

Abstract
Tumor necrosis factor (TNF) is a central mediator of a number of important pathologies such as the systemic inflammatory response syndrome. Administration of high TNF doses induces acute anorexia, metabolic derangement, inflammation, and eventually shock and death. The in vivo effects of TNF are largely mediated by a complex network of TNF-induced cytokines and hormones acting together or antagonistically. Since TNF also induces leptin, a hormone secreted by adipocytes that modulates food intake and metabolism, we questioned the role of leptin in TNF-induced pathology. To address this question, we tested mouse strains that were defective either in leptin gene (ob/ob) or in functional leptin receptor gene (db/db), and made use of a receptor antagonist of leptin. Ob/ob and db/db mice, as well as normal mice treated with antagonist, exhibited increased sensitivity to the lethal effect of TNF. Exogenous leptin afforded protection to TNF in ob/ob mice, but failed to enhance the protective effect of endogenous leptin in normal mice. We conclude that leptin is involved in the protective mechanisms that allow an organism to cope with the potentially autoaggressive effects of its immune system.
Keywords: leptin, tumor necrosis factor, anorexia, shock, obesity
Tumor necrosis factor (TNF) is a pleiotropic cytokine with potent antitumor activity; however, it is also involved in the pathogenesis of many inflammatory diseases. Above all, TNF is a central mediator of the potentially lethal systemic inflammatory response syndrome (1). A single injection of TNF into animals causes acute anorexia, weight loss, metabolic derangement, hypotension, and, at very high doses, shock and death as a result of a widespread systemic inflammatory reaction (2, 3). However, prolonged treatment with lower doses of TNF results in tachyphylaxis and tolerance (4–6). These effects are largely mediated by a complex network of cytokines, hormones, and low-molecular weight mediators induced by TNF (7). This network is not only responsible for the deleterious outcome, but also for the induction of often more complex endogenous protection mechanisms whose function allows the organism to cope with the potentially autoaggressive consequences of immune/inflammatory reactions. Glucocorticoid hormones represent the most powerful antiinflammatory arm (8), whereas some proinflammatory cytokines, such as IL-1 and IL-6, play a dual role by also contributing to protective mechanisms via induction of acute-phase proteins (9). Some acute-phase proteins act as protease inhibitors to limit the destructive effects of TNF-induced proteases (10, 11). The exact significance of each of these mediators is far from established. In this study we wanted to investigate the role of leptin, which was described as being induced by TNF (12, 13). Leptin is the product of the ob gene associated with obesity (14). Mutant mice with a defective leptin gene (ob/ob) or leptin receptor gene (db/db) exhibit hyperphagia, reduced energy expenditure, and obesity (15, 16). Administration of leptin was found to reduce food intake in both normal and ob/ob mice (17, 18). Recently, a specific receptor antagonist of leptin was obtained by introducing a point mutation into the human leptin gene (19). This leptin mutein binds to the leptin receptor but fails to transduce a signal. When injected into normal mice, the leptin antagonist induced weight gain (19). Given the possible link between TNF and leptin on the production level, we speculated on the role of leptin in TNF-induced effects. Using mutant mouse strains and antagonist, we investigated the role of endogenous leptin in TNF-induced lethality.
Materials and Methods
Animals.
Specific pathogen–free female C57BL/6 mice, 8–12 wk old at the beginning of the experiments, were obtained from Charles River Labs. Specific pathogen–free C57BL/6J mice (referred to as wild-type [wt]), C57BL/6-ob mice (referred to as ob/ob), and C57BL/Ks-db mice (further referred to as db/db) were obtained from Harlan/Olac and The Jackson Laboratory. The animals were housed in a temperature-controlled environment with 12-h light/dark cycles and received food and water ad libitum. All experiments were performed according to the European Union Guidelines on Animal Care and Use.
Reagents.
