

Abstract
To examine the role of nuclear factor (NF)-κB in T cell development and activation in vivo, we produced transgenic mice that express a superinhibitory mutant form of inhibitor κB-α (IκB-αA32/36) under the control of the T cell–specific CD2 promoter and enhancer (mutant [m]IκB-α mice). Thymocyte development proceeded normally in the mIκB-α mice. However, the numbers of peripheral CD8+ T cells were significantly reduced in these animals. The mIκB-α thymocytes displayed a marked proliferative defect and significant reductions in interleukin (IL)-2, IL-3, and granulocyte/macrophage colony-stimulating factor production after cross-linking of the T cell antigen receptor. Perhaps more unexpectedly, double positive (CD4+CD8+; DP) thymocytes from the mIκB-α mice were resistant to α-CD3–mediated apoptosis in vivo. In contrast, they remained sensitive to apoptosis induced by γ-irradiation. Apoptosis of wild-type DP thymocytes after in vivo administration of α-CD3 mAb was preceded by a significant reduction in the level of expression of the antiapoptotic gene, bcl-xL. In contrast, the DP mIκB-α thymocytes maintained high level expression of bcl-xL after α-CD3 treatment. Taken together, these results demonstrated important roles for NF-κB in both inducible cytokine expression and T cell proliferation after TCR engagement. In addition, NF-κB is required for the α-CD3–mediated apoptosis of DP thymocytes through a pathway that involves the regulation of the antiapoptotic gene, bcl-xL.
Keywords: nuclear factor κB, inhibitor κB-αA32/36, thymocytes, apoptosis, bcl-xL
The mammalian nuclear factor (NF)-κB1 transcription factors, NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), c-Rel, and RelB are involved in the regulation of immune and inflammatory responses, cellular proliferation, and cell death (for review see references 1–4). NF-κB proteins modulate these diverse biological processes by regulating the expression of a wide variety of genes encoding cytokines and cytokine receptors, chemokines, cell adhesion molecules, cell-surface receptors, hematopoietic growth factors, acute phase proteins, and other transcription factors. Transcriptional activation or repression of NF-κB target genes requires binding of NF-κB dimers to κB DNA binding sites. Although p50/p65 heterodimers are considered the prototypical NF-κB dimer, many different combinations of NF-κB homo- and heterodimers exist (5) and there is evidence to suggest that these different NF-κB dimers regulate the expression of different genes (6).
In most cells, the transcriptional activity of NF-κB proteins is controlled at the posttranslational level by regulated associations with members of the inhibitor (I)κB family of inhibitory proteins. NF-κB proteins are retained in the cytoplasm of unstimulated cells and thus rendered inactive via interactions with one or more of the seven known IκB proteins: IκB-α, IκB-β, IκB-γ, Bcl-3, IκB-ε, p105, and p100 (3, 7). In response to a wide variety of stimuli, including antigen receptor cross-linking, and exposure to cytokines (TNF-α, IL-1), bacterial components (LPS), viruses, ionizing radiation, or oxidative stress, IκB-α and -β proteins are phosphorylated and degraded and cytoplasmic NF-κB dimers released from their inhibitory proteins are rapidly translocated to the nucleus where they regulate the transcription of genes containing κB binding sites (8–11). Recent studies have demonstrated that phosphorylation of Ser32 and Ser36 and subsequent polyubiquitination of IκB-α are essential for the release and nuclear translocation of NF-κB dimers (12–18). Substitution of IκB-α Ser32 and Ser36 by Ala residues prevents phosphorylation and degradation of IκB-α, thereby retaining NF-κB dimers in an inactive form in the cytoplasm.
NF-κB proteins are thought to play important roles in T cell activation and development (19, 20). Functionally important κB binding sites have been identified in a large number of T cell transcriptional regulatory elements, including the IL-2, IL-2Rα, G-CSF, and macrophage inflammatory protein (MIP)-2 promoters (1). Preformed NF-κB proteins are present in the cytoplasm of thymocytes and resting peripheral T cells (21). TCR engagement results in the rapid phosphorylation of IκB-α and the concomitant nuclear migration of active NF-κB dimers (19, 22, 23). Despite these findings, it has been difficult to precisely elucidate the role of NF-κB in regulating T cell development, activation, and apoptosis in vivo. During the last several years, gene targeting experiments have demonstrated that RelB, c-Rel, and NF-κB1 each play important but distinct roles in regulating the development and function of the mammalian immune system. NF-κB1–deficient mice displayed defects in B cell proliferation in response to the mitogen LPS but not to IgM cross-linking (24, 25). RelB is required for the differentiation and/or survival of dendritic cells and thymic medullary epithelial cells and RelB−/− mice displayed severely defective cellular immune responses (26, 27). The immune dysfunction observed in RelB−/− mice was worsened in NF-κB1/RelB double knockout mice, suggesting that the lack of RelB is compensated for by other NF-κB1–containing dimers (28). c-Rel–deficient mice displayed defective B and T cell proliferation in response to mitogen stimulation as well as markedly decreased IL-2 production after TCR engagement (29). In contrast, mice lacking RelA died on embryonic day 15 from massive hepatocyte apoptosis (30, 31). However, progenitor cells derived from RelA−/− mice were able to give rise to normal T cells, suggesting that RelA is not required for T cell development (30, 32).
Recent studies have suggested that, in addition to regulating lymphocyte function, NF-κB proteins might play a critical role in protecting cells against apoptosis. Support for this model came both from the finding of hepatocyte apoptosis in the RelA−/− mice and from experiments demonstrating that overexpression of a dominant-negative form of the IκB-α protein in transgenic mice (33) and several cell lines promoted apoptosis in vitro (34–39). However, other studies have suggested that NF-κB can also function in a proapoptotic fashion. For example, induction of apoptosis in 293 cells after serum withdrawal requires RelA activation (40). Similarly, radiation-induced apoptosis in ataxia telangiectasia cells is suppressed by dominant-negative IκB-α proteins (41) and inhibition of NF-κB activation prevents apoptosis in cultured human thymocytes (42). Finally, NF-κB activation promotes apoptosis in neural cells (43) and T cell hybridomas (44), and high levels of c-Rel induce apoptosis in avian embryos and in bone marrow cells in vitro (45).
Although gene targeting experiments have been useful for identifying the essential nonredundant roles of individual NF-κB transcription factors in T cell development and function, the interpretation of these experiments can be obscured by functional redundancies of related gene products in the mutant animals and by embryonic lethal phenotypes that preclude a complete analysis of lymphoid development and function. Moreover, in some cases it can be difficult to distinguish cell autonomous and non–cell autonomous phenotypes because mutant animals lack expression of the targeted gene in all cell lineages. We and others (33, 46) have used a complementary approach that circumvents these potential problems to study the role of NF-κB in T cell development, activation, and apoptosis in vivo. Specifically, we have generated transgenic mice that express a mutant superinhibitory form of the IκB-α protein under the control of the T cell–specific CD2 promoter and enhancer. This superinhibitory form of IκB-α cannot be phosphorylated on Ser32 and Ser36 and degraded in response to TCR cross-linking and would therefore be expected to constitutively inhibit the nuclear translocation of NF-κB proteins after T cell activation. Studies of the mIκB-α mice revealed several important functions for NF-κB in T cells in vivo. First, NF-κB is required to develop or maintain normal numbers of peripheral CD8+ T cells. Second, NF-κB activation is necessary for both cytokine production and thymocyte proliferation after TCR engagement. Finally and somewhat unexpectedly, NF-κB is required for α-CD3–mediated apoptosis of double positive (DP) thymocytes in vivo via a pathway that involves downregulated expression of the antiapoptotic gene bcl-xL in DP thymocytes.
Materials and Methods
Transgene Construction and Generation of Transgenic Mice.
The construction of the hemagglutinin (HA)-tagged IκB-α cDNA with Ser to Ala substitutions at positions 32 and 36 has been described previously (47). The mutant IκB-α cDNA was inserted into the XhoI site of pTEx (provided by Dr. M. Owen, Imperial Cancer Research Fund, London, UK), which contains the promoter, polyadenylation site, and locus control region elements from the human CD2 gene (48). Transgene DNA was microinjected into the male pronucleus of fertilized single cell embryos of CD1 mice. Microinjected eggs were transferred to pseudopregnant CD1 foster mothers. EcoRI-digested tail DNA from 12–14-d-old pups was hybridized to a radiolabeled 0.8-kb IκB-α–specific probe, and the transgene copy number was determined using a PhosphorImager (Molecular Dynamics).
Cell Culture.
