

Abstract
Survivin is an inhibitor of apoptosis protein that also functions during mitosis. It is expressed in all common tumors and tissues with proliferating cells, including thymus. To examine its role in apoptosis and proliferation, we generated two T cell–specific survivin-deficient mouse lines with deletion occurring at different developmental stages. Analysis of early deleting survivin mice showed arrest at the pre–T cell receptor proliferating checkpoint. Loss of survivin at a later stage resulted in normal thymic development, but peripheral T cells were immature and significantly reduced in number. In contrast to in vitro studies, loss of survivin does not lead to increased apoptosis. However, newborn thymocyte homeostatic and mitogen-induced proliferation of survivin-deficient T cells were greatly impaired. These data suggest that survivin is not essential for T cell apoptosis but is crucial for T cell maturation and proliferation, and survivin-mediated homeostatic expansion is an important physiological process of T cell development.
Keywords: proliferation, apoptosis, T cell development, survivin, IAP
Introduction
The thymus is the major organ of T lymphocyte maturation and differentiation. During development, T cells have to confront sequentially fateful decisions: the pre-TCR checkpoint, TCR α chain rearrangements, positive selection, and negative selection (1, 2). Early T cells are CD4− CD8− (double negative [DN]), which can be subdivided further into DN1, DN2, DN3, and DN4 stages based on their expression of CD25 and/or CD44. The first step of rearrangements involves TCR β chain at the DN2 and DN3 stages. If successful, this results in expression of the pTα/TCR β chain–CD3 pre-TCR complex on the cell surface. Subsequent rounds of proliferation between DN3 and DN4 ensue, accompanied by differentiation into CD4+ CD8+ (double positive [DP]) cells. At the DP stage, TCR α rearrangements take place and functional TCRs can be found on the cell surface. Most DP thymocytes, however, die through negative selection or neglect because their TCRs exhibit either too much or no affinity for the major histocompatibility complex/peptides. Only relatively few DP cells undergo positive selection and differentiate into CD4+ CD8− or CD4− CD8+ (single positive [SP]) cells. These mature cells then migrate to the peripheral immune organs where they carry out their major function in defending the body against foreign invasion. In adult animals, proliferation is thought to occur mainly between DN and DP T cells, whereas little proliferation takes place in the later stages of T cell development. In contrast, proliferation of SP thymocytes has been seen in up to 21-d-old animals (3, 4), the significance of which, however, has not been fully addressed.
In addition to proliferation, programmed cell death also plays a critical role in the development of T cells at multiple stages. Developing DN or DP T cells are destined to die unless a functional pre-TCR or TCR is expressed. Thymocytes bearing “useless” or “self-reactive” receptors are eliminated through an apoptotic process, thereby allowing only ∼3–5% thymocytes to eventually mature and be exported into the periphery. The mechanism of lymphocyte apoptosis can be broadly divided into extrinsic and intrinsic pathways (5, 6). The intrinsic pathway involves the Bcl-2 and inhibitor of apoptosis protein (IAP) family members, whereas apoptosis initiated by the death receptor comprises the extrinsic pathway. The Bcl-2 family members are involved in the process of thymocyte death by neglect and negative selection. Overexpression of Bcl-2 leads to an enlarged thymus and resistance of transgenic DP cells to apoptosis, as assessed by in vitro cell culture. Mutations in Bim, a BH3 pro-apoptotic Bcl-2 family member, or its downstream proteins, Bax and Bak, result in defective negative selection processes (7, 8). In contrast, the role of TNF receptor family members in T cell selection remains controversial. Fas, the prototype TNF receptor family member, is not required for negative selection, except in some special circumstances (9, 10). TRAIL, a TNF-related ligand, was reported recently to be important for negative selection (11), but this could not be reproduced in another study (12). Transgenic mice expressing a FADD dominant negative that blocks signaling of all TNF death receptors, exhibit no defects in negative selection (13).
Survivin, or TIAP, is a member of the IAP family (14–16), a major class of modulators of apoptosis. IAP members are evolutionarily conserved and in Drosophila they comprise the major antiapoptotic proteins. Most IAP members inhibit apoptosis by binding to effector caspases. Release of Smac/Diablo from mitochondria in insulted cells causes IAPs to dissociate from the corresponding caspases, allowing them to be activated by the apical caspase-9 (17, 18). Survivin contains a single baculovirus IAP repeat and with 142 amino acids, it is the smallest IAP member. In contrast to most IAPs, survivin does not have a RING domain at its C terminus but instead contains a coil-coiled region that is presumably required for its subcellular localization (19). Survivin is ubiquitously expressed during embryonic development, suggesting an important role in embryogenesis (20). In normal adult tissues, survivin is expressed abundantly in the thymus, testis, and proliferating cells. Overexpression of survivin can protect cells from both receptor- and injury-induced apoptosis (21–24), whereas introduction of antisense, ribozyme, or dominant negative survivin leads to spontaneous increased apoptosis (25–29). How survivin inhibits apoptosis, however, is controversial. Survivin has been reported to bind to several caspases (20, 21, 30) but its structure fails to reveal a caspase-binding pocket that is found in other IAP family members (31). Survivin was also shown to bind to Smac and this binding appears essential for regulating its antiapoptotic activity (18, 32).
In addition to its purported role in apoptosis, survivin is important for cell cycle progression. Survivin expression is induced in the G2/M phase of the cell cycle (33) and interestingly, it is expressed at high levels in almost all types of human tumors in a cell cycle–independent manner (34). The latter indicates that survivin expression is deregulated in malignancy. Survivin has been shown to localize to centrosomes, centromeres, and microtubule spindles, and interacts with polymerized tubulin and Aurora B kinase, a protein essential for cytokinesis (35, 36). Survivin-deficient mice die in utero by day 4.5 of gestation (36, 37). Survivin-null embryos exhibit disrupted microtubule formation and polyploidy (36), suggesting that survivin is crucial for regulation of cytokinesis. In lower eukaryotes like yeast and Caenorhabditis elegans, the survivin ortholog, Bir1, only plays a role in cell cycle but not apoptosis (38–40).
To assess the potential role of survivin in apoptosis and proliferation, we generated two lines of T cell–specific survivin-deficient mice. We used the Cre-loxP system to conditionally target and inactivate the survivin gene in T cells at various developmental stages (41). Lck-Cre and CD4-Cre transgenic mice that express Cre at different DN stages were used to delete survivin. We found aberrant thymic development in the lck-survivin mice, whereas CD4-survivin mice exhibited normal thymic populations. However, the CD4-survivin–deficient mice have a drastic reduction in peripheral CD4+ and CD8+ T cells, which exhibit an “immature” phenotype. Apoptosis in survivin-deficient T cells is indistinguishable from their wild-type counterparts but they are defective with respect to the ability to undergo proliferation. Analysis of newborn T cell–specific survivin-deficient mice showed that their mature thymocytes fail to populate the peripheral compartment and this in turn results in a compensatory attempt to proliferate. These data suggest that survivin does not play a major role in inhibition of apoptosis of T cells, but rather is crucial for their maturation, homeostasis, and proliferation at multiple stages.
Materials and Methods
Generation of Mice with the Survivin Gene Flanked by loxP Sites.