Recombinant murine TNF (mTNF), produced by Escherichia coli containing an appropriate expression plasmid (20), was purified to apparent homogeneity. The specific activity was 2 × 108 IU/mg as determined in a cytotoxicity assay on L929 cells (21). Reference mTNF (code 88/532) was from the National Institute for Biological Standards and Control (Potters Bar, UK). The endotoxin content was <0.2 ng/mg, as assessed by a chromogenic Limulus amebocyte lysate assay (Coatest; Chromogenix, Stockholm, Sweden). Human wt leptin was produced by baculovirus-infected insect cells (22) and purified on an immunoaffinity column as described previously (19); the endotoxin content amounted to 2.5 ng/mg. R128Q, an antagonist of human leptin, was created by site-directed mutagenesis and selected for its inhibitory activity on leptin sensitive-BAF3 1423 cells (19); the endotoxin content amounted to 2.1 ng/mg. 2A5, a monoclonal antibody directed against human leptin, was purified from hybridoma supernatant (19); the endotoxin content was 0.09 ng/ mg protein. All reagents were diluted in endotoxin-free PBS before injection.
Treatment.
In experiments involving comparison of the sensitivity of different mouse strains, mice were challenged with sublethal doses of mTNF given intravenously. mTNF is lethal in normal mice at a dose of ∼20 μg (23). Survival was monitored for up to 60 h. There were no further deaths during the 1-wk period of follow-up. In experiments assessing the effect of leptin or leptin antagonist in TNF toxicity, mice were pretreated intraperitoneally with either agent in combination with an antibody (twice daily for 2 d and immediately before the challenge with varying doses of mTNF intravenously). Doses of leptin and leptin antagonist R128Q were 100 μg/mouse; doses of antibody 2A5 were 1 mg/mouse or 100 μg/mouse. Leptin and R128Q are cleared from the circulation very rapidly. The antibody 2A5 raised against human leptin also binds to R128Q and dramatically prolongs the biological half-life of both wt and mutant leptin mice. It was also demonstrated that the biological effects of leptin and the antagonist in wt mice are only seen in the presence of antibody (19). For this reason, antibody was coadministered whenever leptin or R128Q were used. The injection volumes were 0.5 ml in the case of intraperitoneal and 0.2 ml in the case of intravenous administration.
Statistics.
The significance of differences in survival time was analyzed by a Log-rank test for curve comparison using a GraphPad Prism computer program (GraphPad Software). In all cases, P < 0.05 was considered to be significantly different.
Results
Mice Lacking Leptin-signaling Are Highly Sensitive to the Lethal Toxicity of mTNF.
To assess the role of endogenous leptin induced by TNF, we first tested mutant mouse strains lacking a functional leptin system. Both ob/ob and db/db mice were challenged with 500 μg/kg mTNF. This dose of TNF, which does not cause lethality in wt mice, resulted in 100 and 80% lethality in ob/ob and db/db mice, respectively (Fig. 1). Therefore, both ob/ob and db/db mice are far more sensitive to the lethal effect of mTNF (P = 0.0001 ob/ob versus wt; P = 0.0006 db/db versus wt). This suggests that a functional leptin system protects against a low dose of mTNF.
Figure 1.

Sensitivity of ob/ob and db/db mice to mTNF-induced lethality. Mice (wt, ob/ob and db/db) were challenged with 500 μg/kg mTNF intravenously. The percentage of survival was plotted as a function of time (hours after challenge). n = 10 (wt) and 5 (ob/ob and db/db). ○, wt; □, ob/ob; ▵, db/db. ****P = 0.0001 (wt versus ob/ob); ***P = 0.0006 (wt versus db/db). The results shown are representative of three independent experiments.
Exogenous Leptin Protects ob/ob Mice from the Lethal Toxicity of mTNF.
We tested whether exogenous leptin can protect ob/ob mice lacking endogenous leptin. Leptin and 2A5 antibody were administered twice daily for 2 d and at the same time as a challenge with mTNF. In agreement with the previous experiment, ob/ob mice exhibited significantly higher sensitivity to TNF as compared with wt mice (Fig. 2; P = 0.0025 versus wt receiving 2A5 alone). Ob/ob mice pretreated with leptin showed resistance comparable to that of wt, and the survival time was significantly longer (P = 0.0079 versus ob/ob receiving 2A5 alone). There was no significant difference between ob/ob mice pretreated with leptin and wt mice. These data confirm our previous evidence that endogenous leptin is protective against the lethal toxicity of TNF and demonstrate that the lack of endogenous leptin can be compensated by administration of exogenous leptin.
