

Abstract
Autoimmune diseases, like rheumatoid arthritis, result from a dysregulation of the immune response culminating in hyperactivation of effector cells leading to immune-mediated injury. To maintain an appropriate immune response and prevent the emergence of autoimmune disease, activation signals must be regulated by inhibitory pathways. Biochemical and genetic studies indicate that the type IIB low-affinity receptor for immunoglobulin (Ig)G (FcγRIIB) inhibits cellular activation triggered through antibody or immune complexes and may be an important component in preventing the emergence of autoimmunity. To investigate the role of FcγRIIB in the development of type II collagen (CII)-induced arthritis (CIA), a model for rheumatoid arthritis in humans, we have examined its contribution in determining the susceptibility to CIA in the nonpermissive H-2b haplotype. H-2b mice immunized with bovine CII do not develop appreciable disease. In contrast, immunization of the FcγRIIB-deficient, H-2b mice with bovine CII induced CIA at an incidence of 42.2%. The maximal arthritis index of the FcγRIIB-deficient mice developing CIA (6.9 ± 3.6) was comparable to that of DBA/1 mice (8.6 ± 1.9), an H-2q strain susceptible for CIA induction. IgG1, IgG2a, and IgG2b antibody responses against CII were elevated in the FcγRIIB-deficient animals, especially in those mice showing arthritis, but less pronounced than DBA/1 mice. Histological examinations of the arthritic paws from FcγRIIB-deficient mice revealed that cartilage was destroyed and bone was focally eroded in association with marked lymphocyte and monocyte/macrophage infiltration, very similar to the pathologic findings observed in DBA/1 mice. These results indicate that a nonpermissive H-2b haplotype can be rendered permissive to CIA induction through deletion of FcγRIIB, suggesting that FcγRIIB plays a critical role in suppressing the induction of CIA.
Keywords: collagen-induced arthritis, autoimmunity, Fc receptor, gene targeting, macrophage
The Fc receptors (FcRs) for Igs constitute a family of hematopoietic cell surface molecules that include receptors which can either stimulate or inhibit cellular responses upon binding of antibody–antigen complexes (for reviews, see references 1–6). Triggering the activation receptors, FcγRI and III or FcεRI elicits a variety of effector functions, including phagocytosis (7–9), antibody-dependent cell-mediated cytotoxicity (10–13), and the release of inflammatory mediators (for reviews, see references 1 and 2). Analysis of FcR-deficient mice has revealed the central roles these receptors play in the mechanism of initiating type I, II, and III hypersensitivity reactions. In vivo, the binding of antibody–antigen complexes to their cognate FcRs is both necessary and sufficient to trigger anaphylaxis (11, 12, 14–16), autoimmune hemolytic anemia and thrombocytopenia (13), the Arthus reaction (17–19), and autoimmune glomerulonephritis (20). In addition, the interaction of cytotoxic antitumor antibodies with FcRs is a necessary prerequisite for mediating the in vivo activity of these molecules (21).
These activation responses are modulated by the type IIB FcR for IgG (FcγRIIB),1 the most widely expressed FcR. FcγRIIB suppresses B cell, mast cell, and macrophage activation triggered by cross-linking B cell receptor (BCR) or FcRs (22–25). Disruption of FcγRIIB by gene targeting resulted in mice with elevated Ig levels in response to both thymus-dependent and thymus-independent antigens, enhanced passive cutaneous anaphylaxis reaction (26), and enhanced immune complex (IC)-mediated alveolitis (25). These studies indicate that FcγRIIB physiologically acts as a negative regulator of IC-triggered activation (26) and may function in vivo to suppress autoimmunity by regulating both B cell responses and effector cell activation.
Collagen-induced arthritis (CIA), a model for rheumatoid arthritis (RA) in humans, is a chronic inflammatory arthropathy that can be induced in susceptible rodents by immunization with native type II collagen (CII [27–31]). The histopathology of this arthritis is characterized by a proliferative synovitis that erodes the adjacent cartilage, ultimately producing articular injury and ankylosis. Detailed investigations of the immune responses to CII have been undertaken to determine the precise sequence of events leading to CIA. The development of arthritis is thought to be associated with the synergistic effect of high levels of cell-mediated and humoral immunity to CII (27, 29, 30). CIA and RA are clearly associated with the MHC region (32), and in mice only H-2q and H-2r haplotypes are susceptible to CIA (33, 34). The responsible gene in the H-2q haplotype has been isolated and codes for the Aq class II molecule (35), which binds peptides derived from CII, thus leading to T cell activation which is of crucial importance for development of arthritis in this model (36, 37). In addition, a strong B cell response is activated in CIA, producing IgG directed towards CII-specific structures (28, 38). There is evidence that these antibodies are directly pathogenic, as shown by transfer experiments (39, 40), as well as synergizing with activated T cells to promote the development of arthritis (41, 42). B cell–deficient mice on a susceptible background do not develop CIA, indicating that B cells play a crucial role for development of CIA (43).