We have previously prepared a targeting vector to inactivate the survivin gene by homologous recombination in embryonic stem (ES) cells (37). To that end, a 6.5-kb EcoRV fragment of the survivin gene encompassing all four exons was deleted and replaced with a loxP-flanked neomycin gene cassette, which was preceded by a unique Xho1 site. To generate a targeting vector in which the survivin gene was intact yet flanked by loxP sites, we first subcloned the 6.5-kb EcoRV fragment into a shuttle vector such that an additional loxP site was placed upstream of exon 1 at the most 5′ EcoRV site. The resultant loxP-EcoRV fragment was subcloned into the XhoI site of the “knockout” targeting vector, and the correct orientation was confirmed. The new targeting vector DNA was linearized with Not1 and electroporated into 129/SvEv ES cells (42) for selection and confirmed for homologously recombined ES cell colonies by Southern blotting using 5′ and 3′ probes. For the 5′ probe, the DNAs were digested with KpnI and EcoRV to detect a 14-kb targeting allele (see Fig. 1 B). For the 3′ end, the DNAs were digested with SacI and SpeI and probed with the 3′ external probe. The expected wild-type and targeted alleles are 7.8 and 7.2 kb, respectively (unpublished data). The neomycin cassette was excised in vitro by transient exposure of positive ES cell clones to cre recombinase. Those ES cell clones in which only the neomycin gene was excised, confirmed by PCR and Southern blotting, were expanded for aggregation and introduction into pseudopregnant female NIH Swiss white mice. Three >90% chimeric male offspring produced germline transmission of the floxed survivin allele. F1 and F2 offspring were intercrossed. Genotyping was performed on tail DNA by PCR and/or Southern blotting. PCR typing was performed using oligonucleotide primer pair Adv17 (5′-caggccgatggtctcagaaata) and Adv18 (5′-ggtttccttcttgctattctgact), which yielded a 408-bp amplicon across the most 5′ loxP site and a 364-bp amplicon from the wild-type allele. Primer pair Adv25 (5′-gatggtgatgaaactagcatctcaccctg) and Adv28 (5′-gcttaagtccacgtcacaatagagc) resulted in a 577-bp amplicon in the targeted allele across the 3′ loxP site and a 386-bp amplicon in the wild-type allele (see Fig. 1 B).
Figure 1.

Generation of T cell–specific survivin-deficient mice. (A) A schematic diagram of the survivin gene locus and its various alleles. The targeting construct has three loxP sites (triangles) flanking the four survivin exons (boxes) and the neomycin gene. After selecting the cells with the appropriate targeting allele, the neomycin gene was removed by transfecting a cre-expressing plasmid. The survivin flox allele mice were then generated and crossed to lck-cre or CD4-cre according to the scheme described in Results, to generate lck-survivin– or CD4-survivin–deficient mice. K, KpnI; S, SacI; E, EcoRV. Horizontal arrows denote the approximate location of the oligonucleotides used for genotyping and analysis in C (Adv17, Adv25, and Adv28). (B) Southern blot and PCR analysis of the flox survivin allele. Left: Southern blot analysis of the ES cells containing loxP sites in the survivin locus. The DNAs were digested with a combination of KpnI and EcoRV and hybridized with a 5′ probe as depicted in A. The presence of a 14-kb band is indicative of the correct targeted allele. Right: PCR analysis of the resulting survivinflox/+ mice after the neomycin gene has been removed (3′ flox sites). (C) PCR analysis of the survivin-deleted allele in various T cell DN sub-populations. The different DN subpopulations were sorted based on their CD25 and CD44 expression: DN1 (CD44+ CD25−), DN2 (CD44+ CD25+), DN3 (CD44− CD25+), DN4 (CD44− CD25−). PCR analysis using three oligonucleotides (Adv17, Adv25, and Adv28 in A) flanking the survivin flox allele was conducted to analyze for the presence of the survivin flox allele (a 577-bp product of Adv25 and Adv28) or the deleted allele (a 420-bp of Adv17 and Adv28). (D) PCR analysis of the survivin-deleted allele in splenic CD4+ T cell populations. Splenic T cells were stained with anti-CD4 and CD8 antibodies and sorted by flow cytometry (99% pure) and subjected to PCR analysis as described in C above. (E) Western blot analysis of survivin expression in purified DP thymocytes. Two survivin isoforms were detectable in the wild-type thymocytes. C, cytoplasmic extracts; N, nuclear extracts. Western blotting using anti–α-tubulin antibodies was performed as a loading control.
Mouse Genotyping.
Mouse genotyping was performed by PCR of the tail DNA. For genotyping Cre-deleted flox survivin allele, three primers, Adv17, Adv25, and Adv28, were used for PCR. For the survivin flox allele, a fragment of 577 bp was generated with the pair of primers Adv25 and Adv 28 as described above. For the deleted allele, a fragment of 420 bp was generated with primers Adv17 and Adv28 (see Fig. 1, A, C, and D). Genotyping of survivin+/− mice has been described (37).
Flow Cytometry Analysis.
Cell suspensions were prepared from thymus, spleen, and lymph nodes. After red blood cell lysis, cells were stained with the indicated antibodies conjugated to PE, FITC, or tricolor (Caltag Laboratories and BD Biosciences). Normal mouse serum was added to eliminate nonspecific staining. Cell cycle analyses were performed by first staining cells with FITC-conjugated CD4 and CD8 antibodies. Cells were then fixed with 70% ethanol for 2 h and digested with 500 μg/ml RNase A (Sigma-Aldrich) at 37°C for 30 min. After washing, the cells were stained with 50 μg/ml propidium iodide followed by flow cytometric analysis (Coulter). For apoptosis assays, cells were stained with 7-amino actinomycin D (7-AAD; Sigma-Aldrich) or FITC–annexin V (BD Biosciences) as previously described (43).
Preparation of Lymphocyte Subpopulations and Western Blot Analysis.
Thymocytes and peripheral lymphocytes were separated into different subpopulations using a cell sorter for genotyping and Western blot analysis. The different DN stages were identified by CD25 and CD44 staining on thymocytes gated on the CD4− CD8− population. The purities of the separated subpopulations were checked by rerunning a portion of purified cells (they ranged from 95 to 99%). For isolation of purified CD4 or CD8 splenocytes, biotin-conjugated CD4 and CD8 antibodies (BD Biosciences) were used. After staining, the splenocytes were incubated with streptavidin microbeads (Miltenyi Biotec) at 4°C for 20 min followed by repeated washing. The splenocytes were run on a MACS cell sorter (Miltenyi Biotec) to isolate CD4+ or CD8+ lymphocytes. Protein lysates were prepared by lysing the cells in a hypotonic buffer as previously described (44). Cytosolic and nuclear fractions were prepared and separated on a 15% SDS-PAGE. Western blot analysis was performed according to a standard protocol using a purified rabbit anti–survivin antibody (22).
Apoptosis Assay.
Primary thymocytes and peripheral lymphocytes were prepared from thymus, spleen, and lymph nodes and stained with 7-AAD as well as PE anti-CD4 and tricolor anti-CD8 on ice for 30 min. Alternatively, cells were stained with FITC–annexin V. Stained cells were then subjected to flow cytometry analysis. For induction of apoptosis, 5 × 106/ml thymocytes or peripheral lymphocytes in 10% FBS/RPMI 1640 were treated at various time points at 37°C with either 1 μg/ml hamster anti–Fas antibody (BD Biosciences), 50 ng/ml recombinant FasL (Qbiogene), 50 nM dexamethasone, a combination of 0.5 mg/ml plate-bound anti-CD3 (500A2) and 0.5 mg/ml anti-CD28 (37N) antibodies, 2 mg/ml Con A (Sigma-Aldrich), or a combination of 10 ng/ml PMA (Calbiochem) and 1 mM ionomycin (Calbiochem). Treated cells were then stained with 7-AAD, PE anti-CD4, and tricolor anti-CD8 followed by flow cytometry analysis.
Carboxy Fluorescein Diacetate Succinimidyl Ester (CFSE).