Figure 2.

Effect of leptin pretreatment in ob/ob mice receiving a lethal challenge with mTNF. Mice (wt and ob/ob) were pretreated as described in Materials and Methods. Doses of 2A5 antibody and leptin were both 100 μg/mouse. Mice were challenged with 500 μg/kg mTNF. The percentage of survival was plotted as a function of time (hours after challenge). For each group, n = 5. ○, 2A5 in wt mice; □, 2A5 in ob/ob mice; ▵, leptin + 2A5 in ob/ob mice. **P = 0.0025 (wt versus ob/ob); *P = 0.0079 (ob/ob-2A5 versus ob/ob-leptin). The results shown are representative of three independent experiments.
Leptin Antagonist Sensitizes Normal Mice to the Lethal Toxicity of TNF.
If sensitization to the toxic effect of TNF in the genetically defective leptin system is fully attributable to leptin (and not to the consequence of secondary effects, such as obesity, elevated levels of insulin, or corticoids), the same sensitization should be obtained in normal lean mice, where the function of leptin is blocked. Therefore, we examined the effect of leptin and leptin antagonist R128Q on the lethal toxicity of mTNF in normal mice. We tested two doses of mTNF that would result in ∼50 and 0% lethality, respectively, in normal mice. Mice were pretreated with leptin or R128Q antagonist, both in the presence of 100 μg 2A5 antibody per mouse. Treatment with R128Q clearly sensitized mice to TNF toxicity, since 60% died even in response to a low dose of mTNF (Fig. 3 A; P = 0.0008, a significant difference compared with the control group receiving 2A5 alone). Moreover, treatment with R128Q and a higher dose of TNF increased the death toll from 50 to 100%, and the survival time was significantly reduced (Fig. 3 B; P = 0.0063, compared with the control group receiving 2A5 alone). On the other hand, exogenous leptin did not provide any further protection, suggesting that the endogenous leptin level is sufficient for a protective effect. The difference observed between mice pretreated with leptin antagonist and those with antibody alone is due to the functional level of leptin. These experiments allow us to conclude that endogenous leptin protects against TNF-induced lethality.
Figure 3.


Effect of leptin and leptin antagonist on mTNF-induced lethality. C57BL/6 mice were pretreated with leptin (100 μg/mouse) or R128Q (100 μg/mouse) in combination with 2A5 antibody (100 μg/ mouse) as described in Materials and Methods. Mice were challenged with a low dose (A, 375 μg/kg) or a high dose (B, 750 μg/kg) of mTNF. The percentage of survival was plotted as a function of time (hours after challenge). Each group consisted of 13 mice. The result is a cumulative sum of two independent experiments. (A) ○, 2A5; □, leptin + 2A5; ▵, R128Q + 2A5; ***P = 0.0008 (2A5 versus R128Q + 2A5); (B) •, 2A5; ▪, leptin + 2A5; ▴, R128Q + 2A5. **P = 0.0063 (2A5 versus R128Q + 2A5).