Single cell suspensions of thymocytes and splenocytes were cultured at 37°C, 5% CO2 in RPMI 1640 medium containing 10% heat inactivated FCS, 100 U/ml penicillin/streptomycin, 2 mM glutamine, 0.1 mM nonessential amino acids, 5.5 × 102 μM β-ME (all from GIBCO BRL). Thymocyte proliferation assays were performed in 96-well plates (Becton Dickinson) that had been coated overnight with α-CD3 mAb (145-2C11; PharMingen) and/or α-CD28 mAb (37.51; PharMingen) at a concentration of 16 μg/ml. Stimulation with PMA (Sigma Chemical Co.) and ionomycin (Calbiochem) was performed at 5 ng/ml and 0.25 μg/ml, respectively. After stimulation for 48 h, cells (0.5 × 106/ml) were pulsed for 18 h at 37°C with [3H]thymidine (1 μCi/ml; Amersham). Cells were transferred onto glass fiber filter mats and radioactive incorporation was measured with a beta scintillation counter.
Fetal Thymic Organ Culture.
Fetuses of CD1 and mIκB-α mice were obtained at embryonic day 17.5 of pregnancy, counting the day of vaginal plug as embryonic day 1. Equally sized thymic lobes were cultured in 24-well Transwell dishes (Costar Corp.) containing complete DMEM-10 medium. The samples were maintained in an atmosphere of 5% CO2 for 72 h and treated with α-CD3 mAb (145-2C11) as described in the text.
ELISA Assays.
For ELISA assays, 96-well plates (Dynatech Labs.) were coated overnight at 4°C with α–IL-2 (JES6-1A12), α–IL-3 (MP2-8F8), or α–GM-CSF (MP1-22E9) mAbs (PharMingen). Serial dilutions of supernatants from thymocytes stimulated in culture for 48 h were added to the antibody-coated plates and incubated for 18 h at 4°C. Biotinylated antibodies against IL-2 (JES5-5H4), IL-3 (MP2-43D11), or GM-CSF (MP1-31G6) were then added to the plates and incubated for 2 h at room temperature. Avidin peroxidase (Sigma Chemical Co.) and ABTS (3-ethylbenzthiazoline-6-sulfonic acid) were added to the wells and enzymatic reactions were quantitated in a microplate reader (Dynatech Labs.). All samples were assayed in triplicate.
Western Blot Analysis.
Cytoplasmic and nuclear thymocyte extracts were prepared as previously described (49). In brief, thymocytes were washed with PBS, incubated in buffer A (10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, and 0.2 mM phenylmethylsulfonyl fluoride) for 10 min at 4°C and centrifuged to separate nuclei (pellet) from cytoplasmic proteins (supernatant). Nuclei were lysed by incubation with buffer C (20 mM Hepes, pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride, 10 mg/ml aprotinin, and 10 mg/ml leupeptin) at 4°C and lysates were cleared by centrifugation. Nuclear and cytoplasmic extracts were frozen at −70°C. Protein concentrations were determined using a commercially available kit (Bio-Rad). For Western blot analysis, 15 μg of cytoplasmic extracts were fractionated by SDS-PAGE in 10% polyacrylamide gels and transferred to PVDF membranes (Millipore). Western blots were probed with a rabbit polyclonal antibody against MAD-3 (1:2,000 dilution; nomenclature of DiDonato et al. [reference 13], No. 644) or an α-HA-specific antibody (1:2,000 dilution; 12CA5). Blots were developed with goat α-rabbit (1:2,000 dilution; Amersham) or rat α-mouse (1:5,000 dilution; Kirkegaard and Perry Labs.) secondary antibodies and a commercially available chemiluminescence kit according to the manufacturer's instructions (Pierce Chemical Co.).
Electrophoretic Mobility Shift Assay.
A double-stranded oligonucleotide, spanning the κB site of the IL-2Rα promoter (50) (5′ GGAACGGCAGGGGAATTCCCCTCTCCTT 3′) was labeled with 32P-nucleotides using the Klenow fragment of DNA polymerase I. Binding reactions contained 2 μg nuclear extract, 20,000 disintegrations per min (0.1–0.5 ng) of radiolabeled oligonucleotide probe, 2 μg poly (dI-dC) in 75 mM KCl, 10 mM Tris, pH 7.5, 1 mM dithiothreitol, 1 mM EDTA, and 4% (vol/ vol) Ficoll in a final volume of 20 μl. After incubation on ice for 30 min, DNA protein complexes were fractionated by electrophoresis in nondenaturing 4% polyacrylamide gels at 120 V for 2.5 h in running buffer (25 mM Tris, 25 mM Boric acid, and 0.5 mM EDTA) with 47 μM β-ME. For competition experiments, 50 ng of unlabeled NF-κB oligonucleotide was added to the binding reaction before addition of the radiolabeled oligonucleotide probe. For antibody supershift experiments, the nuclear extracts were incubated with 1 μl of α-p50 (F056), α-RelB (A277), α-p65 (B127), or α-c-Rel (L036) antibodies (all from Santa Cruz Biotechnology) before starting the binding reactions.
Flow Cytometry.
Single cell suspensions of lymphocytes (106 cells) were washed in PBS and incubated with PE–α-CD4 (RM4-5), FITC–α-CD8 (53-6.7), FITC–α-CD3 (145-2C11), and/or PE–α-TCR-α/β (H57-97) mAbs (PharMingen) in PBS plus 0.1% BSA for 30 min on ice. After staining, the cells were washed in PBS and analyzed on a FACScan® (Becton Dickinson). Each plot represents analysis of >104 events using WinMDI software (Windows Multiple Document Interface, version 2.5). For intracellular staining, 106 thymocytes were stained with Cy-Chrome– α-CD4 (RM4-5) and PE–α-CD8 (53-6.7), and fixed in 4% paraformaldehyde. Fixed thymocytes were washed with 0.03% saponin (Sigma Chemical Co.) and incubated with 100 μl 0.3% saponin, 20 μl blocking serum (Sigma Chemical Co.) and FITC– α-Bcl-xL (7B2.5) (Southern Biotechnology Associates). After 30 min of incubation at 4°C, cells were washed twice in 0.03% saponin followed by one wash in FACS buffer before FACScan® analysis.
Ribonuclease Protection Assays.
Ribonuclease protection assays were performed using a commercially available kit as described by the manufacturer (PharMingen) using the mAPO-2 multiprobe template set, 10 μg thymic RNA, and 0.1 mCi α-[32P]UTP (Amersham). RNAse–protected fragments were resolved by electrophoresis in polyacrylamide gels and visualized by autoradiography (Eastman Kodak Co.).
TUNEL Assays.
TUNEL (Tdt-mediated dUTP nick end labeling) assays were performed on paraffin-embedded sections of thymi according to Gavrieli et al. (51). Photomicrographs were obtained with a Zeiss Axioskop.
Results
Production of mIκB-α Transgenic Mice.
To inhibit inducible NF-κB activity in thymocytes in vivo, we produced transgenic mice that expressed a “superinhibitory” form of IκB-α under the control of the T cell–specific CD2 promoter and enhancer (mIκB-α mice). We chose to use an IκB-α transgene because previous studies have demonstrated that IκB-α plays a critical role in regulating inducible NF-κB expression (for review see references 2–4, 52). The superinhibitory mIκB-αA32/36 transgene containing Ser32 and Ser36 to Ala substitutions cannot be phosphorylated and degraded in response to TCR engagement and would therefore be expected to constituitively inhibit NF-κB activity in both resting and activated thymocytes and T cells. The CD2 promoter and enhancer (48, 53; Fig. 1 A) were used in these studies because (a) they program T cell– specific transgene expression and (b) they are expressed at high levels in all thymocyte subsets including double negative (CD4−CD8−; DN), DP (CD4+CD8+), and single positive (CD4+ or CD8+; SP) cells (54). The transgene construct also contained three copies of an 11-amino acid influenza virus HA epitope tag (55) that allowed us to distinguish the transgene-encoded protein from endogenous murine IκB-α. A line of mIκB-α mice containing ∼12 copies of the transgene was produced by injection of this construct into fertilized CD1 mouse embryos (Fig. 1 B). Homozygous transgenic mice were generated by breeding heterozygous littermates in order to obtain maximal levels of IκB-αA32/36 protein expression, an important consideration in producing a dominant-negative phenotype. A second line of transgenic mice expressing the IκB-αA32/36 cDNA under the control of the proximal lck promoter and the CD2 enhancer was also produced (data not shown). Similar phenotypes were observed in both lines of transgenic mice and are therefore not distinguished in the results described below.