106 splenocytes/ml were labeled with 10 μM CFSE in PBS/0.1% bovine serum albumin at 37°C for 10 min. After washing twice with 10% FBS/RPMI 1640, splenocytes were stimulated with either 0.5 mg/ml plate-bound anti-CD3 and 0.5 mg/ml anti-CD28 antibodies, 2 mg/ml Con A, or a combination of 10 ng/ml PMA and 1 mM ionomycin for 4 d at 37°C as previously described (45). The stimulated splenocytes were harvested from culture plates and then stained with PE anti-CD4 antibody. CD4+ gated lymphocytes were further analyzed for CFSE fluorescence by flow cytometry. Splenocytes were stimulated as described above for cell cycle analysis, except the treatments lasted for 48 h. Stimulated cells were then stained with FITC anti-CD4− and CD8− antibodies, fixed with ethanol, and stained with propidium iodide for flow cytometric analysis on CD4+ or CD8+ gated lymphocytes.
5-Bromo-2-Deoxyuridine (BrdU) Labeling.
For DNA incorporation analysis of thymocyte and peripheral T cells, adult (6–8 wk) or newborn (5–7 d) mice received two intraperitoneal injections of BrdU (Sigma-Aldrich) at 2-h intervals as previously described (41). For each injection, 1 and 0.2 mg BrdU for adult and newborn mice were used, respectively. Mice were killed 1 h after the last injection and thymi and spleens were taken out for cell preparation. Cells were first stained with PE anti-CD4 and tricolor anti-CD8 antibodies. After fixing and permeabilization, cells were stained with FITC anti-BrdU antibody (BD Biosciences) and subjected to flow cytometry.
7-AAD/BrdU Analysis.
BrdU and cell cycle studies were performed according to the published protocol (46) with some modifications. Newborn mice received either four intraperitoneal injections of 0.2 mg BrdU once every 6 h or 3-h BrdU labeling with two injections as described above. Thymocytes and splenocytes were then stained with either PE anti-CD4 or PE anti-CD8 antibodies. After two washes, cells were fixed with 75% ethanol and then permeabilized with 1% paraformaldehyde and 0.01% Tween 20. After digestion with 100 Kuntz/ml DNase I (Sigma-Aldrich), cells were stained with FITC anti-BrdU antibody at 4°C for 30 min. For 7-AAD staining, the cells were washed and incubated with 500 μg/ml RNase A (Sigma-Aldrich) at 37°C for 30 min followed by staining with 7-AAD at 4°C for 30 min.
Results
Generation of T Cell–specific Survivin-deficient Mice.
Three steps were involved in the generation of T cell–specific survivin knockout mice. First, the wild-type survivin exons were replaced by the loxP-flanked gene through specific targeting, resulting in survivinflox/+ mice (Fig. 1, A and B) . Second, lck-cre and CD4-cre mice (41) were crossed with survivin+/− mice in which all four exons have been deleted in one of the survivin alleles (37) to obtain lck-cre/survivin + / − or CD4-cre/survivin + / − mice. Lastly, survivinflox/− mice were bred to lck-cre/survivin+/− or CD4-cre/survivin+/− mice to generate lck-cre/survivinflox/− and CD4-cre/survivinflox/− mice, respectively (henceforth termed lck-survivin and CD4-survivin). Their survivin expressing littermates (survivinflox/−, cre + /survivin+/−, or cre +/survivin flox/+) were used as controls. Lck-Cre and CD4-Cre transgenic mice express the Cre recombinase in DN2 and DN3 T cells, respectively (41, 47). Recombination of the loxP sites is usually complete by DN3 for lck-Cre mice and DN4 for CD4-Cre mice. As TCR β checkpoint and proliferation occur between DN3 and DN4, these two lines of mice offer us the opportunity to assess the requirement of survivin for early β checkpoint and late T cell events in DP and mature T cells.
Survivinflox/−, lck-survivin, and CD4-survivin mice were all born alive and appeared healthy. Thymocytes were collected from 4–8-wk-old mice and separated into DN1, DN2, DN3, and DN4 subpopulations based on their CD44 and/or CD25 expression (>96% purity). PCR analysis with the appropriate oligonucleotides can detect a 577-bp product that represents the undeleted loxP survivin allele and a 420-bp product that represents the deleted allele. As shown in Fig. 1, C and D, deletion starts at DN3 for CD4-survivin and is complete by the DN4 stage. At this stage, the upper 577-bp product is undetectable. In lck-survivin mice, however, deletion initiates earlier at the DN2 and increases through the DN3 and DN4, but the upper loxP wild-type band remains in DN4 and in mature peripheral T cells (Fig. 1, C and D). This may reflect preferential proliferation and differentiation of those thymocytes with incomplete deletion of survivin. Using the identical lck-cre transgenic line, complete deletion was detected in Notch-deficient mice (47). To see whether the extent of DNA deletion corresponded to the loss of survivin protein, we performed Western blot analysis to detect survivin in the DP population. No survivin was detected in DP thymocytes from CD4-survivin mice (Fig. 1 E). However, consistent with the PCR results, a small amount of survivin protein was seen in lck-survivin DP thymocytes (Fig. 1 E).
Aberrant Thymic Development in lck-survivin Mice.
When 4–8-wk-old lck-survivin mice were analyzed, we found their thymic cellularities to be about one eighth of those in their littermate controls (1.38 × 107 ± 0.39 vs. 11.33 × 107 ± 1.22). Spleen weight and total cell numbers of lck-survivin mice were comparable to the wild-type controls, whereas the lymph nodes were 50% smaller (2.06 × 107 ± 0.28 vs. 4.64 × 107 ± 0.78). T cell subpopulations in thymus, spleen, and lymph nodes were examined. Severe impairment of T cell development was observed in lck-survivin mice with most of the thymocytes being DN, indicating that the progression of thymocytes from DN to DP stages was blocked (Fig. 2) . This is also reflected by the decrease in the absolute cell number of DP in lck-survivin mice (Fig. 2 A). Interestingly, in spleens and lymph nodes, mature T cells from lck-survivin mice were reduced but not drastically so (Fig. 2 A). This is most likely due to normal development of survivin+ cells. Indeed, peripheral T cells of lck-survivin mice contain significant undeleted flox survivin allele (Fig. 1 D). The ratio of CD4 to CD8 T cells was comparable to that of the wild-type controls and as expected (Fig. 2 B), the B cell and macrophage populations in lck-survivin mice were not affected (unpublished data).
Figure 2.

Analysis of lck-survivin mice. (A) The absolute cell numbers of DN, DP, SP (CD4+ CD8− or CD8+ CD4−), and peripheral T cells in lck-survivin mice and the littermate controls (n = 12). (B) CD4/CD8 ratio of thymocyte SP and peripheral T cells. (C) Left: CD4 versus CD8 flow cytometric analysis of lck-survivin thymocytes and the littermate controls. Middle: Flow cytometric analysis of lck-survivin thymocytes gated on CD4− CD8− DN cells. Numbers indicate the percentages of the corresponding populations. Right: Flow cytometric analysis of CD25+ DN thymocytes from lck-survivin and control thymocytes. Numbers indicate the percentages of the corresponding population. E, small cells; L, large cells.
To examine the DN thymocytes in more detail, we stained them with CD44 and CD25 antibodies. lck-survivin thymocytes exhibited a lower percentage of DN4 (CD44− CD25−), with a corresponding increase in DN3 (CD44− CD25+) cells (Fig. 2 C, middle). This indicates that loss of survivin affected DN3 to DN4 proliferation stages of T cell development. To see if pre-TCR signaling is intact, we gated the DN CD25+ cells and separated them into small (E) and large (L) cells. Previous studies have shown that the E cells consist of mostly G0/G1 cells, whereas the L cells are mostly cycling, having received the pre-TCR signals (48, 49). Cells from lck-survivin mice exhibit a larger percentage of L cells (Fig. 2 C, right), suggesting that pre-TCR signals are intact in these cells. The increased number of L cells suggests that loss of survivin does not affect the G1 to S checkpoints but are arrested later in the cell cycle.