In this study, we demonstrate that FcγRIIB-deficient (FcγRIIB−/−) mice on a nonpermissive background (H-2b) become susceptible to CIA induction upon immunization with CII. The histopathological characteristics of the arthritic paws were similar to those observed in CIA-susceptible DBA/1 mice (H-2q). FcγRIIB−/− animals show augmented anti-CII IgG production, as well as elevated release of proinflammatory mediators by macrophages stimulated with IgG ICs, suggesting a mechanism for CIA induction in a nonpermissive background. These results suggest that FcγRIIB normally suppresses the emergence of autoimmune disease, and its modulation could be a factor in determining susceptibility and disease severity in the pathogenesis of RA.
Materials and Methods
Animals.
FcγRIIB−/− mice were generated in the 129/SvJ (H-2b) and C57BL/6 (H-2b) hybrid background as described previously (26). These mice and their wild-type counterparts (129/ BL6 hybrids) were kept and bred in the Animal Unit of The Institute of Development, Aging and Cancer, an environmentally controlled and specific pathogen–free facility. DBA/1 and C57BL/6 mice were obtained from Charles River Japan, Inc. All experiments were performed on 8–12-wk-old, age-matched male mice.
Induction of Arthritis.
Bovine CII was obtained from Collagen Gijutsu-kenshukai (Tokyo, Japan) and dissolved at a concentration of 4 mg/ml in 0.02 M Tris/0.15 M NaCl (pH 8.0) at 4°C. Mice were immunized at the tail base with 200 μg of CII emulsified in CFA containing Mycobacterium tuberculosis strain H37Rv (Wako Pure Chemical Industries Ltd.) and boosted at the same location with 200 μg CII plus IFA (Wako Pure Chemical Industries Ltd.) 21 and 42 d later. The mice were observed for the development of arthritis starting from day 16 after immunization and bled periodically for anti-CII antibody determination. The clinical severity of arthritis was quantified according to the following scoring system: 0, no change; 1, swelling in one joint (digitus, wrist, or ankle); 2, swelling in more than one joint or mild inflammation of paws; 3, severe swelling of the entire paw and/or ankylosis. Each paw was graded, so that each mouse could achieve a maximum score of 12. At the end of the experiment, joints were prepared for histopathology. Joints were examined for erosions, pannus formation, and synovium infiltrates.
Assay for Detection of Serum Anti-CII Antibodies.
Serum antibody titers were measured by modification of an ELISA assay described previously (44). In brief, a 96-well microplate (Falcon; Becton Dickinson Labware) was coated with 50 μl/well of a 20 μg/ml solution of CII in PBS at 4°C overnight, washed three times with PBS containing 0.05% Tween 20 and 0.1% BSA, and then blocked with 250 μl/well of PBS containing 0.2% BSA at 4°C overnight. The diluted serum (1:400–20,000) was added at 50 μl/well and allowed to react at 4°C overnight. The wells were washed three times with PBS containing 0.05% Tween 20, incubated with 50 μl of a 1:200 dilution of goat anti–mouse IgG1, IgG2a, IgG2b, or IgM coupled to horseradish peroxidase (Sigma Chemical Co.) at 4°C for 2 h, washed three times with PBS containing 0.05% Tween 20, and developed at room temperature for 30 min with 0.1 ml of TrueBlue Peroxidase Substrate (Kirkegaard & Perry Labs). The OD450 was read using a microplate reader (Biolumin 960; Molecular Dynamics).
Cytokine Production.
Mice were injected intraperitoneally with 1 ml of 5% thioglycollate, and peritoneal exudate cells were harvested 4 d later. The cells were suspended in DMEM supplemented with 10% heat-inactivated FCS, to a concentration of 106 cells/ml. The cells were plated in 24-well culture plates (Sumilon; Sumitomo Bakelite Co., Tokyo, Japan) at 1 ml/well and incubated for 1 h at 37°C in 95% air, 5% CO2. Nonadherent cells were removed by rinsing the monolayers with PBS, and the purified macrophages were subjected to the determination of IL-1α release. SRBCs derivatized with TNP were coated with mouse anti-TNP IgG1 (G1 in reference 44), and then used for the stimulation of macrophages as described previously (12). For the analysis of IL-1α production, the culture supernatant was collected and cytokine production determined using an ELISA plate (Endogen, Inc.) according to the manufacturer. For the determination of cytokine production by lymph node cells, 11 d after CII immunization single-cell suspensions from pooled inguinal and popliteal lymph nodes from the immunized mice were made. The cells (106 cell/well) were cultured in 96-well plates (Falcon; Becton Dickinson Labware) with heat-denatured CII (100 μg/ well). After 72 h, the supernatants were collected and subjected to determination for IFN-γ production using an ELISA plate (Endogen, Inc.) according to the manufacturer. As a control, cells were stimulated with LPS (5 μg/ml, O111:B4; Sigma Chemical Co.) and IFN-γ (100 U/ml; Biosource International).