Figure 1.


Production of mIκB-α transgenic mice. (A) Schematic illustration of the IκB-αA32/36 transgene. Nucleotide and amino acid sequences corresponding to amino acids 31 to 37 from the wild-type human IκB-α cDNA (94) and the mutant IκB-αA32/36 are shown above the schematic representation of the transgene construct. Site-specific mutations within the IκB-αA32/36 cDNA sequence are underlined and the resulting Ser to Ala substitutions are circled. The HA epitope (HA), CD2 promoter (CD2-Pr), polyadenylation signal (poly A), and the CD2 enhancer/LCR (CD2 Enh) are also shown. (B) Southern blot analysis of tail DNA from wild-type (WT) and mIκB-α transgenic (Tg) mice was performed with a radiolabeled 0.8-kb IκB-α cDNA probe (see A) that hybridizes to both the transgene (IκB-αA32/36) and the endogenous IκB-α gene. Size markers in kilobases are shown to the right of the autoradiogram.
Endogenous IκB-α is phosphorylated and degraded after treatment of T cells with the NF-κB inducer, TNF-α (47). To analyze the relative levels of endogenous and transgene-encoded IκB-α and to study the differential regulation of the two proteins in response to TNF-α, we performed Western blot analyses using whole cell extracts from mIκB-α and control thymocytes that had been treated for different times with TNF-α (Fig. 2 A). Basal levels of the IκB-αA32/36 protein as detected with both the α-HA and α-IκB-α (α-MAD) antibodies slightly exceeded levels of the endogenous IκB-α protein in the mIκB-α thymocytes. In both wild-type and mIκB-α thymocytes, endogenous IκB-α was almost completely degraded after 15 min of TNF-α treatment and remained undetectable until ∼30 min after treatment. As previously reported (9, 15), endogenous IκB-α was reexpressed between 30 and 60 min after TNF-α treatment (data not shown). This reexpression reflects NF-κB–mediated activation of the IκB-α promoter. In marked contrast, levels of the transgene-encoded IκB-αA32/36 were unchanged by TNF-α treatment at all time points (Fig. 2 A). Activation of mIκB-α thymocytes with PMA plus ionomycin or TCR cross-linking resulted in a similar degradation of endogenous IκB-α, whereas the levels of IκB-αA32/36 remain unaffected (data not shown).
Figure 2.


IκB-α and NF-κB expression in the mIκB-α transgenic mice. (A) Western blot analysis of IκB-α expression in thymocytes from mIκB-α transgenic mice after treatment with TNF-α. Thymocytes from wild-type (WT) and mIκB-α transgenic (Tg) mice were incubated with TNF-α (15 ng/ml) for the indicated times and cytoplasmic extracts were separated by electrophoresis in a denaturing polyacrylamide gel. Proteins were transferred to a PDVF membrane and immunoblotted with either a murine HA-specific antibody (α-HA) or a rabbit polyclonal antibody specific for IκB-α (α-MAD) (13). The positions of the endogenous IκB-α and IκB-αA32/36 transgene-encoded proteins are shown to the left of the autoradiogram. Size markers in kilodaltons are shown to the right of the autoradiogram. (B) EMSA of NF-κB expression in transgenic thymocytes after stimulation in vivo with α-CD3 mAb. Thymic nuclear extracts (2 μg) from mice treated for 3 h with a single intraperitoneal injection of α-CD3 mAb or PBS (control) were analyzed by EMSA with a radiolabeled κB oligonucleotide probe. For antibody supershift experiments, thymic nuclear extracts were preincubated with antibodies specific for NF-κB p50, p65, c-Rel, or RelB. For cold competition experiments, binding reactions contained a 50-fold molar excess of unlabeled competitor oligonucleotide. The positions of bands corresponding to binding of NF-κB p50 homodimers and p65/p50 as well as c-Rel/p50 and RelB/p50 heterodimers are indicated, as are “supershifted” complexes containing p50, p65, c-Rel, and RelB proteins (•).
To analyze the effects of IκB-αA32/36 expression on the nuclear translocation of NF-κB proteins after T cell activation, we performed electrophoretic mobility shift assays (EMSAs) using nuclear extracts prepared from both resting thymocytes and thymocytes activated in vivo by intraperitoneal injection of an α-CD3 mAb (Fig. 2 B). As reported previously, nuclear extracts from unstimulated wild-type and mIκB-α thymocytes both contained p50 homodimers that bound to the radiolabeled κB probe (22, 56). The presence of such p50 homodimers in uninduced thymic nuclear extracts probably reflected the fact that p50 binds to IκB proteins with lower affinity and therefore is not quantitatively retained in the cytoplasm of either wild-type or mIκB-α thymocytes (57). Because p50 homodimers do not contain a transcriptional activation domain they are thought to repress κB-dependent gene expression (58). After activation with α-CD3 mAb, the level of p50 homodimers increased equivalently in the wild-type and mIκB-α thymocyte nuclear extracts. In addition, an inducible low mobility κB binding complex appeared in the nuclear extracts of wild-type thymocytes. Antibody supershift experiments demonstrated that this complex contained predominantly p50/p65 heterodimers as well as smaller amounts of c-Rel– and RelB-containing heterodimers (Fig. 2 B). The induction of this low mobility κB binding activity was markedly inhibited after α-CD3–mediated activation of the mIκB-α thymocytes (Fig. 2 B). Taken together, these Western blotting and EMSA experiments demonstrated that thymocytes from the mIκB-α transgenic animals expressed high levels of cytoplasmic IκB-αA32/36 protein that was not degraded after TCR engagement in vivo. Moreover, constitutive expression of the IκB-αA32/36 protein significantly reduced the induction of nuclear p50/ p65, c-Rel/p50, and RelB/p50 NF-κB heterodimers after either TNF-α or α-CD3–mediated activation of the mIκB-α thymocytes.
Normal Thymocyte Ontogeny in the mIκB-α Transgenic Mice.
To investigate T cell development in the mIκB-α mice, thymocytes and peripheral T cells from transgenic and wild-type animals were analyzed by flow cytometry using antibodies specific for a variety of developmentally regulated T cell surface antigens (Fig. 3). Thymi from the mIκB-α animals contained normal populations of DN, DP, and SP cells. Levels of CD3 and TCR-α/β expression were also indistinguishable on wild-type and mIκB-α thymocytes (Fig. 3 B), as were expression of TCR-γ/δ and heat-shock antigen (data not shown). Thus, expression of IκB-αA32/36 did not appear to perturb thymocyte ontogeny. Total numbers of SP peripheral T cells in both the spleen and the lymph nodes were normal in mIκB-α mice as were the numbers of splenic CD4+ T cells. However, as described previously by Boothby at al. and Esslinger et al. (33, 46), the numbers of peripheral CD8+ T cells were reduced by ∼50% in mIκB-α mice (Fig. 3 C).
Figure 3.



T cell development in mIκB-α transgenic mice. Single cell suspensions of thymocytes from mIκB-α transgenic (Tg) and wild-type (WT) mice were stained with PE–α-CD4 and FITC–α-CD8 (A), or PE–α-TCR-α/β and FITC–α-CD3 (B) antibodies and analyzed by flow cytometry. Percentages of cells in the individual subpopulations are shown in the lower right quadrant. (C) Splenocytes from mIκB-α transgenic (Tg) and wild-type (WT) mice were stained with 7-amino actinomycin D, PE–α-CD4, and FITC–α-CD8, and were analyzed by flow cytometry. Total numbers (× 106 cells) of live CD4+ and CD8+ splenic T cells were determined from at least three mice in each group. The data is shown as mean ± SD.
Impaired Proliferation and Cytokine Production by mIκB-α Transgenic Thymocytes.
To assess the effects of IκB-αA32/36 expression on cytokine production and proliferation, thymocytes from mIκB-α and wild-type control littermates were activated in vitro for 72 h with PMA plus ionomycin, α-CD3 mAb, or α-CD3 plus α-CD28 mAbs. When compared with wild-type thymocytes, proliferation of the mIκB-α thymocytes was reduced by 63% in response to PMA plus ionomycin and by 78% after activation with α-CD3 mAb (Fig. 4). Previous studies have suggested that CD28 costimulation may function, at least in part, through an NF-κB–dependent pathway (59). Therefore, we tested the ability of CD28 costimulation to rescue the defect in α-CD3–mediated thymocyte proliferation seen in the mIκB-α thymocytes. Interestingly, the observed reductions in thymocyte proliferation were partially, but not completely, rescued by α-CD28 costimulation (Fig. 4). Thus, the CD28 signaling pathway can at least partially bypass the requirement for NF-κB activation in thymocyte activation by TCR engagement.