Discussion
The finding that endogenous leptin plays a protective role against TNF-induced lethality suggests an entirely new property to be attributed to this protein, namely immunomodulatory and antiinflammatory effects. Recently, it was also reported that genetically obese rodents were more susceptible to endotoxin-induced liver injury (24). This could be explained by increased sensitivity to TNF in the absence of a functional leptin system as shown here. Although increased production of certain proinflammatory cytokines by leptin is reported (24, 25), these results rather point to a possible impairment of the function of these cytokines by leptin. A recent finding that leptin upregulates suppressors of cytokine signaling 3 (26) supports such a speculation. The protective role of leptin reminds us of another class of TNF-induced hormones, namely glucocorticoids. Also in this case, mice devoid of a functional glucocorticoid system (by adrenalectomy or administration of a receptor antagonist) are highly sensitized to the toxic effects of TNF (27, 28). The fact that exogenous leptin is able to reverse the increased sensitivity of mice lacking an active leptin system, and that it is ineffective in further decreasing the sensitivity of normal mice, indicates that the leptin levels after TNF induction are high enough to obtain a maximal effect. This is also similar to the situation with glucocorticoids, where adding exogenous hormone could not improve survival in sepsis; on the contrary, blocking the action of this hormone by administering a receptor antagonist, even hours after TNF administration, increased the lethality (28). TNF activates the hypothalamus pituitary-adrenal axis (HPAA), resulting in a release of glucocorticoids, which in turn attenuate the immune/inflammatory reaction (29). Both leptin and glucocorticoids induced by TNF itself seem to exert a regulatory negative feedback to prevent the potentially harmful consequences of the proinflammatory cascade. The presence of a bidirectional relationship is suggestive of a coordinated function. Glucocorticoids elevate the leptin level (30), whereas leptin elicits an effect on HPAA and alters the circulating glucocorticoid levels (31). Besides, leptin signaling might lead to an enhanced level of another antiinflammatory hormone, α-melanocyte-stimulating hormone (α-MSH; reference 32). It is highly likely that the protective effect of leptin is mediated by α-MSH, which was shown to protect against endotoxin-induced liver injury and mortality (33, 34). The action of α-MSH resembles that of glucocorticoids in that it inhibits the production of proinflammatory mediators (33, 35). Thus a picture emerges that leptin, acting on two parallel antiinflammatory axes, modulates the defence of the body against the TNF-induced inflammatory cascade (Fig. 4). Both α-MSH and corticotrophins are derived from the proopiomelanocortin gene and partly share common receptor subtypes. Thus these two arms are also interrelated. One of the receptors for α-MSH, melanocortin-1 receptor, is found on neutrophils and macrophages, and is postulated to mediate the antiinflammatory effects of α-MSH (33, 35). However, the antiinflammatory effect of α-MSH involves both central and peripheral signals (36), suggesting an involvement of yet another receptor subtype that mediates central signaling. Quite intriguingly, it was demonstrated that melanocortin-4 receptor, another MSH receptor, mediated the anorectic effect of leptin (37). Whether or not this receptor type also plays a role in the potentially antiinflammatory function of leptin should be addressed in the future. The fact that db/db mice lacking