Proliferation of Lymph Node Cells.
For cell proliferation assays, male mice were immunized with 500 μg CII emulsified in CFA intradermally in both hind footpads, the neck, and at the base of the tail. Inguinal, popliteal, and axillary lymph nodes from the immunized mice were obtained 10 d after immunization. The tissue was minced through sterile wire mesh, resulting in single cell suspensions. Cells (5 × 105/well) from immunized mice were cultured in 96-well, flat-bottomed microplates (Falcon; Becton Dickinson Labware) in the absence or presence of 5, 50, or 100 μg/ml of CII at 37°C in 5% CO2 for 4 d. During the final 18 h of culture, cells were pulsed with 0.5 μCi of [3H]TdR. Cells were harvested on glass fiber filters by using an automated sample harvester (Packard Japan). The incorporated radioactivity was measured with a scintillation spectrometer (Aloka Co. Ltd.). The results of the [3H]TdR incorporation assay were expressed as the mean cpm ± SD of triplicate determinations from each of the three lymph node cell preparations derived from different mice.
Histological Study.
The mice were killed with an overdose of diethyl ether. Their arthritic paws were removed and fixed in 10% neutral buffered formalin. The tissues were decalcified in a 5% EDTA-2Na solution. The joints were then embedded in paraffin. The specimens were cut into 6-μm sections and stained with hematoxylin and eosin.
Statistical Analysis.
Statistical differences between groups for onset of arthritis, the arthritic index, the mean maximum arthritis score, serum levels of antibodies, and T cell proliferation were calculated using Student's t test; differences in the frequency of arthritis were calculated using Fisher's test. P < 0.05 was considered significant.
Results
FcγRIIB− /− Mice in an H-2b Background Are Susceptible to CIA.
Immunization of DBA/1 mice (H-2q) with CII results in typical and progressive polyarthritis in parallel with the production of high levels of anticollagen antibody, as described (27). Neither arthritis nor high levels of antibody are induced in BALB/c (H-2d), C3H/He (H-2k), or C57BL/6 (H-2b) mice (27, 42). Many lines of evidence indicate that CIA susceptibility is restricted to only two H-2 alleles, H-2q and H-2r (33, 34). Although the FcγRIIB−/− mice were generated on H-2b background (25), a haplotype not susceptible to CIA induction, we set out to determine if deletion of this inhibitory receptor would convert a nonsusceptible strain of mice into a susceptible one. FcγRIIB-deficient male mice were immunized with CII/ CFA and then boosted with CII/IFA, and monitored for the occurrence of arthritis in comparison to age and sex-matched H-2b wild-type or DBA/1 mice. Three separate experiments were conducted with similar results as summarized in Table I. Fig. 1 shows the time course and severity of CIA in one such experiment. FcγRIIB-deficient mice develop arthritis with a time course and severity comparable to DBA/1 mice when immunized with CII. Although the incidence of arthritis in FcγRIIB-deficient mice was lower than DBA/1 (42.2 vs. 95.2%), it was dramatically enhanced compared with wild-type H-2b mice (42.2 vs. 7.0%). In those mice that developed arthritis, the mean onset of disease for FcγRIIB−/− mice was comparable to that in DBA/1 controls (35.3 vs. 33.2). Similarly, the mean maximal arthritic index of the mutant animals (6.9 ± 3.6) was also comparable to DBA/1 controls (8.6 ± 1.9).
Table I.
Summary of the CIA Course in FcγRIIB−/− Mice
| Mice | Incidence* (%) | Onset ‡ | Arthritic index‡ | No. of arthritic paws* (%) | ||||
|---|---|---|---|---|---|---|---|---|
| (d) | ||||||||
| Wild-type | 3/43 (7.0) | 50.5 ± 7.6 | 2.3 ± 1.9 | 7/172 (4.1) | ||||
| FcγRIIB−/− | 19/45 (42.2) | 35.3 ± 12.5 | 6.9 ± 3.6 | 48/180 (26.7) | ||||
| DBA/1 | 40/42 (95.2) | 33.2 ± 8.6 | 8.6 ± 1.9 | 113/168 (67.3) |
Mice were immunized with CII in CFA as described in Materials and Methods and monitored for signs of arthritis. Data are given as number and percentage of diseased mice for the incidence, as means ± SD for onset and arthritic index, and as number and percentage of arthritic paws. Arthritic indices are expressed as the maximal scores reached by each arthritic mouse during the course of CIA.