Normal Thymic Development but Reduced Peripheral T Cells in CD4-survivin Mice.
In contrast to lck-survivin mice, the thymic cellularities in CD4-survivin mice were similar to those of their littermate controls (14.55 ± 2.48 × 107 vs. 18.06 ± 1.46 × 107). There were also no significant differences in the total cell numbers of spleens and lymph nodes of CD4-survivin and control mice (4.28 ± 0.62 × 107 vs. 4.82 ± 0.45 × 107 for lymph nodes and 12.28 ± 2.35 × 107 vs. 14.81 ± 2.61 × 107 for spleen). Flow cytometry analysis using CD4, CD8, and CD3 antibodies showed a normal profile and comparable absolute cell numbers of DN, DP, and SP thymocytes from CD4-survivin mice and their control littermates (Fig. 3 and unpublished data). The difference in the phenotype between CD4-survivin and lck-survivin mice is most likely due to the timing of survivin gene deletion and the fact that pre-TCR–mediated proliferation takes place between DN3 and DN4 stages (48). Although deletion in lck-survivin starts early, survivin deletion in CD4-survivin mice does not start until DN3 and is complete only in DN4 (see above) and hence most DN3 cells still express survivin required for DN3 to DN4 proliferation.
Figure 3.

Analysis of CD4-survivin mice. (A) The absolute cell numbers of DN, DP, SP (CD4+ CD8− or CD8+ CD4−), and peripheral T cells in 4–8-wk-old CD4-survivin mice and the littermate controls (n = 14). (B) CD4/CD8 ratio of thymocyte SP and peripheral T cells. (C) CD4 versus CD8 flow cytometric analysis of CD4-survivin thymocytes and the littermate controls. Numbers indicate the percentages of the corresponding T cell populations.
Although the numbers of DP and SP thymocytes in CD4-survivin mice were normal, it was surprising to note that the mature T cells in the spleen and lymph nodes were reduced significantly (Fig. 3 A, right). The reduction was particularly severe for CD8+ CD4− T cells. This is reflected in the ratio of CD4 to CD8 T cells, which is normally 2:1. In CD4-survivin peripheral organs, the ratio was ∼4:1 (Fig. 3 B).
Survivin-deficient T Cells in Periphery Are Less Mature.
The apparent reduction of peripheral T cells in CD4-survivin mice might be due to increased apoptosis or lack of proliferation (see below). We reasoned that if apoptosis were involved, the number of peripheral T cells in younger survivin-deficient animals would be the same as in the wild-type controls. Once in the periphery, they would then gradually disappear. In cases where mature T cells were depleted of their TCR α gene, CD8+ cells die with a half-life of 16 d, whereas CD4+ cells die with a half-life of 46 d (50). Staining with annexin V did not show any increase in apoptosis of the T cells of newborn or adult survivin-deficient mice (see below). In contrast, flow cytometric analysis showed that the reduction of peripheral T cells in CD4-survivin mice was even more pronounced in newborn (0–1-wk-old) mice. Although the subpopulations and cell numbers of the newborn survivin-deficient thymocytes are identical to their littermate controls (Fig. 4 A), splenic and lymph node CD4+ CD8− and CD4− CD8+ T cells were reduced by up to 14-fold (Fig. 4, A and B; CD4-survivin mice: CD4 cells: 2.68 ± 0.55 × 105, CD8 cells: 0.94 ± 0.21 × 105; wild-type littermate controls: CD4 cells: 27.05 ± 1.71 × 105, CD8 cells: 13.20 ± 0.49 × 105). As proliferation of SP thymocytes is known to occur between 1 and 21 d after birth (3), these data suggest that survivin deficiency affects this proliferative step, leading to a reduced number of peripheral T cells.
Figure 4.

Reduced number of peripheral T cells in young mice and their elevated HSA level. (A) The absolute cell numbers of DP, SP (CD4+ CD8− or CD8+ CD4−), thymocytes, and peripheral T cells in newborn CD4-survivin mice and the littermate controls (n = 5). (B) Representative flow cytometric profile of splenocytes from 0–1-wk-old CD4-survivin mice. The experiments have been repeated at least three times with similar findings. (C) CD8 versus HSA flow cytometric profile of peripheral T cells from CD4-survivin and control littermates. (D) Impaired CD25 activation marker in CD4-survivin peripheral T cells. Splenic cells from CD4-survivin mice and their littermate controls (WT) were stimulated with anti-CD3/CD28 antibodies for 18 h and stained with anti-CD4 and anti-CD25 antibodies. The CD4+ gated cell profiles are shown here.
To examine the activation status after homeostatic proliferation and to see if peripheral CD4+ CD8− and CD4− CD8+ from CD4-survivin mice are phenotypically mature, staining using several activation and maturation markers was performed. These markers include CD25 IL-2Rα and CD69, which is transiently elevated in positively selecting T cells and heat-stable antigen (HSA or CD24), which is down-regulated as T cells mature. No significant differences in CD69 and CD25 levels on the thymocytes and unstimulated peripheral T cells were observed between CD4-survivin mice and their littermate controls (unpublished data and see below). However, a significant proportion of peripheral T cells in CD4-survivin mice were HSA+, whereas wild-type animals have few HSA+ T cells in the periphery. In newborn mice, HSAhigh CD8+ T cells were nearly double that of the littermate controls (Fig. 4 C, left). In older animals, the difference is more pronounced. Although 5-wk-old wild-type animals contain few HSA+ T cells, CD4-survivin mice have significant numbers of HSA+ peripheral T cells. This is particularly pronounced in the CD8+ CD4− population, where close to 40% of the cells are HSA+ (Fig. 4 C). Some of these differences in the HSA levels can also be detected in the SP thymocyte population (unpublished data). These data suggest that survivin-deficient peripheral T cells are phenotypically semimature. They are also functionally immature as stimulation through their TCR complex did not lead to up-regulation of the activation marker CD25 (IL-2Rα; Fig. 4 D).
Survivin-deficient T Cells Do Not Exhibit Increased Sensitivity to Apoptotic Stimuli.
No massive cell death was observed in CD4-survivin DP thymocytes. Staining of thymocytes or peripheral T cells with 7-AAD or annexin V did not reveal any significant changes in the percentage of apoptotic cells (Fig. 5 and unpublished data). Differentiation into SP thymocytes also proceeds normally, suggesting that loss of survivin does not affect positive selection and steady-state apoptosis in vivo. To examine the process of death by neglect and induced cell death in the absence of survivin, we examined thymocytes and peripheral T cells of CD4-survivin mice for their propensity to undergo apoptosis and in response to outside stimuli. Baseline apoptosis ex vivo of cells from CD4-survivin and heterozygous controls is similar. As shown in Fig. 5, the absence of survivin has no effect on the propensity of thymocytes to undergo apoptosis in vitro. CD4-survivin–deficient thymocytes cultured in vitro for 12 h die at the same rate as survivin+ thymocytes. Similar results were also found when thymocytes were stimulated to undergo apoptosis by addition of an antibody specific to Fas, dexamethasone, etoposide, or a combination of PMA and ionomycin (Fig. 5 A and unpublished data). No differences were observed either when soluble FasL recombinant protein was added for a different period of time (Fig. 5 B). At a higher dose of dexamethasone, peripheral T cells undergo apoptosis. Survivin-deficient peripheral cells showed the same propensity for dexamethasone- or etoposide-induced cell death as the wild-type cells (Fig. 5 C). As expected, the addition of anti-CD3/CD28 antibodies had no effect on apoptosis of peripheral T cells. We conclude that survivin is not essential for the inhibition of apoptosis in T lymphocytes.
Figure 5.