the functional long form of the leptin receptor (38) are also sensitized to TNF indicates that signaling through this type of receptor is essential for the protective effect. The leptin receptor belongs to the class-I cytokine receptor family (16), which suggests an ancestral link between the cytokine system and leptin. Furthermore, leptin itself is structurally related to the cytokines (39). It is intriguing that there is an overlap between the activities of TNF and leptin (Fig. 4). Both TNF and leptin activate the HPAA and melanocortin system (35), and it appears that leptin provides a redundant way to ensure protection. On the other hand, HPAA exerts a negative feedback on the production of TNF and a positive feedback on leptin production. In this way, the protective pathway induced by TNF can be amplified in the absence of TNF.
Figure 4.
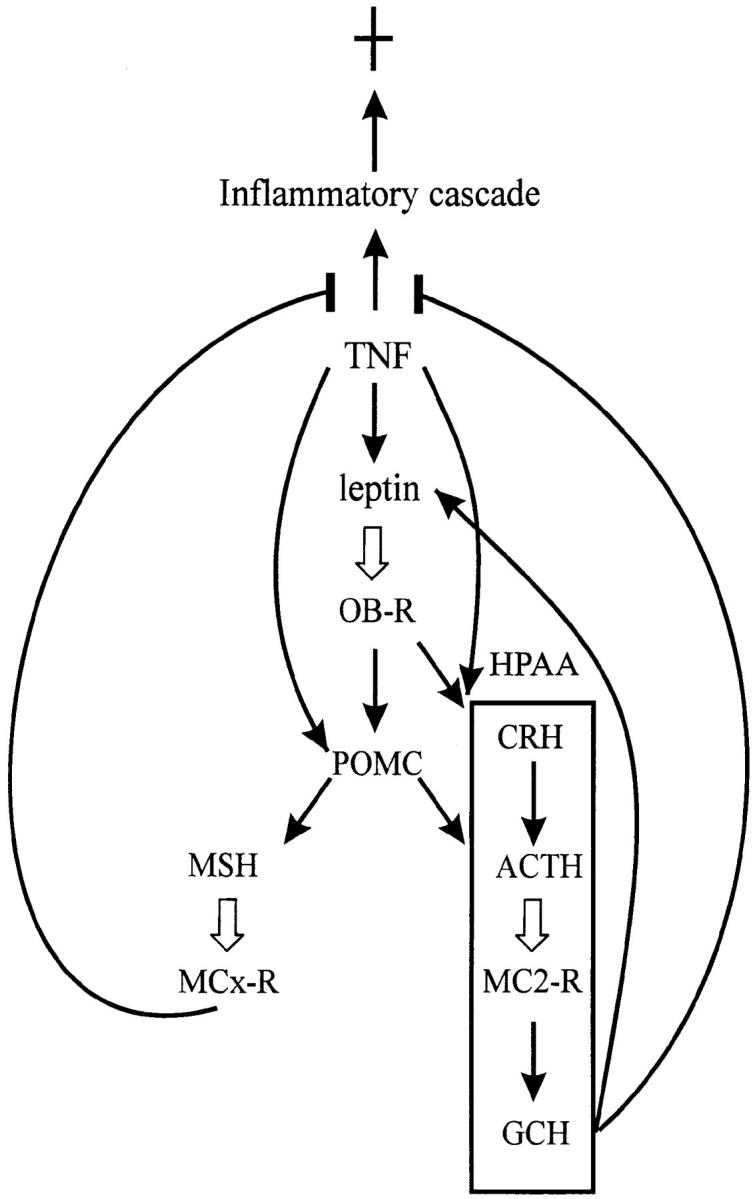
Immune neuroendocrine network involving TNF and leptin. TNF induces a proinflammatory cascade on the one hand and an antiinflammatory protection mechanism on the other hand. → , induction; ¢, inhibition; ⇒ , binding to a receptor. GCH, glucocorticoid hormone; OB-R, leptin receptor long form; MC2-R, melanocortin-2 receptor; MCx-R, melanocortin-1 receptor or possibly other subtypes.
We also found that TNF can still induce weight loss in the absence of a functional leptin system (results not shown). This rules out the mediation of leptin in this effect. Recently, a similar result on endotoxin-induced anorexia was published using ob/ob and db/db mice (39). In this case, TNF is presumably a principal mediator, and the study confirms our finding. Therefore, TNF and leptin seem to control the body weight independently. It is not unlikely that both induce either a common mediator or partly common signaling pathways. Recently, a defective melanocortin-4 receptor was shown to cause agouti-type obesity (40). Considering the role of this receptor in leptin-induced anorexia (37), it is extremely tempting to speculate that it is part of the common pathway. It should be noted that anorectic factors, such as melanocortins and corticotrophin- releasing factor, can be independently activated by TNF and leptin (Fig. 4). Here again, factors involved in the immune/neuroendocrine network form a partly common and partly distinct network.
In conclusion, the finding of a protective role for leptin against TNF toxicity extends the picture of communication between the immune system and the neuroendocrine system. It is attractive to speculate that the leptin system, with its structural relationship to the cytokine system (41) and functional similarity to glucocorticoids, evolved to form a partly redundant and partly distinct regulatory network to maintain homeostasis both under physiological and pathological conditions. Future studies will clarify the molecular basis underlying the communication among cytokine/glucocorticoid/leptin systems.
Acknowledgments
This work was supported by the Fonds voor Wetenschappelijk Onderzoek—Vlaanderen and the Algemene Spaar- en Lijfrentekas. Wim Waelput is a Fellow with the Vlaams Instituut voor de Bevordering van het Wetenschappelijk-technologisch Onderzoek in de Industrie.
References
- 1.Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 2.Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, III, Zentella A, Albert JD, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 3.Tracey, K.J. 1992. The acute and chronic pathophysiologic effects of TNF: mediation of septic shock and wasting (cachexia). In Tumor Necrosis Factors. The Molecules and Their Emerging Role in Medicine. B. Beutler, editor. Raven Press, New York, NY. 255–273.