Statistical analyses were performed using Fisher's test: P < 0.001 between wild-type and FcγRIIB−/− mice, and between FcγRIIB−/− and DBA/1 mice.
Statistical analyses were performed using Student's t test: P < 0.05 between wild-type and FcγRIIB−/− mice; not significant between FcγRIIB−/− and DBA/1 mice.
Figure 1.

Development of CIA in mice by disruption of FcγRIIB expression. Incidence of arthritis (A) and severity of clinical signs (B) in FcγRIIB−/− mice (•, n = 16), wild-type H-2b mice of 129/B6 hybrid background (⋄, n = 13), and DBA/1 mice (□, n = 11) after immunization with CII in CFA as described in Materials and Methods. Results are expressed as a percentage of arthritic mice with the arthritic index 5 (A) and as the mean arthritic scores in each group on a given day during the course of CIA (B). Representative data from three separate experiments with similar results are shown.
Histopathological Features of CIA in FcγRIIB− /− Mice.
Histopathological features of the CIA induced in FcγRIIB−/− mice were examined (Fig. 2). The joints of nonarthritic wild-type mice appeared histologically normal, with no significant inflammatory cell infiltration or cartilage–bone destruction (Fig. 2 D). In contrast, the arthritic lesions of the FcγRIIB−/− mice showed massive lymphocytic and monocyte/macrophage infiltration associated with cartilage–bone destruction (Fig. 2 E) similar to that observed in DBA/1 immunized animals (Fig. 2 F). Thus, the results obtained by histopathologic examination of FcγRIIB−/− mice immunized with CII verified a destructive arthritis, which is qualitatively similar to the arthritis induced in DBA/1 mice.
Figure 2.
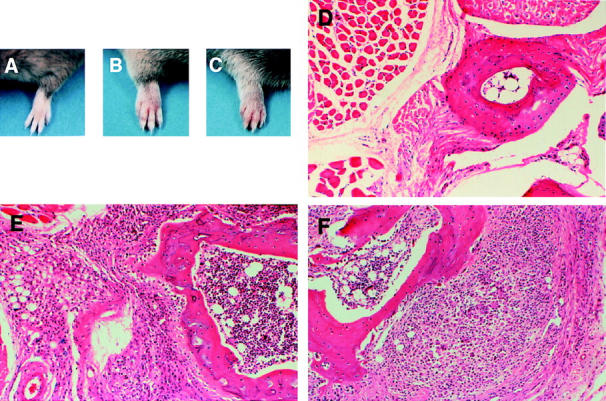
Clinical and histologic presentation of CIA in FcγRIIB−/− and DBA/1 mice. (A–C) The appearance of a normal forepaw from a CII-immunized wild-type mouse (A) contrasted with arthritic paws from an FcγRIIB−/− animal (B) and a positive control DBA/1 mouse (C). (D–F) Cross-sections of the forefoot from a normal wild-type mouse (D) compared with an arthritic joint from FcγRIIB−/− (E) and DBA/1 animals (F). Original magnifications: ×50 (D), ×80 (E), ×80 (F). D illustrates normal cartilage–bone without inflammation, whereas E and F show marked mononuclear cell infiltration with cartilage–bone destruction.
Anti-CII Antibody Levels in CIA-induced FcγRIIB− /− Mice.
Antibodies specific for CII play a major role in the pathogenesis of CIA (28, 38–40). We determined the collagen-specific IgG1, IgG2a, IgG2b, and IgM antibody production in the sera of FcγRIIB−/− and DBA/1 immunized mice. Data derived from sera taken periodically during the experiment are presented in Fig. 3. The mean of all mice of different groups is presented regardless of whether or not the mice had developed arthritis (Fig. 3, A–D). As described previously, FcγRIIB−/− mice have higher antibody levels in response to both thymic-dependent and -independent antigens. As expected, FcγRIIB−/− mice had higher anti-CII antibody titers than those of wild-type mice for all isotypes tested. However, these responses to CII were lower than those observed in DBA/1 mice. The augmented anti-CII IgG responses in arthritic FcγRIIB−/− mice were more pronounced compared with those of nonarthritic wild-type mice (Fig. 3, E–G). Therefore, this enhanced antibody response to CII in the FcγRIIB−/− mice could contribute to the emergence of CIA in this nonpermissive strain.