Apoptosis proceeds normally in the absence of survivin. (A) Thymocytes from CD4-survivin and their littermate controls were incubated for 12 h in the absence of any stimuli or with anti-Fas antibody, dexamethasone (dex), or a combination of PMA phorbol ester and ionomycin (n = 4). Percent apoptotic cells were measured by staining with 7-AAD. The same experiments have been performed with annexin V with similar results. 0, unstimulated fresh thymocytes. (B) Percent thymocytes that were undergoing apoptosis were measured after 12, 24, or 48 h of incubation with 50 ng/ml soluble FasL recombinant protein (Qbiogene). Thymocytes were taken from CD4-survivin mice or their wild-type littermates (WT). (C) Percent apoptotic CD4 splenic T cells were measure by 7-AAD staining after 12 h of incubation in the absence (−) or presence of either 1 μM dexamethasone (dex), 10 μg/ml etoposide (ET), or a combination of anti-CD3/CD28 antibodies.
Proliferation and Cell Cycle Progression Are Blocked in Survivin-deficient T Cells.
The preceding data in lck-survivin mice suggest that they have a defect in T cell proliferation associated with an early developmental checkpoint. To determine the requirement for survivin in mature T cells, we prepared splenocytes or lymph node cells from CD4-survivin–deficient and control mice, labeled them with CFSE, and cultured them at 37°C for 4 d. Aliquots were treated with anti-CD3/CD28, Con A, or PMA/ionomycin to induce proliferation. These cells were then labeled with anti-CD4 antibody and the incorporation of CFSE was used to assess cell proliferation and cell division. As shown in Fig. 6 A, although wild-type control cells underwent several rounds of division as evidenced by the decreasing CFSE peaks, survivin-deficient T cells failed to complete even one division. The addition of the strongly mitogenic PMA/ionomycin also failed to induce cell division.
Figure 6.

Defective cell cycle progression of survivin-deficient T cells. (A) CFSE incorporation of Con A or PMA/ionomycin-stimulated T cells from adult animals. Numbers indicate the number of cell divisions of stimulated T cells. (B) Propidium iodide staining of PMA/ionomycin or Con A–stimulated T cells from adult or newborn mice. The experiments have been repeated more than three times with similar results.
To identify which step of cell cycle progression is affected, a similar experiment was performed, but the cells were stained with propidium iodide to assess DNA content. As shown in Fig. 6 B, the loss of survivin led to decreasing S phase. This was more evident in T cells from newborn animals (Fig. 6 B). When exposed to PMA and ionomycin, survivin-deficient T cells accumulated in the G2/M phase (Fig. 6 B). However, in cells stimulated with Con A, survivin-deficient cells did not accumulate at the G2/M phases but seemed to exhibit defects at the S phase. In vivo BrdU labeling studies of homeostatic proliferating thymocytes indicated that survivin deficiency did not lead to a G2/M block and resulted in an incomplete S phase (see below). These data suggest that in addition to the association with Aurora B mitotic kinase (35), which is required for cytokinesis, survivin might also participate in the replication checkpoint by binding with a yet to be identified protein.
Expansion of SP Thymocytes for Peripheral Homeostasis.
To investigate further the mechanism underlying the low T cell number in the periphery despite a normal thymocyte profile in survivin-deficient mice, we conducted BrdU labeling experiments in newborn and adult mice to measure the rate of DNA synthesis. BrdU was injected intraperitoneally into newborn and adult mice and the extent of BrdU incorporation into recently replicating thymocytes and peripheral T cells was assessed 3 h later. Although adult thymocytes did not proliferate and hence contained few BrdU+ cells, thymocytes from newborn mice exhibited a significant number of BrdU+ cells (Fig. 7 A). This is consistent with the notion that SP thymocytes undergo homeostatic proliferation in newborn animals before export to the periphery (3). As the peripheral compartment is filled up, the rate of proliferation drops. In survivin-deficient mice, however, BrdU incorporation was not diminished. If any, a reproducible increase in BrdU labeling was detected instead (Fig. 7 A). This effect was more pronounced in CD8+ cells than CD4+ cells (percent BrdU incorporation in CD8+ SP cells: CD4-survivin: 27.42 ± 1.78%, wild-type littermates: 18.2 ± 2.86%; percent BrdU incorporation in CD4+ cells: CD4-survivin: 7.53 ± 3.94%, wild-type littermates: 5.495 ± 1.03%; n = 4). These observations were substantiated by FACS® analysis of the side and forward scatter plots of these survivin-deficient SP thymocytes, which revealed a similar increase in the proportion of larger than normal CD8+ and CD4+ SP thymocytes (Fig. 7 B). Interestingly, splenic CD8+ cells, which usually are not cycling, incorporated BrdU when survivin was deleted (Fig. 7 A, right). These data suggest that survivin-deficient T cells could synthesize DNA and indeed replicated at a higher rate than wild-type T cells, but CFSE and propidium iodide analysis suggests that they could not complete the cell cycle.
Figure 7.
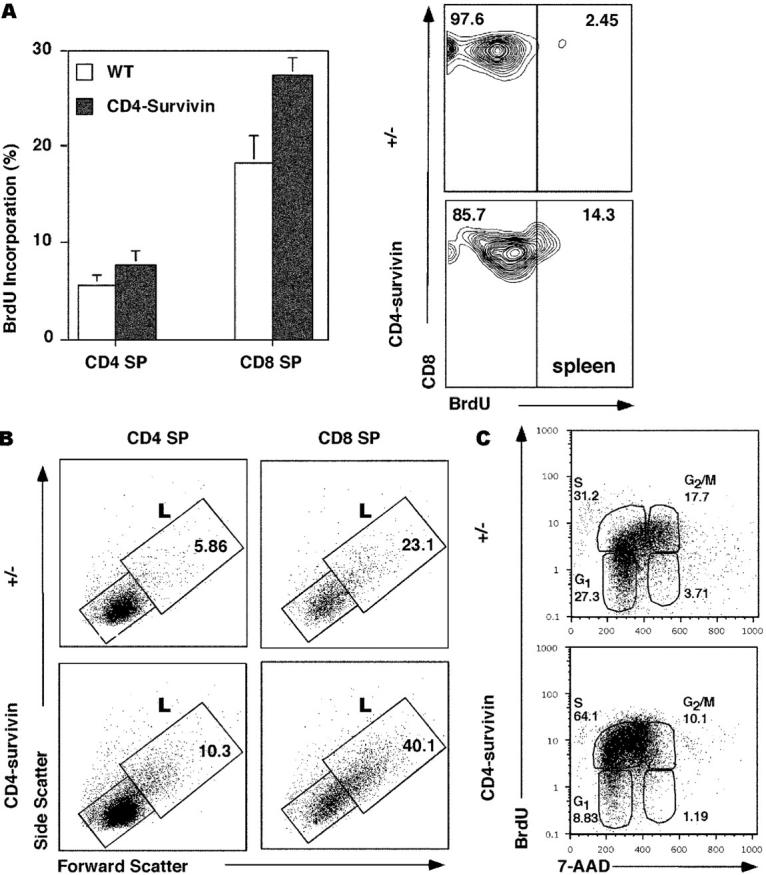
BrdU incorporation study of CD4-survivin–deficient mice. (A) BrdU labeling of SP thymocytes and peripheral T cells from CD4-survivin mice and their littermate (+/−) controls. Mice were injected intraperitoneally with BrdU and 3 h later flow cytometric analyses were conducted for the respective T cell populations: CD4 or CD8 SP thymocytes and spleen CD8+ cells. (B) Side and forward scatter analysis of CD4 or CD8 SP thymocytes from CD4-survivin and littermate (+/−) controls. The percentages of the large (L) cells are indicated. (C) Mice were injected with BrdU every 6 h in a 25-h period before their thymocytes were harvested and stained with either anti-CD4 or anti-CD8 antibodies followed by anti-BrdU antibodies and 7-AAD. The BrdU versus 7-AAD profiles of CD4− gated thymocytes, which include CD8+ CD4− SP and DN cells, are shown. Similar findings were also observed for CD8− gated thymocytes.