- 4.Fraker DL, Stovroff MC, Merino MJ, Norton JA. Tolerance to tumor necrosis factor in rats and the relationship to endotoxin tolerance and toxicity. J Exp Med. 1988;168:95–105. doi: 10.1084/jem.168.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tracey KJ, Wei H, Manogue KR, Fong Y, Hesse DG, Nguyen HT, Kuo GC, Beutler B, Cotran RS, Cerami A, Lowry SF. Cachectin/tumor necrosis factor induces cachexia, anemia, and inflammation. J Exp Med. 1988;167:1211–1227. doi: 10.1084/jem.167.3.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi N, Brouckaert P, Fiers W. Mechanism of tolerance to tumor necrosis factor: receptor-specific pathway and selectivity. Am J Physiol. 1995;269:R398–R405. doi: 10.1152/ajpregu.1995.269.2.R398. [DOI] [PubMed] [Google Scholar]
- 7.Brouckaert P, Fiers W. Tumor necrosis factor and the systemic inflammatory response syndrome. Curr Top Microbiol Immunol. 1996;216:167–187. doi: 10.1007/978-3-642-80186-0_8. [DOI] [PubMed] [Google Scholar]
- 8.Munck, A., and P.M. Guyre. 1991. Glucocorticoids and immune function. In Psychoimmunology. R. Ader, D. Felten, and N. Cohen, editors. Academic Press, New York, NY. 447–479.
- 9.Castell JV, Gómez-Lechón MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242:237–239. doi: 10.1016/0014-5793(89)80476-4. [DOI] [PubMed] [Google Scholar]
- 10.Libert C, Brouckaert P, Fiers W. Protection by α1-acid glycoprotein against tumor necrosis factor-induced lethality. J Exp Med. 1994;180:1571–1575. doi: 10.1084/jem.180.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libert C, Van Molle W, Brouckaert P, Fiers W. α1-antitrypsin inhibits the lethal response to TNF in mice. J Immunol. 1996;157:5126–5129. [PubMed] [Google Scholar]
- 12.Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ, III, Flier JS, Lowell BB, Fraker DL, Alexander HR. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med. 1997;185:171–175. doi: 10.1084/jem.185.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, Feingold KR. Endotoxin and cytokines induce expression of leptin, the obgene product, in hamsters. J Clin Invest. 1996;97:2152–2157. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obesegene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 15.Zhang B, Graziano MP, Doebber TW, Leibowitz MD, White-Carrington S, Szalkowski DM, Hey PJ, Wu M, Cullinan CA, Bailey P, et al. Down-regulation of the expression of the obese gene by an antidiabetic thiazolidinedione in Zucker diabetic fatty rats and db/dbmice. J Biol Chem. 1996;271:9455–9459. doi: 10.1074/jbc.271.16.9455. [DOI] [PubMed] [Google Scholar]
- 16.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 17.Frederich RC, Lollmann B, Hamann A, Napolitano-Rosen A, Kahn BB, Lowell BB, Flier JS. Expression of obmRNA and its encoded protein in rodents. Impact of nutrition and obesity. J Clin Invest. 1995;96:1658–1663. doi: 10.1172/JCI118206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 19.Verploegen SABW, Plaetinck G, Devos R, Van der Heyden J, Guisez Y. A human leptin mutant induces weight gain in normal mice. FEBS Lett. 1997;405:237–240. doi: 10.1016/s0014-5793(97)00192-0. [DOI] [PubMed] [Google Scholar]
- 20.Tavernier, J., L. Fransen, A. Marmenout, J. Van der Heyden, R. Müller, M.R. Ruysschaert, A. Van Vliet, R. Bauden, and W. Fiers. 1987. Isolation and expression of the genes coding for mouse and human tumor necrosis factor (TNF) and biological properties of recombinant TNF. In Lymphokines, vol. 13: Molecular Cloning and Analysis of Lymphokines. D.R. Webb and D.V. Goeddel, editors. Academic Press, Orlando, FL. 181–198.
- 21.Ruff, M.R., and G.E. Gifford. 1981. Tumor necrosis factor. In Lymphokines, vol. 2. E. Pick, editor. Academic Press, New York, NY. 235–272.