Figure 3.

Concentration of anti-CII antibodies in sera from mice immunized with CII. The mean ± SD antibody levels of IgG1 (A), IgG2a (B), IgG2b (C), and IgM (D) subclasses, for all animals, and the mean ± SD of antibody levels (E–H) of arthritic DBA/1 (□) and FcγRIIB−/− (•) mice and of nonarthritic wild-type mice (⋄) are shown. **P < 0.01.
Proliferative Response of Lymph Node Cells from CII-primed FcγRIIB− /− Mice.
Since CIA is dependent on dysregulation of both humoral and cell-mediated responses, we determined whether the absence of FcγRIIB altered the phenotype of the cell-mediated immune response to CII. Therefore, we compared the specific proliferative responses and cytokine production of CII-primed lymph node cells derived from FcγRIIB−/−, wild-type H-2b, and DBA/1 mice. As shown in Fig. 4, antigenic stimulation with CII induced higher levels of proliferation in DBA/1 animals and similar lower levels of specific proliferation in FcγRIIB−/− and wild-type animals. Similar results were obtained when IFN-γ production was used as a measure of specific T cell stimulation. These results indicate that disruption of FcγRIIB does not appreciably modify the antigen-specific T cell response in nonpermissive animals and is not likely to account for the susceptibility of these animals to CIA.
Figure 4.

Proliferation and IFN-γ production of anticollagen lymph node cells in response to CII. (A) Lymph node cells (5 × 105/well) were stimulated in vitro with 5, 50, or 100 μg/ml heat-denatured CII (dCII) for 4 d. Proliferative response was determined by uptake of [3H]TdR pulsed for the final 18 h of culturing. (B) Each of the culture supernatants at the end of the experiment in A was collected and assessed for the IFN-γ content by ELISA. **P < 0.01 compared with wild-type mice.
IL-1α Production Is Enhanced in FcγRIIB− /− Macrophages Stimulated with IgG-opsonized Antigen.
At later stages of autoimmune arthritis, local synthesis of cytokines such as IL-1, TNF, and other inflammatory mediators is likely to be responsible for the progression from inflammation to a destructive arthritis. Supporting this notion are studies showing that anti-TNF antibodies or an IL-1 receptor antagonist reduce cytokine production by synovium cells from RA patients (45; for a review, see reference 31), and ameliorated arthritis in DBA/1 mice (46, 47). In several phases of joint inflammation, macrophages secrete chemoattractants for polymorphonuclear cells and monocytes (IL-6, IL-1, GM-CSF, monocyte chemoattractant protein 1, and macrophage inflammatory protein 1α) and upregulate integrins and vascular adhesion molecules through their production of IL-1 and TNF-α (31). Deletion of FcγRIIB decreases the threshold of IC necessary to trigger mast cell and macrophage activation in vitro and in vivo (25) and could contribute to the development of CIA in nonsusceptible H-2 backgrounds by either lowering threshold response or increasing the total cytokine response. To determine if macrophages derived from FcγRIIB−/− animals showed enhanced release of inflammatory mediators upon stimulation, we determined the levels of IL-1α produced upon stimulation with IgG-opsonized SRBCs. As shown in Fig. 5, thioglycollate-elicited peritoneal macrophages from FcγRIIB−/− mice released quantitatively more IL-1α than those from wild-type controls and at levels comparable to macrophages derived from DBA/1 mice. Thus, the absence of FcγRIIB makes macrophages more sensitive to stimulation with IgG ICs, and results in a higher level of secretion of a proinflammatory mediator.
Figure 5.

Secretion of IL-1α by peritoneal macrophages stimulated with IC. Thioglycollate-elicited peritoneal macrophages from FcγRIIB−/− (black bars) and wild-type (white bars) mice and DBA/1 mice (stippled bars) were stimulated with IgG1- opsonized SRBCs as described in Materials and Methods. The culture supernatant was analyzed for the IL-1α content by ELISA.
Discussion
Autoimmune disease results from the dysregulation of the normal immune response, resulting in the loss of tolerance to self-antigens, augmented T and B cell responses, and inappropriate activation of effector cell pathways. Disruption of the ability to generate T or B cell responses blocks the development of autoimmunity and autoimmune disease, while disruption of effector cell pathways attenuates disease development. However, identification of the genetic components that modulate these central pathways which could confer susceptibility to the development of disease has been stymied by the complex multigenic nature of these disorders. It has been known for some time that the MHC haplotype is one such susceptibility factor in both human and animal systems. In the murine model of RA, CIA, H-2 haplotype determines the susceptibility of an animal to the development of disease. In this study, we demonstrate that the inhibitory FcR for IgG, FcγRIIB, is another susceptibility gene, functioning to suppress the development of CIA in nonsusceptible hosts. Deletion of FcγRIIB converts a nonsusceptible H-2b animal to one susceptible to the development of CIA. The mechanism by which deletion of FcγRIIB results in susceptibility to CIA involves augmentation of both antibody and effector cell responses, supporting a threshold model for autoimmune disease.