Figure 4.

Defective proliferation of mIκB-α thymocytes in response to mitogenic stimuli. Wild-type (WT) and mIκB-α transgenic (Tg) thymocytes were incubated with PMA (5 ng/ml) plus ionomycin (0.25 μg/ml; P + I), or stimulated with immobilized α-CD3 or α-CD3 plus α-CD28 antibodies (16 μg/ml) for 60 h at 37°C. Proliferation was measured by [3H]thymidine incorporation.
Many cytokine genes including IL-2, IL-3, and GM-CSF are potential targets for NF-κB (60). However, the role of NF-κB in regulating activation-specific cytokine expression in vivo, particularly the expression of the IL-2 gene, remains controversial. Therefore, we compared cytokine production by the wild-type and mIκB-α thymocytes after activation with α-CD3 plus α-CD28 mAbs (Fig. 5). As compared with wild-type thymocytes, the mIκB-α thymocytes displayed significantly decreased production of IL-2, IL-3, and GM-CSF after α-CD3 plus α-CD28 activation (Fig. 5). IL-2 production was most significantly reduced (28% of wild-type levels), whereas GM-CSF and IL-3 production were less dramatically inhibited (Fig. 5). Taken together, these experiments demonstrated that NF-κB is required both for the induction of multiple cytokines and for normal thymocyte proliferation after TCR cross-linking.
Figure 5.

Reduced cytokine production by mIκB-α transgenic thymocytes. Cultured thymocytes from wild-type (white bars) and transgenic IκB-αA32/36 mice (black bars) were stimulated with immobilized α-CD3 plus α-CD28 antibodies (16 μg/ml). The levels of IL-2, GM-CSF, and IL-3 in the culture supernatants were assayed by ELISA 48 h after stimulation.
DP IκB-αA32/36 Thymocytes Are Protected against α-CD3– mediated Apoptosis In Vivo.
Systemic administration of α-CD3 mAb has been shown to result in the rapid depletion of >90% of DP thymocytes by apoptosis (61). NF-κB positively regulates the expression of death-rescuing genes, thereby preventing TNF-α–induced apoptosis at least in certain cell types (34–39). Based upon these findings, it was reasonable to hypothesize that DP thymocytes from the mIκB-α mice that failed to activate NF-κB normally would display increased apoptosis after systemic administration of α-CD3 mAb. To directly test the role of NF-κB in mediating the α-CD3–mediated apoptotic death of DP thymocytes, mIκB-α and wild-type animals were injected intraperitoneally with α-CD3 mAb and thymocyte subsets were assessed by flow cytometry 48 h after injection. As shown in Fig. 6 and consistent with previous reports (62, 63), treatment of wild-type animals with α-CD3 mAb resulted in dose-dependent reductions in the size of the DP thymocyte population. Wild-type animals treated with 40 μg of α-CD3 mAb displayed a 98% reduction in DP thymocytes within 48 h after α-CD3 administration. Somewhat surprisingly, this α-CD3–mediated loss of DP thymocytes was completely and reproducibly prevented in the mIκB-α mice even after administration of 40 μg of α-CD3 mAb (Fig. 6). Similar reductions in DP thymocyte apoptosis were observed at both 24 and 48 h after injection and after administration of either 20 or 40 μg of α-CD3 mAb (Fig. 6 and data not shown).
Figure 6.


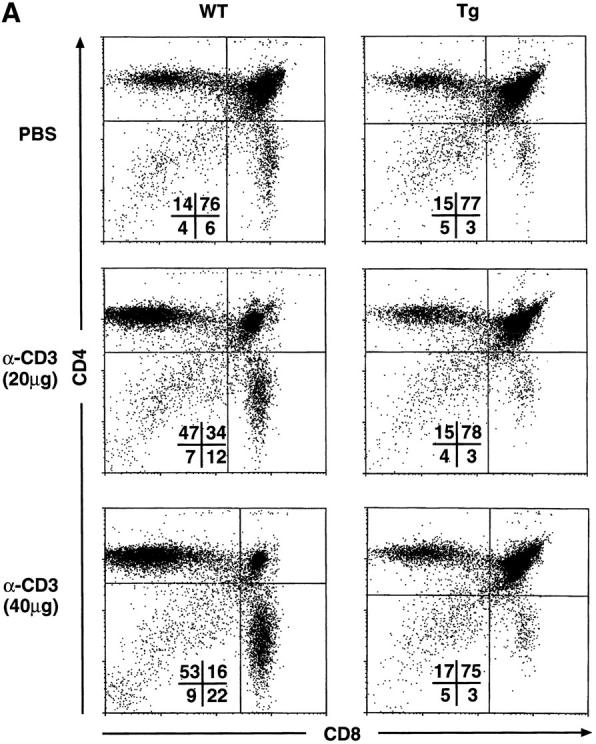
Decreased α-CD3–mediated deletion of DP thymocytes in mIκB-α transgenic mice. (A) 3–7-wk-old wild-type (WT) or mIκB-α transgenic mice (Tg) were injected intraperitoneally with 200 μl of PBS containing either 0 (Control), 20, or 40 μg of α-CD3 mAb. Freshly isolated thymocytes from these animals were analyzed by flow cytometry with PE–α-CD4 and FITC–α-CD8 48 h after α-CD3 administration. (B) Absolute numbers of thymocytes in wild-type (n = 3) and mIκB-α transgenic (n = 4) animals 48 h after intraperitoneal injection with PBS or 40 μg of α-CD3 mAb. The data are shown as mean ± SEM. (C) 3–7-wk-old wild-type (WT) or mIκB-α transgenic mice (Tg) were treated with whole body γ irradiation (500 RADs). Freshly isolated thymocytes from these animals were analyzed by flow cytometry with PE–α-CD4 and FITC–α-CD8 12 h after γ irradiation.
To confirm the protective effects of IκB-αA32/36 expression on DP thymocyte apoptosis in vivo, we performed TUNEL assays on thymi from wild-type and mIκB-α mice after intraperitoneal injection of α-CD3 mAb (Fig. 7). Large numbers of apoptotic cells were seen in wild-type thymi after treatment with α-CD3 mAb as compared with littermates treated with an isotype-matched control antibody. In contrast, the frequency of apoptotic cells in the mIκB-α thymi was dramatically reduced as compared with that observed in wild-type thymi after treatment with similar amounts of α-CD3 mAb (Fig. 7).
Figure 7.
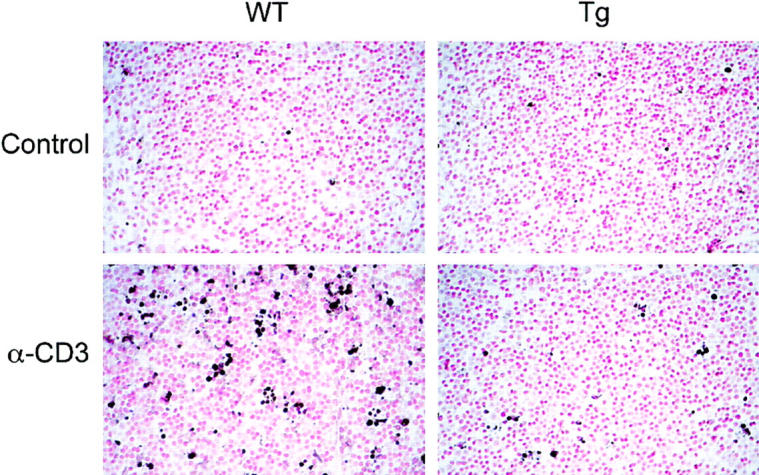
Decreased α-CD3–mediated thymocyte apoptosis in mIκB-α transgenic mice. Wild-type (WT) or mIκB-α transgenic (Tg) mice were injected intraperitoneally with 40 μg of α-CD3 mAb or an isotype-matched control antibody. Thymi were harvested and sections were analyzed for apoptotic lymphocytes by TUNEL assay. Each photomicrograph is representative of at least four different sections taken from each thymus. Original magnification: ×400.
DP IκB-αA32/36 Thymocytes Remain Sensitive to γ Irradiation–induced Apoptosis In Vivo.