To further examine the cell cycle status of survivin-deficient thymocytes in vivo, 7-AAD was used to assess thymocyte DNA content after BrdU injection of newborn animals. Mice were analyzed 3 h after injection or after 25 h with four BrdU injections. As shown in Fig. 7 C, the BrdU versus 7-AAD profiles of CD8+ SP cells and some DN thymocytes (DN cells are the same between CD4-survivin and their wild-type littermates) revealed thymocytes in the G1/G0 state (BrdU−/7-AADlow) and some in the G2/M state (BrdU+/7-AADhigh). Proliferating cells were BrdU+ and exhibited an increasing amount of DNA content as they progressed through the different states of the cell cycle. Survivin-deficient thymocytes showed increased BrdU incorporation, even when labeled only for 3 h (unpublished data), suggesting that they have no problem at the G1 to S checkpoint. In contrast to wild-type cells, however, they accumulate at the state between 2N and 4N DNA contents (Fig. 7 C), indicating that they are blocked at the S phase of the cell cycle, before cells have undergone mitosis.
Discussion
We initiated our studies of survivin in T cell development with the notion that this unique IAP might play a dual role in proliferation and apoptosis. Based on the observation that weaker cells are purged actively during development, Raff (51) and Abrams (52) proposed the existence of a class of proteins with the characteristics of “proliferation–apoptosis coupler.” These types of protein are essential for an organism as they balance the process of proliferation versus apoptosis and can signal the cells to die if mitogen signals are not properly received. One such example is FADD, an adaptor protein for the TNF death receptor family. Cells with FADD deficiency are not only defective in TNF death receptor-mediated apoptosis, but also exhibit proliferation defects (53). The regions involved in apoptosis and proliferation have been mapped to two separate domains in FADD (45). Based on previously published work, survivin could also be a proliferation–apoptosis coupler. Survivin is expressed highly during the G2/M phases of the cell cycle and associates with the microtubule in the centrosomes (33). Loss of survivin expression by antisense oligonucleotides or disruption of survivin–microtubule interaction in HeLa cells leads to increased apoptosis and cell cycle arrest (25, 33). Survivin, caspase-3, and p21 cell cycle inhibitor were found to colocalize to centrosomes and perturbation of survivin expression by antisense technology led to increased caspase-3 activity and cytokinesis dysfunction (25, 33).
In contrast to the results obtained using antisense technology, however, here we show that survivin is not a proliferation–apoptosis coupler in vivo, at least in T cells. Similar to FADD deficiency (43, 53), the absence of survivin in developing T cells leads to a block in the DN to DP transition, and mature survivin-deficient T cells exhibit cell cycle defects at multiple stages. Interestingly, the block of DN T cells in lck-survivin mice occurs at a later stage than FADD-deficient mice. DN3 L cells can still be found in the absence of survivin, suggesting that FADD functions upstream of survivin (45, 53). In contrast to FADD, loss of survivin has no effect on cell death in T cells. It is possible that redundant functions of other antiapoptotic proteins in T cells can compensate for the loss of survivin. Alternatively, survivin does not have a strong antiapoptotic activity in vivo. Consistent with this, loss of function mutation of the survivin ortholog in Caenorhabditis elegans does not affect apoptosis, but exhibits defective cytokinesis (39, 40).
In addition to its role in early T cell development, survivin is also surprisingly essential for maturation and homeostasis of SP T cells. 0–8-wk-old CD4-survivin mice have normal thymic profiles and cell numbers, but their peripheral T cells are greatly reduced. This is especially true for CD8+ CD4− cells, but a similar situation was also seen for the CD4+ CD8− population. The effect is more pronounced in newborn animals. Although the proliferation stage at the DN3 to DN4 transition has been well characterized, the requirement of proliferation at the later stages of T cell development remains unclear. Proliferation of SP cells before migration to the peripheral tissues takes place actively early in life up to 21 d of age (3, 54–56). Adult animals exhibit some but little SP proliferation (3, 4, 57). Here we show that the proliferation in newborn animals is essential for complete T cell maturation and peripheral T cell compartment. Survivin-deficient peripheral T cells are defective in cell cycle progression and exhibit an immature T cell phenotype. The large size and increased number of thymocytes with >2 N DNA content in newborn survivin-deficient mice suggest that the G1 to S checkpoint is not affected and the block occurs at the later phases of the cell cycle. BrdU+ cells are increased in these cells after BrdU labeling experiment, suggesting that normal homeostatic negative feedback is being affected in the absence of survivin. The presence of mature T cells in the peripheral compartment is known to negatively regulate proliferation of SP thymic cells (54). In bone marrow chimera experiments where mature lymph node T cells were mixed with the bone marrow cells before transfer, proliferation of donor-derived SP thymocytes 18 d after transfer was inhibited (54). Thus, in survivin-deficient mice, the normal homeostatic proliferation to fill up the lymphopenic newborn peripheral compartment is severely compromised. This in turn contributes to a compensatory increase in the number of cycling thymic cells. These data underline the importance of proliferation after T cells have undergone positive selection in neonates, where the process of homeostatic expansion is crucial for the final stages of T cell development.
Interestingly, there seems to be more than one proliferative block for survivin-deficient T cells. In vitro, PMA/ionomycin-stimulated survivin-deficient lymphocytes accumulated at the G2/M phase, in accordance with the results shown by others in tumor cell lines and embryos (33, 36, 58). However, our 7-AAD/BrdU labeling data surprisingly indicated that thymocyte proliferation is blocked at the S phase. Con A–stimulated peripheral survivin−/− T cells similarly showed an S phase block (Fig. 6). The difference could be explained by the strong signals conferred by PMA/ionomycin that somehow bypass the S phase block normally seen in milder or physiological conditions. Polyploidy of survivin-deficient cells was described previously for survivin−/− embryos and HeLa cells (36, 58). This was the result of some survivin-deficient cells exiting mitosis without proper chromosome segregation. Consistent with an earlier block of survivin-deficient T cells in vivo, however, little or no polyploidy cells were detected in CD4-survivin– or lck-survivin–deficient mice (Fig. 6 and unpublished data). Most survivin-deficient peripheral cells presumably were exported from the thymus without undergoing proliferation, consistent with the view that thymic SP proliferation is not required for export but is part of homeostatic expansion to fill the “lymphopenic” neonatal peripheral compartment.
Mature T cell homeostatic expansion has been studied in the past by transferring small numbers of mature T cells into lymphopenic hosts (either RAG-1−/− or SCID mice). Upon expansion, they express CD44 and acquire a memory-like phenotype. Homeostatic proliferation requires TCR signals, IL-7, and possibly IL-15 (59–61). Spleen and lymph nodes of newborn mice are “empty” and support lymphopenia-induced proliferation (55, 62). Naive T cells transferred to newborn animals proliferate with the same requirement for MHC/peptide and acquisition of the CD44 memory marker as homeostatic proliferation in adult lymphopenic hosts (55, 62, 63). Survivin is most likely required for lymphopenic proliferation as well because newborn mice are essentially lymphopenic and stimulation of mature T cells in vitro with anti-TCR antibodies did not result in cell division. We showed that homeostatic thymocyte proliferation is blocked in vivo in the absence of survivin despite an enhanced attempt to proliferate. Increased BrdU incorporation was also detected in the splenic T cells of survivin-deficient mice, suggesting that these cells attempt to proliferate to compensate for the lack of peripheral T cells. Further studies will be necessary to understand how survivin-mediated proliferation of neonatal T cells is regulated and how peripheral T cell homeostasis is achieved.