Association of arthritis with high levels of autoantibodies has highlighted the importance of the anticollagen antibody responses in inducing arthritis. Antiserum or purified IgG antibody to CII can transfer arthritis to the susceptible DBA/1 mice (39). This passively transferred arthritis exhibits the histopathologic characteristics of the early lesions of disease induced through immunization of susceptible hosts. The resulting disease is transient and less severe than the disease induced in immunized DBA/1 mice, suggesting that anti-CII antibodies alone are not sufficient to give rise to the full range of lesions that characterize CIA. In contrast, a typical arthritis could be induced by adoptive transfer of anti-CII antibody from arthritic DBA/1 mice together with T cells from DBA-1 mice presensitized with heat-denatured collagen (42). These results indicate the crucial importance of the synergy between humoral and cell-mediated immunities in the pathogenesis of typical arthritis (42).
A strong B cell response is activated in CIA, producing IgG directed towards CII-specific structures. There is evidence that these antibodies are pathogenic, as exemplified by transfer experiments, and promote T cell–mediated arthritis development. In contrast, levels of anti-CII autoantibodies in serum do not correlate with CIA development, as high levels can be detected in nondiseased mice. Thus, the role of B cells in both the priming and effector phases of the disease is unclear. Svensson et al. (43) reported that the B cell–deficient mice of the CIA-susceptible strains B10.Q and B10.RIII (H-2r) are resistant to CIA induction, although the anti-CII T cell reactivity does not differ between B cell–deficient and B cell–sufficient mice, thus indicating a crucial role for B cells in the induction of arthritis. In the present report, we show that the anti-CII IgG antibody response is enhanced in FcγRIIB−/− mice, especially in those mice exhibiting arthritis (Fig. 2), suggesting that the relatively high anti-CII IgG level could be one of the pathogenic factors, although unlikely by itself to explain the induction of disease in the H-2b background.
RA is an autoimmune disease in which macrophages are believed to play a central role (48, 49). We found that macrophages from FcγRIIB−/− mice were hyperresponsive to stimulation with IgG ICs, leading to augmented release of a proinflammatory mediator, IL–1α (Fig. 5), that is able to upregulate integrins and vascular adhesion molecules. At later stages of autoimmune arthritis, local synthesis of cytokines is probably responsible for progression of inflammation to a destructive arthritis (46, 47). Thus, the heightened sensitivity of macrophages to ICs is likely another pathogenic factor making FcγRIIB−/− mice more susceptible to CIA than control mice.
The present study thus suggests that the development of autoimmune disease represents the dysregulation of both humoral and effector pathways. The contribution of each component may be below a critical threshold to result in the development of disease, as has been suggested by the genetic studies in the NZB/NZW F1 autoimmune glomerulonephritis model (20). FcγRIIB is a pleiotropic receptor, functioning to downregulate both B cell and effector cell responses. The finding that deletion of FcγRIIB converts nonsusceptible H-2b mice into susceptible animals for CIA suggests that a similar role may be found in other autoimmune disease models and in human susceptibility to autoimmune disease. Therefore, strategies that result in the upregulation of this receptor and its signaling would represent potential new therapeutic approaches to the treatment of autoimmune diseases.
Acknowledgments
We are grateful to Drs. M. Sasano (Santen Pharmaceuticals Co.) and H. Ohmori (Okayama University) for helpful discussions, and to N. Takagi for secretarial support.
This work is supported by research grants from the Ministry of Education, Science, Sports and Culture of Japan, and Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Corporation (JST) (to T. Takai), and by grants from the National Institutes of Health and the Juvenile Diabetes Foundation (to J.V. Ravetch).