Like α-CD3 treatment, γ irradiation of the thymus is known to cause DP thymocyte apoptosis in vivo (64). However, recent studies have suggested that these different apoptotic stimuli may use distinct signaling pathways to regulate programmed cell death. Given the resistance of mIκB-α DP thymocytes to α-CD3–mediated apoptosis, it was of interest to determine the sensitivity of these cells to γ irradiation in vivo. As shown in Fig. 6 C, DP thymocytes from the mIκB-α mice remained fully sensitive to γ irradiation–induced apoptosis in vivo. Thus, expression of the IκB-αA32/36 transgene protected DP thymocytes from apoptosis in response to α-CD3 but failed to protect these cells against γ irradiation– induced cell death.
DP mIκB-α Thymocytes Are Resistant to α-CD3–mediated Apoptosis in Fetal Thymic Organ Culture.
The administration of α-CD3 mAbs to mice in vivo can effect thymocytes either directly or indirectly via the activation of peripheral blood CD3+ T cells. To attempt to distinguish these possibilities, we tested the susceptibility of mIκB-α thymocytes to α-CD3– mediated apoptosis in fetal thymic organ culture (FTOC) (Fig. 8). Wild-type thymocytes were killed efficiently by 3 d of FTOC treatment with an α-CD3 mAb. In contrast, DP thymocytes from the mIκB-α mice were relatively resistant to α-CD3–mediated killing in FTOC. These results suggested that at least some of the resistance to α-CD3–mediated DP thymocyte apoptosis seen in the mIκB-α mice reflects a thymus-specific effect of transgene expression.
Figure 8.

Decreased α-CD3–mediated deletion of DP thymocytes in FTOCs from mIκB-α transgenic mice. Thymi from embryonic day 17.5 wild-type (WT) or mIκB-α transgenic (Tg) mice were cultured for 72 h on semipermeable membranes in DMEM-10 plus 10% FCS in the absence (untreated) or presence of 10 μg/ml α-CD3 mAb (α-CD3). Thymocytes were analyzed by flow cytometry with PE–α-CD4 and FITC– α-CD8. Percentages of cells in the individual subpopulations are shown in the lower left quadrant.
DP mIκB-α Thymocytes Fail To Downregulate Bcl-xL after α-CD3 Treatment In Vivo
The Bcl-2 family of proteins is comprised of multiple members that function either as death agonists (Bax, Bak, Bcl-XS, and Bad) or death antagonists (Bcl-2, Bcl-xL, Bcl-xγ, Mcl-1, A1, and Bcl-w) (65). Two antiapoptotic members of this family, Bcl-xL and Bcl-2, are expressed in a reciprocal pattern during thymocyte development. Bcl-2 is expressed at high levels in immature DN thymocytes. Its expression is downregulated in DP cells and it is then reexpressed as these DP cells mature to SP thymocytes and peripheral T cells (66, 67). Conversely, Bcl-xL is expressed at low or undetectable levels in immature DN cells. Its expression is significantly upregulated in DP thymocytes, and it is then downregulated as these cells progress to the SP stage of thymocyte ontogeny (68, 69). Interestingly, DP thymocytes from transgenic mice that overexpress Bcl-2 or Bcl-xL are protected from multiple proapoptotic stimuli, including α-CD3 treatment in vivo (69, 70). Given these findings, it was logical to postulate that altered expression of Bcl-2-family genes in the mIκB-α thymocytes might account for their decreased susceptibility to proapoptotic stimuli. Accordingly, we used an RNAse protection assay to directly monitor the expression of Bcl-2–related genes in thymocytes from wild-type and mIκB-α transgenic mice. As shown in Fig. 9 A, both basal and α-CD3–treated levels of bfl-1, bak, bax, bcl-2, and bad mRNAs were equivalent in wild-type and mIκB-α thymocytes. Similarly, we failed to detect differential expression of mRNAs encoding Fas, Fas-L, TRAF, TRADD, Fadd, RIP, and FLICE in either unstimulated or α-CD3–treated wild-type and mIκB-α thymocytes (data not shown). Basal levels of bcl-xL were equivalent in wild-type and mIκB-α thymocytes. However, after α-CD3 treatment, the expression of bcl-xL was significantly decreased in the wild-type cells but was maintained at pretreatment levels in the mIκB-α thymocytes (Fig. 9 A).
Figure 9.




An NF-κB–mediated pathway downregulates the expression of bcl-xL after α-CD3 administration in vivo. (A) Expression of Bcl-2–related genes in thymocytes after administration of α-CD3 mAb. Wild-type (WT) or mIκB-α transgenic mice (IκB-αA32/36) were injected intraperitoneally with 200 μl of PBS containing 0 (−) or 40 μg of α-CD3 mAb and thymocyte RNA was prepared 24 or 48 h after treatment. Expression of Bcl-2–related mRNAs was quantitated by RNAse protection assay. L32 is a control RNA that is present at equivalent levels in each of the samples assayed. Note the specific reduction in bcl-xL mRNA levels in the WT thymocytes that was prevented in the mIκB-α thymocytes. (B) Western blot analysis of Bcl-xL expression in wild-type (WT) or mIκB-α (IκB-αA32/36) thymocytes from mice injected intraperitoneally with 40 μg of α-CD3 mAb for the times shown. Equivalent levels of expression of a slow mobility nonspecific control band demonstrate equal loading of the gel. (C and D) Expression of Bcl-xL in viable thymocytes from wild-type (WT) or mIκB-α (IκB-αA32/36) transgenic mice 24 h after intraperitoneal injection with 40 μg of α-CD3 mAb (dotted lines) or an isotype-matched control antibody (Control Ab; solid lines). Viable thymocytes (as determined by forward and side scatter gating) were analyzed by flow cytometry with Cy-Chrome–α-CD4 and PE–α-CD8 (see inserts for FACS® profiles). Thymocytes were fixed, permeabilized, and stained with an FITC– α-Bcl-xL mAb. Levels of intracellular Bcl-xL in specific subpopulations of thymocytes were analyzed by gating on DP (C) and CD4+ SP cells (D). Note the reduction in intracellular Bcl-xL levels seen in the wild-type DP thymocytes after α-CD3 treatment, which was observed neither in the mIκB-α DP thymocytes nor in the wild-type or mIκB-α SP thymocytes from these same animals.
Because Bcl-xL is the predominant antiapoptotic protein expressed in DP thymocytes, the preferential loss of Bcl-xL from wild-type thymocytes after α-CD3 administration might have simply reflected the death of these DP cells. To confirm that the pattern of Bcl-xL protein expression paralleled that of the mRNA and to assess the possibility that the observed changes in bcl-xL mRNA expression reflected the preferential death of the DP thymocytes in the wild-type animals, we performed Western blot analyses on thymocyte extracts 18 and 39 h after α-CD3 treatment in vivo. As shown in Fig. 9 B, levels of Bcl-xL protein were significantly reduced in wild-type thymocytes at both 18 and 39 h after administration of α-CD3 mAb. In contrast, levels of Bcl-xL protein were maintained at unstimulated levels in the mIκB-α thymocytes at both time points. Because there is not a significant loss of DP thymocytes 18 h after α-CD3 treatment, these results indicated that the loss of Bcl-xL protein precedes thymocyte apoptosis and is prevented in the mIκB-α DP thymocytes. We confirmed this result by performing intracellular staining of viable DP thymocytes using Bcl-xL–specific antibodies (Fig. 9 C). 24 h after α-CD3 administration, viable DP thymocytes displayed a significant decrease in intracellular Bcl-xL as measured by flow cytometry. In contrast, DP thymocytes from the mIκB-α mice failed to downregulate intracellular Bcl-xL expression. In control experiments, SP CD4+ thymocytes from these same wild-type and mIκB-α animals both failed to demonstrate altered levels of intracellular Bcl-xL after α-CD3 administration (Fig. 9 D). Taken together, these results demonstrated that the in vivo administration of α-CD3 mAb resulted in the specific downregulation of the antiapoptotic gene bcl-xL in DP thymocytes and that this reduced expression of Bcl-xL was, in turn, associated with subsequent apoptotic cell death. In contrast, DP thymocytes from mIκB-α mice failed to downregulate Bcl-xL and were protected from α-CD3–mediated programmed cell death.
Discussion
In the studies described in this report, we have used transgenic mice expressing a superinhibitory form of IκB-α to study the function of NF-κB in T cells in vivo. The dominant-negative approach used in these studies circumvents potential problems of functional redundancy and embryonic lethality that can complicate the analysis of gene targeting experiments. In addition, because the transgene is only expressed in thymocytes and T cells we can conclude that the observed defects in T cell function are thymocyte or T cell autonomous. Our results have revealed several important roles for NF-κB proteins in T cell development and function. First in agreement with previous reports (33, 46) we showed that NF-κB is required for the development and/or survival of normal numbers of peripheral CD8+ T cells, for the inducible expression of multiple cytokine genes, and for normal T cell proliferation in response to TCR-mediated T cell activation. However, our studies have also revealed an unexpected and novel role for NF-κB proteins in DP thymocyte apoptosis in response to α-CD3 mAb administration in vivo.
