37.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen LA, Schmidt RE, Sandor M, Capel PJ, Daeron M, et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 38.Hopken UE, Lu B, Gerard NP, Gerard C. Impaired inflammatory responses in the reverse arthus reaction through genetic deletion of the C5a receptor. J Exp Med. 1997;186:749–756. doi: 10.1084/jem.186.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J Exp Med. 1997;85:1977–1985. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sylvestre DL, Ravetch JV. A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity. 1996;5:387–390. doi: 10.1016/s1074-7613(00)80264-2. [DOI] [PubMed] [Google Scholar]