Abbreviations used in this paper
- BCR
B cell receptor
- CII
collagen type II
- CIA
collagen-induced arthritis
- FcγRI
FcγRIIB, and FcγRIII, type I high-affinity Fc receptor for IgG, type IIB, and type III low-affinity receptors for IgG, respectively
- IC
immune complex
- RA
rheumatoid arthritis
References
- 1.Ravetch JV, Kinet J-P. Fc receptors. Annu Rev Immunol. 1991;9:457–492. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 2.Metzger H. The receptor with high affinity for IgE. Immunol Rev. 1992;125:37–48. doi: 10.1111/j.1600-065x.1992.tb00624.x. [DOI] [PubMed] [Google Scholar]
- 3.Fridman WH, Bonnerot C, Daëron M, Amigorena S, Teillaud JL, Sautes C. Structural bases of Fcγ receptor functions. Immunol Rev. 1992;125:49–76. doi: 10.1111/j.1600-065x.1992.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 4.van de Winkel JGJ, Capel PJA. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunol Today. 1993;14:215–221. doi: 10.1016/0167-5699(93)90166-I. [DOI] [PubMed] [Google Scholar]
- 5.Ravetch JV. Fc receptors: rubor redux. Cell. 1994;78:553–560. doi: 10.1016/0092-8674(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 6.Takai, T., and J.V. Ravetch. 1999. Fc receptor genetics and the manipulation of genes in the study of FcR biology. In Immunoglobulin Receptors and Their Physiological and Pathological Roles in Immunity. J.G.J. van de Winkel and P.M. Hogarth, editors. Kluwer Academic Publishers Group, Netherlands. In press.
- 7.Greenberg S, Chang P, Silverstein SC. Tyrosine phosphorylation is required for Fc receptor–mediated phagocytosis in mouse macrophages. J Exp Med. 1993;177:529–534. doi: 10.1084/jem.177.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VLJ, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcγ receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavin AL, Bames N, Dijstelbloem HM, Hogarth PM. Identification of the mouse IgG3 receptor: implications for antibody effector function at the interface between innate and adaptive immunity. J Immunol. 1998;160:20–23. [PubMed] [Google Scholar]
- 10.Fanger NA, Voigtlaender D, Liu C, Swink S, Wardwell K, Fisher J, Graziano RF, Pfefferkorn LC, Guyre PM. Characterization of expression, cytokine regulation, and effector function of the high affinity IgG receptor Fc gamma RI (CD64) expressed on human blood dendritic cells. J Immunol. 1997;158:3090–3098. [PubMed] [Google Scholar]
- 11.Hazenbos WLW, Gessner JE, Hofhuis FMA, Kuipers H, Meyer D, Heijnen IAFM, Schmidt RE, Sandor M, Capel PJA, Däeron M, et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in FcγRIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 12.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 13.Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 14.Dombrowicz D, Flamand V, Brigman KK, Koller BH, Kinet J-P. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor α chain gene. Cell. 1993;75:969–976. doi: 10.1016/0092-8674(93)90540-7. [DOI] [PubMed] [Google Scholar]
- 15.Miyajima I, Dombrowicz D, Martin TR, Ravetch JV, Kinet JP, Galli SJ. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and FcγRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J Clin Invest. 1997;99:901–914. doi: 10.1172/JCI119255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dombrowicz D, Flamand V, Miyajima I, Ravetch JV, Galli SJ, Kinet J-P. Absence of FcεRI α chain results in upregulation of FcγRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between FcεRI and FcγRIII for limiting amounts of FcR β and γ chain. J Clin Invest. 1997;99:915–925. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 18.Sylvestre DL, Ravetch JV. A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity. 1996;5:387–390. doi: 10.1016/s1074-7613(00)80264-2. [DOI] [PubMed] [Google Scholar]
- 19.Sylvestre D, Clynes R, Ma M, Warren H, Carroll MC, Ravetch JV. Immunoglobulin G–mediated inflammatory responses develop normally in complement-deficient mice. J Exp Med. 1996;184:2385–2392. doi: 10.1084/jem.184.6.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 21.Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci USA. 1998;95:652–656. doi: 10.1073/pnas.95.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daëron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, Fridman WH. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of FcγRIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. 1995;3:635–646. doi: 10.1016/1074-7613(95)90134-5. [DOI] [PubMed] [Google Scholar]
- 23.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of FcγRIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 24.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 25.Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. Modulation of immune complex– induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J Exp Med. 1999;189:179–185. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 27.Courtenay JS, Dallman MJ, Dayan AD, Marten A, Mosedale B. Immunization against heterologous type II collagen induces arthritis in mice. Nature. 1980;283:666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 28.Holmdahl R, Jansson L, Gullberg D, Rubin K, Forsberg PO, Klareskog L. Incidence of arthritis and autoreactivity of anti-collagen antibodies after immunization of DBA/1 mice with heterologous and autologous collagen II. Clin Exp Immunol. 1985;62:639–646. [PMC free article] [PubMed] [Google Scholar]