Defective Thymocyte Proliferation and Cytokine Production in the mIκB-α Mice.
TCR engagement leads to the precisely orchestrated transcriptional induction of >100 new genes whose expression together determine the activated T cell phenotype. Several inducible transcription factors have been implicated as critical early regulators of activation-specific T cell gene expression. These include NF-AT, CREB, AP1, and NF-κB (60). Three of these, NF-AT, CREB, and NF-κB, are present in an inactive form in resting T cells and are activated by posttranslational phosphorylation or dephosphorylation in response to TCR cross-linking. The precise role of each of these transcription factors in regulating T cell activation remains unclear because many T cell genes contain binding sites for multiple inducible transcription factors, and because it has been difficult to extrapolate from the results of transient transfection assays performed in immortalized T cell lines to in vivo transcriptional regulatory pathways. Thus, for example, NF-AT, AP1, and NF-κB transcription factors can each bind to functionally important sites in the IL-2 promoter in vitro (71–74). Moreover, transient transfection assays have suggested that each of these transcription factors may play an important role in regulating the expression of this T cell cytokine (75–76). However, gene targeting experiments have thus far failed to demonstrate a critical role for NF-AT in positively regulating IL-2 expression and T cell proliferation in normal T cells (77, 78). To circumvent these problems, we used a genetic approach involving overexpression of IκB-αA32/36 in transgenic mice to directly study the role of NF-κB proteins in the development and activation of normal T cells in vivo. Our results demonstrated that NF-κB is required for the inducible expression of the IL-2, IL-3, and GM-CSF genes and for subsequent T cell proliferation in response to TCR stimulation. These findings are consistent with similar defects in cytokine production and T cell proliferation observed in mice lacking the c-Rel protein (29) or expressing a constitutively active IκB-α transgenic protein containing an NH2-terminal deletion (33).
Previous studies have suggested that the CD28 costimulatory pathway functions to activate cytokine gene transcription via an NF-κB–mediated signaling pathway (59, 79–81). Interestingly, however, we and others (33) found that the inhibitory effects of IκB-αA32/36 on α-CD3–mediated thymocyte proliferation could be partially reversed by α-CD28–mediated costimulation. This suggests that at least some of the costimulatory effects of the CD28 pathway are mediated via one or more NF-κB–independent pathways. In this regard, it is of interest that α-CD28 costimulation has also been reported to increase IL-2 expression by stabilizing IL-2 mRNA (82). It will be of interest to determine if this mRNA-stabilizing effect of CD28 signaling is regulated via an NF-κB–independent pathway.
The mechanism responsible for the observed reduction in peripheral CD8+ T cells in the mIκB-α mice remains unclear. It may reflect unique roles for NF-κB in the signaling pathways required for the maturation or export of mature CD8+ thymocytes. Alternatively, it may reflect a defect in the survival of CD8+ T cells in the periphery of the mIκB-α mice. In this regard, it is of interest that the survival of peripheral naive SP CD8+ T cells has been reported to require continuous stimulation by class I MHC. Thus, it is possible that this class I MHC–mediated signal is transmitted through an NF-κB–dependent pathway (83).
The Role of NF-κB in DP Thymocyte Apoptosis.
Pro grammed cell death plays an important role in shaping the T cell repertoire during thymocyte ontogeny. More than 95% of immature DP thymocytes die before they exit the thymus (84). Thymocytes that express TCRs with low avidity for antigen plus self-MHC fail to be positively selected and die by “neglect”, whereas self-reactive DP T cells are deleted during the process of negative selection after high affinity engagement of their TCRs by self-antigenic peptides and MHC (85, 86). Our results demonstrate that NF-κB proteins are necessary positive mediators of DP thymocyte apoptosis in response to α-CD3 administration in vivo. These findings were somewhat surprising given several recent studies that have reported that NF-κB proteins are potent inhibitors of TNF-α– and IL-1–mediated apoptosis in fibroblasts (36–39, 87). However, our findings are consistent with previous reports demonstrating that NF-κB proteins can positively regulate apoptosis in certain cell types in vitro including thymocytes and T cell hybridomas (42, 44). The different findings of these studies may reflect the fact that NF-κB can regulate the expression of distinct proapoptotic and antiapoptotic programs in different cell lineages, at different developmental stages of a single lineage, and/or in response to different extracellular signals.
At least three different apoptotic pathways have been identified in thymocytes: (a) a TCR-mediated pathway (63, 88); (b) a glucocorticoid-responsive pathway (89, 90); and (c) a γ irradiation–sensitive pathway (64). Recent evidence has suggested that these pathways can be distinguished at a molecular level. Thus, for example, γ irradiation–induced thymocyte apoptosis requires p53 and does not occur in p53-deficient thymocytes. In contrast, α-CD3–mediated thymocyte apoptosis is p53 independent (64, 91). Our results are consistent with such a model in that the mIκB-α DP thymocytes were protected from α-CD3–induced apoptosis but remained sensitive to programmed cell death induced by γ irradiation. Thus, we would suggest that NF-κB is not required for p53-dependent thymocyte apoptosis, but is a critical positive regulator of at least one p53-independent pathway of programmed cell death in DP thymocytes. In this regard, it will be of interest to study DP thymocyte apoptosis in p53-deficient, mIκB-α transgenic mice.
Our results suggest that NF-κB mediates α-CD3–mediated apoptosis in wild-type DP thymoctyes by downregulating the expression of the antiapoptotic gene bcl-xL. Because bcl-xL is the predominant antiapoptotic gene expressed in DP thymocytes (68, 69) it is not surprising that its downregulation would predispose these cells to apoptosis. Such a model is also consistent with previous studies that have demonstrated that constitutive expression of a bcl-xL transgene in DP thymocytes protects them from α-CD3–mediated apoptosis (69). Recently, an alternatively spliced form of bcl-xL called bcl-xγ has been shown to be expressed in DP thymocytes and to be regulated in response to TCR stimulation (92). Because the Bcl-xL and Bcl-xγ proteins are virtually identical in size and are both recognized by the anti-Bcl-xL antibodies and probes used in our studies, we cannot distinguish them by RNAse protection, Western blotting, or intracellular flow cytometry. Thus, it is possible that one or both isoforms of Bcl-x contribute to the antiapoptotic effects of the IκB-αA32/36 transgene. The effect of NF-κB on Bcl-xL expression in DP thymocytes may be direct or indirect. That is, NF-κB proteins might bind directly to the bcl-xL promoter and downregulate its activity, or alternatively, might control the expression of other (positive and/or negative) regulators of the bcl-xL gene in thymocytes. In this regard, it will be of interest to further characterize the bcl-xL promoter and to study its regulation by NF-κB proteins.
It is important to emphasize that at present, we cannot conclude that the proapoptotic effects of NF-κB in DP thymocytes are thymocyte autonomous. For example, it is possible that α-CD3 administration causes peripheral T cell activation resulting in cytokine expression that in a paracrine fashion causes DP thymocyte toxicity (93). In such a model, decreased cytokine production by peripheral mIκB-α T cells in response to α-CD3 treatment could account for the decreased DP thymocyte apoptosis seen in these animals. Indeed our results demonstrate decreased cytokine production by thymocytes after TCR cross-linking (Fig. 5). However, this non–thymocyte-autonomous model is less likely to account for the resistance to α-CD3–induced thymocyte death seen in the mIκB-α mice because DP thymocytes from the mIκB-α mice are also resistant to α-CD3–mediated killing in FTOC. Thus, it would appear more likely that NF-κB expression in DP thymocytes is directly required for α-CD3–induced apoptosis.
During the last several years, there has been considerable interest in developing inhibitors of NF-κB for the treatment of both autoimmune diseases and cancer. In the case of cancer, such inhibitors might function to increase the sensitivity of tumor cells to apoptotic cell death induced both by cytokines such as TNF-α and by CTLs. In patients with autoimmune disease, such inhibitors might function by suppressing T cell activation in response to self-antigens. Our finding that NF-κB can function to potentiate apoptosis in certain cell types and specifically in DP thymocytes raises several potential concerns about the systemic administration of NF-κB inhibitors. First, it is possible that some tumor cells and/or virus-infected cells, like DP thymocytes, may actually demonstrate decreased apoptosis in response to NF-κB inhibitors. Thus, it is possible that potent NF-κB inhibitors might potentiate tumorgenesis in some tissues and might also reduce host responses against viral pathogens by decreasing susceptibility to apoptosis. Similarly, our finding that NF-κB inhibitors protect DP thymocytes from apoptosis suggests the possibility that the administration of such inhibitors to patients with autoimmune diseases might result in a failure in the negative selection of some DP thymocytes, thereby potentially increasing the generation of autoreactive T cell clones. Given these possibilities, it will be important to carefully assess the effects of NF-κB inhibitors on the apoptotic potential of different human tumor and virus-infected cells and on the negative selection of DP thymocytes before their wide-spread use in human therapy.