- 22.Guisez Y, Faché I, Campfield LA, Smith FJ, Farid A, Plaetinck G, van der Heyden J, Tavernier J, Fiers W, Burn P, Devos R. Efficient secretion of biologically active recombinant OB protein (leptin) in Escherichia coli, purification from the periplasm and characterization. Protein Expr Purif. 1998;12:249–258. doi: 10.1006/prep.1997.0836. [DOI] [PubMed] [Google Scholar]
- 23.Brouckaert P, Libert C, Everaerdt B, Fiers W. Selective species specificity of tumor necrosis factor for toxicity in the mouse. Lymphokine Cytokine Res. 1992;11:193–196. [PubMed] [Google Scholar]
- 24.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, Klein AS, Bulkley GB, Bao C, Noble PW, et al. Leptin regulates proinflammatory immune responses. FASEB (Fed Am Soc Exp Biol) J. 1998;12:57–65. [PubMed] [Google Scholar]
- 26.Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 27.Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1708–1712. doi: 10.1084/jem.167.5.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brouckaert P, Everaerdt B, Fiers W. The glucocorticoid antagonist RU38486 mimics interleukin-1 in its sensitization to the lethal and interleukin-6-inducing properties of tumor necrosis factor. Eur J Immunol. 1992;22:981–986. doi: 10.1002/eji.1830220416. [DOI] [PubMed] [Google Scholar]
- 29.Sharp BM, Matta SG, Peterson PK, Newton R, Chao C, McAllen K. Tumor necrosis factor-α is a potent ACTH secretagogue: comparison to interleukin-1β. Endocrinology. 1989;124:3131–3133. doi: 10.1210/endo-124-6-3131. [DOI] [PubMed] [Google Scholar]
- 30.Slieker LJ, Sloop KW, Surface PL, Kriauciunas A, LaQuier F, Manetta J, Bue-Valleskey J, Stephens TW. Regulation of expression of obmRNA and protein by glucocorticoids and cAMP. J Biol Chem. 1996;271:5301–5304. doi: 10.1074/jbc.271.10.5301. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thornton JE, Cheung CC, Clifton DK, Steiner RA. Regulation of hypothalamic proopiomelanocortin mRNA by leptin in ob/obmice. Endocrinology. 1997;138:5063–5066. doi: 10.1210/endo.138.11.5651. [DOI] [PubMed] [Google Scholar]
- 33.Lipton JM, Ceriani G, Macaluso A, McCoy D, Carnes K, Biltz J, Catania A. Antiinflammatory effects of the neuropeptide α-MSH in acute, chronic, and systemic inflammation. Ann NY Acad Sci. 1994;741:137–148. doi: 10.1111/j.1749-6632.1994.tb39654.x. [DOI] [PubMed] [Google Scholar]
- 34.Chiao H, Foster S, Thomas R, Lipton J, Star RA. α-melanocyte-stimulating hormone reduces endotoxin-induced liver inflammation. J Clin Invest. 1996;97:2038–2044. doi: 10.1172/JCI118639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Star RA, Rajora N, Huang J, Stock RC, Catania A, Lipton JM. Evidence of autocrine modulation of macrophage nitric oxide synthase by α-melanocyte-stimulating hormone. Proc Natl Acad Sci USA. 1995;92:8016–8020. doi: 10.1073/pnas.92.17.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macaluso A, McCoy D, Ceriani G, Watanabe T, Biltz J, Catania A, Lipton JM. Antiinflammatory influences of α-MSH molecules: central neurogenic and peripheral actions. J Neurosci. 1994;14:2377–2382. doi: 10.1523/JNEUROSCI.14-04-02377.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW. Melanocortin receptors in leptin effects. Nature. 1997;390:349. doi: 10.1038/37016. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/dbmice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 39.Faggioni R, Fuller J, Moser A, Feingold KR, Grunfeld C. LPS-induced anorexia in leptin-deficient (ob/ob) and leptin receptor-deficient (db/db)mice. Am J Physiol. 1997;273:R181–R186. doi: 10.1152/ajpregu.1997.273.1.R181. [DOI] [PubMed] [Google Scholar]
- 40.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 41.Madej T, Boguski MS, Bryant SH. Threading analysis suggests that the obesegene product may be a helical cytokine. FEBS Lett. 1995;373:13–18. doi: 10.1016/0014-5793(95)00977-h. [DOI] [PubMed] [Google Scholar]