Acknowledgments
We thank Christopher Wilson for the generous gift of lck-Cre and CD4-cre transgenic mice, Saskia Pollefeyt and Astrid DeVriese for preparing the floxed survivin gene targeting vector, Sue Sohn for critical reading of the manuscript and technical advice, Hector Nolla for help in flow cytometric analysis, Zi-Chun Hua for discussion, the ES cell and transgenic core facility personnel at the Center for Transgene Technology and Gene Therapy, and the Flanders Ineruniversity Institute for Biotechnology for technical assistance.
This work was supported by the Henry Wheeler postdoctoral fellowship (to Z. Xing), by grants from the National Institutes of Health (CA75162), and the Flanders Fund for Scientific Research, Belgium (G.0086.02).
Abbreviations used in this paper: 7-AAD, 7-amino actinomycin D; BrdU, 5-bromo-2-deoxyuridine; CFSE, carboxy fluorescein diacetate succinimidyl ester; DN, double negative; DP, double positive; ES, embryonic stem; HSA, heat-stable antigen; IAP, inhibitor of apoptosis protein; SP, single positive.
References
- 1.von Boehmer, H. 1994. Positive selection of lymphocytes. Cell. 76:219–228. [DOI] [PubMed] [Google Scholar]
- 2.Sohn, S.J., A. Rajpal, and A. Winoto. 2003. Apoptosis during lymphoid development. Curr. Opin. Immunol. 15:209–216. [DOI] [PubMed] [Google Scholar]
- 3.Ceredig, R. 1990. Intrathymic proliferation of perinatal mouse alpha beta and gamma delta T cell receptor-expressing mature T cells. Int. Immunol. 2:859–867. [DOI] [PubMed] [Google Scholar]
- 4.Ernst, B., C.D. Surh, and J. Sprent. 1995. Thymic selection and cell division. J. Exp. Med. 182:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krammer, P.H. 2000. CD95's deadly mission in the immune system. Nature. 407:789–795. [DOI] [PubMed] [Google Scholar]
- 6.Opferman, J.T., and S.J. Korsmeyer. 2003. Apoptosis in the development and maintenance of the immune system. Nat. Immunol. 4:410–415. [DOI] [PubMed] [Google Scholar]
- 7.Bouillet, P., J.F. Purton, D.I. Godfrey, L.C. Zhang, L. Coultas, H. Puthalakath, M. Pellegrini, S. Cory, J.M. Adams, and A. Strasser. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 415:922–926. [DOI] [PubMed] [Google Scholar]
- 8.Rathmell, J.C., T. Lindsten, W.X. Zong, R.M. Cinalli, and C.B. Thompson. 2002. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat. Immunol. 3:932–939. [DOI] [PubMed] [Google Scholar]
- 9.Singer, G.G., and A.K. Abbas. 1994. The fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1:365–371. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto, H., C.D. Surh, and J. Sprent. 1998. A role for fas in negative selection of thymocytes in vivo. J. Exp. Med. 187:1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamhamedi-Cherradi, S.E., S.J. Zheng, K.A. Maguschak, J. Peschon, and Y.H. Chen. 2003. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL(−/−) mice. Nat. Immunol. 4:255–260. [DOI] [PubMed] [Google Scholar]
- 12.Cretney, E., A.P. Uldrich, S.P. Berzins, A. Strasser, D.I. Godfrey, and M.J. Smyth. 2003. Normal thymocyte negative selection in TRAIL-deficient mice. J. Exp. Med. 198:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newton, K., A.W. Harris, M.L. Bath, K.G.C. Smith, and A. Strasser. 1998. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 17:706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deveraux, Q.L., and J.C. Reed. 1999. IAP family proteins-suppressors of apoptosis. Genes Dev. 13:239–252. [DOI] [PubMed] [Google Scholar]
- 15.Verhagen, A.M., E.J. Coulson, and D.L. Vaux. 2001. Inhibitor of apoptosis proteins and their relatives: IAPs and other BIRPs. Genome Biol. 2:reviews3009. [DOI] [PMC free article] [PubMed]
- 16.Altieri, D.C. 2003. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer. 3:46–54. [DOI] [PubMed] [Google Scholar]
- 17.Verhagen, A.M., P.G. Ekert, M. Pakusch, J. Silke, L.M. Connolly, G.E. Reid, R.L. Moritz, R.J. Simpson, and D.L. Vaux. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 102:43–53. [DOI] [PubMed] [Google Scholar]
- 18.Du, C.Y., M. Fang, Y.C. Li, L. Li, and X.D. Wang. 2000. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 102:33–42. [DOI] [PubMed] [Google Scholar]
- 19.Ambrosini, G., C. Adida, and D.C. Altieri. 1997. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3:917–921. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi, K., M. Hatano, M. Otaki, T. Ogasawara, and T. Tokuhisa. 1999. Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation. Proc. Natl. Acad. Sci. USA. 96:1457–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamm, I., Y. Wang, E. Sausville, D.A. Scudiero, N. Vigna, T. Oltersdorf, and J.C. Reed. 1998. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 58:5315–5320. [PubMed] [Google Scholar]
- 22.Conway, E.M., S. Pollefeyt, J. Cornelissen, I. DeBaere, M. Steiner-Mosonyi, K. Ong, M. Baens, D. Collen, and A.C. Schuh. 2000. Three differentially expressed survivin cDNA variants encode proteins with distinct antiapoptotic functions. Blood. 95:1435–1442. [PubMed] [Google Scholar]
- 23.Grossman, D., P.J. Kim, O.P. Blanc-Brude, D.E. Brash, S. Tognin, P.C. Marchisio, and D.C. Altieri. 2001. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J. Clin. Invest. 108:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawamura, K., N. Sato, J. Fukuda, H. Kodama, J. Kumagai, H. Tanikawa, Y. Shimizu, and T. Tanaka. 2003. Survivin acts as an antiapoptotic factor during the development of mouse preimplantation embryos. Dev. Biol. 256:331–341. [DOI] [PubMed] [Google Scholar]
- 25.Li, F., E.J. Ackermann, C.F. Bennett, A.L. Rothermel, J. Plescia, S. Tognin, A. Villa, P.C. Marchisio, and D.C. Altieri. 1999. Pleiotropic cell division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1:461–465. [DOI] [PubMed] [Google Scholar]
- 26.Ambrosini, G., C. Adida, G. Sirugo, and D.C. Altieri. 1998. Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting. J. Biol. Chem. 273:11177–11182. [DOI] [PubMed] [Google Scholar]
- 27.Jiang, X., C. Wilford, S. Duensing, K. Munger, G. Jones, and D. Jones. 2001. Participation of survivin in mitotic and apoptotic activities of normal and tumor-derived cells. J. Cell. Biochem. 83:342–354. [DOI] [PubMed] [Google Scholar]
- 28.Choi, K.S., T.H. Lee, and M.H. Jung. 2003. Ribozyme-mediated cleavage of the human survivin mRNA and inhibition of antiapoptotic function of survivin in MCF-7 cells. Cancer Gene Ther. 10:87–95. [DOI] [PubMed] [Google Scholar]
- 29.Mesri, M., N.R. Wall, J. Li, R.W. Kim, and D.C. Altieri. 2001. Cancer gene therapy using a survivin mutant adenovirus. J. Clin. Invest. 108:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasof, G.M., and B.C. Gomes. 2001. Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem. 276:3238–3246. [DOI] [PubMed] [Google Scholar]