Acknowledgments
We thank C. Clendenin for help with the production of transgenic mice, J. Auger for technical assistance with FACS® analyses, and P. Lawrey and L. Gottschalk for help with the preparation of the manuscript and illustrations.
Abbreviations used in this paper
- EMSA
electrophoretic mobility shift assay
- FTOC
fetal thymic organ culture
- HA
hemagglutinin
- IκB-α
inhibitor κB-α
- mIκB-α
mutant IκB-α
- NF-κB
nuclear factor κB
- TUNEL
Tdt-mediated dUTP nick end labeling
Footnotes
Thore Hettmann is a Terry Fox Research Fellow. This work was supported in part by a grant from the NIAID to J.M. Leiden (2 R37 A129637-07) and by the Cancer Center of the University of Chicago, and by a grant from the NIAID to M. Karin (R01 AI43477-01).
References
- 1.Grilli M, Chiu JJ-S, Lenardo MJ. NF-κB and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionary conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 5.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 6.Kunsch C, Ruben S, Rosen C. Selection of optimal κB-Rel DNA-binding motifs: interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 8.Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown K, Park S, Kanno T, Franzoso G, Siebenlist U. Mutual regulation of the transcriptional activator NF-kappa B and its inhibitor, I kappa B-alpha. Proc Natl Acad Sci USA. 1993;90:2532–2536. doi: 10.1073/pnas.90.6.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature. 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 11.Thompson J, Phillips R, Erdjument-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 12.Alkalay I, Yaron A, Hatzubai A, Jung S, Avraham A, Gerlitz O, Pashut-Lavon L, Ben-Neriah Y. In vivo stimulation of IκB phosphorylation is not sufficient to activate NF-κB. Mol Cell Biol. 1995;15:1294–1301. doi: 10.1128/mcb.15.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiDonato JA, Mercurio F, Karin M. Phosphorylation of I kappa B alpha precedes but is not sufficient for its dissociation from NF-kappa B. Mol Cell Biol. 1995;15:1302–1311. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin YC, Brown K, Siebenlist U. Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha: signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B. Proc Natl Acad Sci USA. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 16.Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO (Eur Mol Biol Organ) J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scherer DC, Brockman JA, Chen Z, Maniatis T, Ballard DW. Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc Natl Acad Sci USA. 1995;92:11259–11263. doi: 10.1073/pnas.92.24.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 19.Sen J, Venkataraman L, Shinkai Y, Pierce JW, Alt FW, Burakoff SJ, Sen R. Expression and induction of nuclear factor-kappa B-related proteins in thymocytes. J Immunol. 1995;154:3213–3221. [PubMed] [Google Scholar]
- 20.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-kappa B activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 21.Feuillard J, Dargemont C, Ferreira V, Tarantino N, Debre P, Raphael M, Korner M. Nuclear Rel-A and c-Rel protein complexes are differentially distributed within human thymocytes. J Immunol. 1997;158:2585–2591. [PubMed] [Google Scholar]
- 22.Chen D, Rothenberg EV. Molcular basis for developmental changes in Interleukin-2 gene inducibility. Mol Cell Biol. 1993;13:228–237. doi: 10.1128/mcb.13.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang S-M, Tran A-C, Grilli M, Lenardo MJ. NF-κB subunit regulation in nontransformed CD4+T-lymphocytes. Science. 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 24.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 25.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B cells from p50/ NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 26.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relBis required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 27.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 28.Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. p50-NF-κB complexes partially compensate for the absence of RelB severely increased pathology in p50−/−relB−/−double knockout mice. J Exp Med. 1997;185:1359–1370. doi: 10.1084/jem.185.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-relproto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 30.Doi TS, Takahashi T, Tagushi O, Azuma T, Obata Y. NF-κB RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative response. J Exp Med. 1997;185:953–961. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 32.Horwitz BH, Scott ML, Cherry SR, Bronson RT, Baltimore D. Failure of lymphopoiesis after adoptive transfer of NF-κB-deficient fetal liver cells. Immunity. 1997;6:765–772. doi: 10.1016/s1074-7613(00)80451-3. [DOI] [PubMed] [Google Scholar]
- 33.Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-κB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arsura M, Wu M, Sonnenshein GE. TGFβ1 inhibits NF-κB/Rel activity inducing apoptosis of B cells: transcriptional activation of IκBα. Immunity. 1996;5:31–40. doi: 10.1016/s1074-7613(00)80307-6. [DOI] [PubMed] [Google Scholar]
- 35.Wu M, Arsura M, Bellas RE, FitzGerald MJ, Lee H, Schauer S, Sherr DH, Sonnenshein GE. Inhibition of c-myc expression induces apoptosis of WEHI 231 murine B cells. Mol Cell Biol. 1996;16:5015–5025. doi: 10.1128/mcb.16.9.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 37.Van Antwerp D, Martin S, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 38.Wang C-Y, Mayo MW, Baldwin AS. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–786. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 39.Liu Z-g, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 40.Grimm S, Bauer MK, Baeuerle PA, Schulze K - Osthoff. Bcl-2 down-regulates the activity of transcription factor NF-kappaB induced upon apoptosis. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jung M, Zhang Y, Lee S, Dritschilo A. Correction of radiation sensitivity in ataxia telangiectasia by a truncated IκB-α. Science. 1995;268:1619–1621. doi: 10.1126/science.7777860. [DOI] [PubMed] [Google Scholar]
- 42.Bessho R, Matsubara K, Kubot M, Kuwakado K, Hirota H, Wakazono Y, Wei Y, Lin, Okuda A, Kawai M, Nishikomoro R, Heike T. Pyrrolidine dithiocarbamate, a potent inhibitor of nuclear factor κB (NF-κB) activation, prevents apoptosis in human promyelocytic leukemia HL-60 cells and thymocytes. Biochem Pharmacol. 1994;48:1883–1889. doi: 10.1016/0006-2952(94)90586-x. [DOI] [PubMed] [Google Scholar]
- 43.Grilli M, Pizzi M, Memo M, Spano PF. Neuroproduction by aspirin and sodium salicylate through blockade by NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 44.Ivanov VN, Lee RK, Podack ER, Malek TR. Regulation of Fas-dependent activation-induced T cell apoptosis by cAMP signaling: a potential role for transcription factor NF-κB. Oncogene. 1997;14:2455–2464. doi: 10.1038/sj.onc.1201088. [DOI] [PubMed] [Google Scholar]
- 45.Abbadie C, Kabrun N, Bouali F, Smardova J, Stehelin D, Banderbunder B, Enrietto PJ. High level of c-Rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. . Cell. 1993;75:899–912. doi: 10.1016/0092-8674(93)90534-w. [DOI] [PubMed] [Google Scholar]
- 46.Esslinger CW, Wilson A, Sordat B, Beermann F, Jongeneel CV. Abnormal T cell development induced by targeted overexpression of IκBα. J Immunol. 1997;158:5075–5078. [PubMed] [Google Scholar]
- 47.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lake RA, Wotton D, Owen MJ. A 3′ transcriptional enhancer regulates tissue-specific expression of the human CD2 gene. EMBO (Eur Mol Biol Organ) J. 1990;9:3129–3136. doi: 10.1002/j.1460-2075.1990.tb07510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C-Y, Bassuk AG, Boise LH, Thompson CB, Bravo R, Leiden JM. Activation of the granulocyte-macrophage colony-stimulating factor promoter in T cells requires cooperative binding of Elf-1 and AP-1 transcription factors. Mol Cell Biol. 1994;14:1153–1159. doi: 10.1128/mcb.14.2.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ballard DW, Walker WH, Doerre S, Sista P, Molitor JA, Dixon EP, Peffer NJ, Hannink M, Greene WC. The v-rel oncogene encodes a kappa B enhancer binding protein that inhibits NF-kappa B function. Cell. 1990;63:803–814. doi: 10.1016/0092-8674(90)90146-6. [DOI] [PubMed] [Google Scholar]