- 29.Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. J Exp Med. 1977;146:857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trentham DE, Townes AS, Kang AH, David JR. Humoral and cellular sensitivity to collagen in type II collagen–induced arthritis in rats. J Clin Invest. 1978;61:89–96. doi: 10.1172/JCI108929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 32.Nabozny GH, Baisch JM, Cheng S, Cosgrove D, Griffiths MM, Luthra HS, David CS. HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: a novel model for human polyarthritis. J Exp Med. 1996;183:27–37. doi: 10.1084/jem.183.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wooley PH, Luthra HS, Stuart JM, David SC. Type II collagen–induced arthritis in mice. I. Major histocompatibility complex (I-region) linkage and antibody correlates. J Exp Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wooley PH, Luthra HS, Griffiths MM, Stuart JM, Huse A, David CS. Type II collagen-induced arthritis in mice. IV. Variations in immunogenetic regulation provide evidence for multiple arthritogenic epitopes on the collagen molecule. J Immunol. 1985;135:2443–2451. [PubMed] [Google Scholar]
- 35.Brunsberg U, Gustafson K, Jansson L, Michaëlsson E, Ährlund-Richter L, Pettersson S, Mattsson R, Holmdahl R. Expression of transgenic class II Ab gene confers susceptibility to collagen-induced arthritis. Eur J Immunol. 1994;24:1698–1702. doi: 10.1002/eji.1830240736. [DOI] [PubMed] [Google Scholar]
- 36.Ranges GE, Sriram S, Cooper SM. Prevention of type II collagen–induced arthritis by in vivo treatment with anti-L3T4. J Exp Med. 1985;162:1105–1110. doi: 10.1084/jem.162.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiocchia G, Boissier MC, Fournier C. Therapy against murine collagen-induced arthritis with T cell receptor Vβ-specific antibodies. Eur J Immunol. 1991;21:2899–2905. doi: 10.1002/eji.1830211202. [DOI] [PubMed] [Google Scholar]
- 38.Mo J, Scheynius A, Holmdahl R. Antibody recognition of mouse cartilage in vivo; epitope- and idiotype-specific binding and inhibition. Scand J Immunol. 1994;39:122–130. doi: 10.1111/j.1365-3083.1994.tb03350.x. [DOI] [PubMed] [Google Scholar]
- 39.Stuart JM, Dixon FJ. Serum transfers of collagen-induced arthritis in mice. J Exp Med. 1983;158:378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmdahl R, Andersson M, Goldshmidt TJ, Gustafsson K, Jansson L, Mo JA. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 41.Holmdahl R, Vingsbo C, Malmström V, Jansson L, Holmdahl M. Chronicity of arthritis induced with homologous type II collagen (CII) in rats is dependent on anti-CII B-cell activation. J Autoimmun. 1994;7:739–752. doi: 10.1006/jaut.1994.1058. [DOI] [PubMed] [Google Scholar]
- 42.Seki N, Sudo Y, Yoshioka T, Sugihara S, Fujitsu T, Sakuma S, Ogawa T, Hamaoka T, Senoh H, Fujiwara H. Type II collagen-induced murine arthritis. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. J Immunol. 1988;140:1477–1484. [PubMed] [Google Scholar]
- 43.Svensson L, Jirholt J, Holmdahl R, Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA) Clin Exp Immunol. 1998;111:521–526. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohmori H, Hase N, Hikida M, Takai T, Endo N. Enhancement of antigen-induced interleukin 4 and IgE production by specific IgG1 in murine lymphocytes. Cell Immunol. 1992;145:299–310. doi: 10.1016/0008-8749(92)90333-k. [DOI] [PubMed] [Google Scholar]
- 45.Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNFα antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 46.Thorbecke GJ, Shah R, Leu CH, Kuruvilla AP, Hardison AM, Palladino MA. Involvement of endogenous tumour necrosis factor α and transforming growth factor β during induction of collagen type arthritis in mice. Proc Natl Acad Sci USA. 1992;89:7375–7379. doi: 10.1073/pnas.89.16.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams RO, Mason LJ, Feldmann M, Maini RN. Synergy between anti-CD4 and anti-TNF in the amelioration of established collagen-induced arthritis. Proc Natl Acad Sci USA. 1994;91:2762–2766. doi: 10.1073/pnas.91.7.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996;39:115–124. doi: 10.1002/art.1780390116. [DOI] [PubMed] [Google Scholar]
- 49.van Lent, P.L., A.E. Holthuysen, L. van den Bresselaar, N. van Rooijen, L.B. van de Putte, and W.B. van den Berg. 1995. Role of macrophage-like synovial lining cells in localization and expression of experimental arthritis. Scand. J. Rheumatol. 24(Suppl. 101):83–89. [DOI] [PubMed]