- 31.Verdecia, M.A., H. Huang, E. Dutil, D.A. Kaiser, T. Hunter, and J.P. Noel. 2000. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat. Struct. Biol. 7:602–608. [DOI] [PubMed] [Google Scholar]
- 32.Song, Z., X. Yao, and M. Wu. 2003. Direct interaction between survivin and Smac is essential for the anti-apoptotic activity of survivin during Taxol-induced apoptosis. J. Biol. Chem. 278:23130–23140. [DOI] [PubMed] [Google Scholar]
- 33.Li, F., G. Ambrosini, E. Chu, J. Plescia, S. Tognin, P. Marchisio, and D. Altieri. 1998. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 396:580–584. [DOI] [PubMed] [Google Scholar]
- 34.Lu, C.-D., D.C. Altieri, and N. Tanigawa. 1998. Expression of a novel antiapoptosis gene, survivin, correlated with tumor cell apoptosis and p53 accumulation in gastric carcinomas. Cancer Res. 58:1808–1812. [PubMed] [Google Scholar]
- 35.Bolton, M.A., W. Lan, S.E. Powers, M.L. McCleland, J. Kuang, and P.T. Stukenberg. 2002. Aurora B kinase exists in a complex with survivin and INCENP and its kinase activity is stimulated by survivin binding and phosphorylation. Mol. Biol. Cell. 13:3064–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uren, A.G., L. Wong, M. Pakusch, K.J. Fowler, F.J. Burrows, D.L. Vaux, and K.H.A. Choo. 2000. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 10:1319–1328. [DOI] [PubMed] [Google Scholar]
- 37.Conway, E.M., S. Pollefeyt, M. Steiner-Mosonyi, W. Luo, A. Devriese, F. Lupu, F. Bono, N. Leducq, F. Dol, P. Schaeffer, et al. 2002. Deficiency of survivin in transgenic mice exacerbates Fas-induced apoptosis via mitochondrial pathways. Gastroenterology. 123:619–631. [DOI] [PubMed] [Google Scholar]
- 38.Uren, A.G., T. Beilharz, M.J. O'Connell, S.J. Bugg, R. van Driel, D.L. Vaux, and T. Lithgow. 1999. Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc. Natl. Acad. Sci. USA. 96:10170–10175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller, L.K. 1999. An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol. 9:323–328. [DOI] [PubMed] [Google Scholar]
- 40.Fraser, A.G., C. James, G.I. Evan, and M.O. Hengartner. 1999. Caenorhabditis elegans inhibitor of apoptosis protein (IAP) homologue BIR-1 plays a conserved role in cytokinesis. Curr. Biol. 9:292–301. [DOI] [PubMed] [Google Scholar]
- 41.Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. Perez-Melgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, et al. 2001. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15:763–774. [DOI] [PubMed] [Google Scholar]
- 42.Schoonjans, L., V. Kreemers, S. Danloy, R.W. Moreadith, Y. Laroche, and D. Collen. 2003. Improved generation of germline-competent embryonic stem cell lines from inbred mouse strains. Stem Cells. 21:90–97. [DOI] [PubMed] [Google Scholar]
- 43.Kabra, N.H., C. Kang, L.C. Hsing, J. Zhang, and A. Winoto. 2001. T cell-specific FADD-deficient mice: FADD is required for early T cell development. Proc. Natl. Acad. Sci. USA. 98:6307–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sternsdorf, T., K. Jensen, D. Zuchner, and H. Will. 1997. Cellular localization, expression, and structure of the nuclear dot protein 52. J. Cell Biol. 138:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hua, Z.C., S.J. Sohn, C. Kang, D. Cado, and A. Winoto. 2003. A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity. 18:513–521. [DOI] [PubMed] [Google Scholar]
- 46.Shapiro, H.M. 1995. Practical Flow Cytometry. Wiley-Liss Inc., New York. 326 pp.
- 47.Wolfer, A., A. Wilson, M. Nemir, H.R. MacDonald, and F. Radtke. 2002. Inactivation of Notch1 impairs VDJbeta rearrangement and allows pre-TCR-independent survival of early alpha beta Lineage Thymocytes. Immunity. 16:869–879. [DOI] [PubMed] [Google Scholar]
- 48.Hoffman, E.S., L. Passoni, T. Crompton, T.M.J. Leu, D.G. Schatz, A. Koff, M.J. Owen, and A.C. Hayday. 1996. Productive T-cell receptor beta-chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev. 10:948–962. [DOI] [PubMed] [Google Scholar]
- 49.Hayday, A.C., D.F. Barber, N. Douglas, and E.S. Hoffman. 1999. Signals involved in gamma/delta T cell versus alpha/beta T cell lineage commitment. Semin. Immunol. 11:239–249. [DOI] [PubMed] [Google Scholar]
- 50.Polic, B., D. Kunkel, A. Scheffold, and K. Rajewsky. 2001. How alpha beta T cells deal with induced TCR alpha ablation. Proc. Natl. Acad. Sci. USA. 98:8744–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raff, M.C. 1992. Social controls on cell survival and cell death. Nature. 356:397–400. [DOI] [PubMed] [Google Scholar]
- 52.Abrams, J.M. 2002. Competition and compensation: coupled to death in development and cancer. Cell. 110:403–406. [DOI] [PubMed] [Google Scholar]
- 53.Zhang, J., D. Cado, A. Chen, N.H. Kabra, and A. Winoto. 1998. Absence of Fas-mediated apoptosis and T cell receptor-induced proliferation in FADD-deficient mice. Nature. 392:296–300. [DOI] [PubMed] [Google Scholar]
- 54.Le Campion, A., F. Vasseur, and C. Penit. 2000. Regulation and kinetics of premigrant thymocyte expansion. Eur. J. Immunol. 30:738–746. [DOI] [PubMed] [Google Scholar]
- 55.Min, B., R. McHugh, G.D. Sempowski, C. Mackall, G. Foucras, and W.E. Paul. 2003. Neonates support lymphopenia-induced proliferation. Immunity. 18:131–140. [DOI] [PubMed] [Google Scholar]
- 56.Ichii, H., A. Sakamoto, M. Hatano, S. Okada, H. Toyama, S. Taki, M. Arima, Y. Kuroda, and T. Tokuhisa. 2002. Role for Bcl-6 in the generation and maintenance of memory CD8+ T cells. Nat. Immunol. 3:558–563. [DOI] [PubMed] [Google Scholar]
- 57.Le Campion, A., B. Lucas, N. Dautigny, S. Leaument, F. Vasseur, and C. Penit. 2002. Quantitative and qualitative adjustment of thymic T cell production by clonal expansion of premigrant thymocytes. J. Immunol. 168:1664–1671. [DOI] [PubMed] [Google Scholar]
- 58.Carvalho, A., M. Carmena, C. Sambade, W.C. Earnshaw, and S.P. Wheatley. 2003. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J. Cell Sci. 116:2987–2998. [DOI] [PubMed] [Google Scholar]
- 59.Lodolce, J.P., D.L. Boone, S. Chai, R.E. Swain, T. Dassopoulos, S. Trettin, and A. Ma. 1998. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 9:669–676. [DOI] [PubMed] [Google Scholar]
- 60.Jameson, S.C. 2002. Maintaining the norm: T-cell homeostasis. Nat. Rev. Immunol. 2:547–556. [DOI] [PubMed] [Google Scholar]
- 61.Seddon, B., P. Tomlinson, and R. Zamoyska. 2003. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 4:680–686. [DOI] [PubMed] [Google Scholar]
- 62.Le Campion, A., C. Bourgeois, F. Lambolez, B. Martin, S. Leaument, N. Dautigny, C. Tanchot, C. Penit, and B. Lucas. 2002. Naive T cells proliferate strongly in neonatal mice in response to self-peptide/self-MHC complexes. Proc. Natl. Acad. Sci. USA. 99:4538–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adkins, B., T. Williamson, P. Guevara, and Y. Bu. 2003. Murine neonatal lymphocytes show rapid early cell cycle entry and cell division. J. Immunol. 170:4548–4556. [DOI] [PubMed] [Google Scholar]