- 51.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finco TS, Baldwin AS. Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 53.Lang G, Wotton D, Owen MJ, Sewell WA, Brown MH, Mason DY, Crumpton MJ, Kioussis D. The structure of the human CD2 gene and its expression in transgenic mice. EMBO (Eur Mol Biol Organ) J. 1988;7:1675–1682. doi: 10.1002/j.1460-2075.1988.tb02995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Owen MJ, Jenkinson EJ, Brown MH, Sewell WA, Krissansen GW, Crumpton MJ, Owen JJ. Murine CD2 gene expression during fetal thymus ontogeny. Eur J Immunol. 1988;18:187–189. doi: 10.1002/eji.1830180129. [DOI] [PubMed] [Google Scholar]
- 55.Kolodziej PA, Young RA. Epitope tagging and protein surveillance. Methods Enzymol. 1991;194:508–516. doi: 10.1016/0076-6879(91)94038-e. [DOI] [PubMed] [Google Scholar]
- 56.Moore NC, Girdlestone J, Anderson G, Owen JT, Jenkinson EJ. Stimulation of thymocytes before and after positive selection results in the induction of different NF-κB/Rel protein complexes. J Immunol. 1995;155:4653–4660. [PubMed] [Google Scholar]
- 57.Nolan GP, Fujita T, Bhatia K, Huppi C, Liou HC, Scott ML, Baltimore D. The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol Cell Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plaksin D, Baeuerle PA, Eisenbach L. κBF1 (p50 NF-kappa B homodimer) acts as a repressor of H-2κB gene expression in metastatic tumor cells. J Exp Med. 1993;177:1651–1662. doi: 10.1084/jem.177.6.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verweij CL, Geerts M, Aarden LA. Activation of interleukin-2 gene transcription via the T-cell surface molecule CD28 is mediated through an NF-κB-like response element. J Biol Chem. 1991;266:14179–14182. [PubMed] [Google Scholar]
- 60.Crabtree GR, Clipstone NA. Signal transmission between the plasma mambrane and nucleus of T-lymphocytes. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y, Bissonnette RP, Parfrey N, Szalay M, Kubo RT, Green DR. In vivo administration of mononuclear antibodies to the CD3 T cell receptor complex induces cell death (apoptosis) in immature thymocytes. J Immunol. 1991;146:3340–3346. [PubMed] [Google Scholar]
- 62.Tokoro Y, Tsuda S, Tanaka S, Nakauchi H, Takahama Y. CD3-induced apoptosis of CD4+CD8+thymocytes in the absence of clonotypic T cell antigen receptor. Eur J Immunol. 1996;26:1012–1017. doi: 10.1002/eji.1830260509. [DOI] [PubMed] [Google Scholar]
- 63.Smith CA, Williams AGT, Kingston R, Jenkinson EJ, Owen JJT. Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature. 1989;337:181–184. doi: 10.1038/337181a0. [DOI] [PubMed] [Google Scholar]
- 64.Lowe SW, Schmitt EM, Smith SW, Osborne B, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 65.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 66.Gratiot-Deans J, Ding L, Turka LA, Nunez G. bcl-2 proto-oncogene expression during human T cell development. Evidence for biphasic activation. J Immunol. 1993;151:83–91. [PubMed] [Google Scholar]
- 67.Veis DJ, Sentman CL, Bach EA, Korsmeyer SJ. Expression of the Bcl-2 protein in murine and human thymocytes and in peripheral T lymphocytes. J Immunol. 1993;151:2546–2554. [PubMed] [Google Scholar]
- 68.Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, Thompson CB. Bcl-x regulates the survival of double-positive thymocytes. Proc Natl Acad Sci USA. 1995;92:4763–4767. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grillot DA, Merino R, Nunez G. Bcl-XLdisplays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J Exp Med. 1995;182:1973–1983. doi: 10.1084/jem.182.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chao DT, Linette GP, Boise LH, White LS, Thompson CB, Korsmeyer SJ. Bcl-XLand Bcl-2 repress a common pathway of cell death. J Exp Med. 1995;182:821–828. doi: 10.1084/jem.182.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Durand DB, Shaw J-P, Bush MR, Replogle RE, Belagave R, Crabtree GR. Characterization of antigen receptor response elements within the interleukin-2 enhancer. Mol Cell Biol. 1988;8:1715–1720. doi: 10.1128/mcb.8.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ballard DW, Bohnlein E, Siekevitz M, Greene WC. κB-specific DNA binding proteins: a role in the regulation of human interleukin-2 gene expression. Science. 1989;244:457–459. doi: 10.1126/science.2497518. [DOI] [PubMed] [Google Scholar]
- 73.Serfling E, Avots A, Neumann M. The architecture of the interleukin-2 promoter: a reflection of T lymphocyte activtion. Biochem Biophys Acta. 1995;1263:181–200. doi: 10.1016/0167-4781(95)00112-t. [DOI] [PubMed] [Google Scholar]
- 74.Jain J, Loh C, Rao A. Transcriptional regulation of the interleukin 2 gene. Curr Opin Immunol. 1995;7:333–342. doi: 10.1016/0952-7915(95)80107-3. [DOI] [PubMed] [Google Scholar]
- 75.Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science. 1988;241:202–205. [PubMed] [Google Scholar]
- 76.Jain J, Miner Z, Rao A. Analysis of the preexisting and nuclear forms of nuclear factor of activated T cells. J Immunol. 1993;151:837–848. [PubMed] [Google Scholar]
- 77.Hodge MR, Ranger AM, de la Brousse FC, Hoey T, Grusby M, Glimcher LH. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity. 1996;4:397–405. doi: 10.1016/s1074-7613(00)80253-8. [DOI] [PubMed] [Google Scholar]
- 78.Xanthoudakis S, Viola JPB, Shaw KTY, Luo C, Wallace JD, Bozza PD, Luk DC, Curran T, Rao A. An enhanced immune response in mice lacking the transcription factor NFAT1. Science. 1996;272:892–895. doi: 10.1126/science.272.5263.892. [DOI] [PubMed] [Google Scholar]
- 79.Lai JH, Tan TH. CD28 signaling causes a sustained down-regulation of I kappa B alpha which can be prevented by the immunosuppressant rapamycin. J Biol Chem. 1994;269:30077–30080. [PubMed] [Google Scholar]
- 80.Bryan RG, Li Y, Lai JH, Van M, Rice NR, Rich RR, Tan TH. Effect of CD28 signal transduction on c-Rel in human peripheral blood T cells. Mol Cell Biol. 1994;14:7933–7942. doi: 10.1128/mcb.14.12.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghosh P, Tan TH, Rice NR, Sica A, Young HA. The interleukin 2 CD28-responsive complex contains at least three members of the NF kappa B family: c-Rel, p50, and p65. Proc Natl Acad Sci USA. 1993;90:1696–1700. doi: 10.1073/pnas.90.5.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1998;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 83.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 84.Osborne BA. Apoptosis and maintenance of homeostasis in the immune system. Curr Opin Immunol. 1996;8:245–254. doi: 10.1016/s0952-7915(96)80063-x. [DOI] [PubMed] [Google Scholar]
- 85.Allen PM. Peptides in negative and positive selection: a delicate balance. Cell. 1994;76:593–596. doi: 10.1016/0092-8674(94)90497-9. [DOI] [PubMed] [Google Scholar]
- 86.Nossal GJV. Negative selection of lymphocytes. Cell. 1994;76:229–232. doi: 10.1016/0092-8674(94)90331-x. [DOI] [PubMed] [Google Scholar]
- 87.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 88.Jenkinson EJ, Kingston R, Jenkinson EJ, Owen JJT. Antigen-induced apoptosis in developing T cells: a mechanism for negative selection of the T cell repertoire. Eur J Immunol. 1989;19:2175–2181. doi: 10.1002/eji.1830191132. [DOI] [PubMed] [Google Scholar]
- 89.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–557. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 90.Cohen JJ, Duke RC. Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. J Immunol. 1984;123:38–45. [PubMed] [Google Scholar]
- 91.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 92.Yang X-F, Weber GF, Cantor H. A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity. 1997;7:629–639. doi: 10.1016/s1074-7613(00)80384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lenardo MJ. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–891. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 94.Haskill S, Beg AA, Tompkins SM, Morris JS, Yurochko AD, Sampson-Johannes A, Mondal K, Ralph P, Baldwin AS., Jr Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell. 1991;65:1281–1289. doi: 10.1016/0092-8674(91)90022-q. [DOI] [PubMed] [Google Scholar]