

Abstract
Voltage-gated Na+ channels exhibit two forms of inactivation, one form (fast inactivation) takes effect on the order of milliseconds and the other (slow inactivation) on the order of seconds to minutes. While previous studies have suggested that fast and slow inactivation are structurally independent gating processes, little is known about the relationship between the two. In this study, we probed this relationship by examining the effects of slow inactivation on a conformational marker for fast inactivation, the accessibility of a site on the Na+ channel III–IV linker that is believed to form a part of the fast inactivation particle. When cysteine was substituted for phenylalanine at position 1304 in the rat skeletal muscle sodium channel (μl), application of [2-(trimethylammonium)ethyl]methanethiosulfonate (MTS-ET) to the cytoplasmic face of inside-out patches from Xenopus oocytes injected with F1304C RNA dramatically disrupted fast inactivation and displayed voltage-dependent reaction kinetics that closely paralleled the steady state availability (h∞•) curve. Based on this observation, the accessibility of cys1304 was used as a conformational marker to probe the position of the fast inactivation gate during the development of and the recovery from slow inactivation. We found that burial of cys1304 is not altered by the onset of slow inactivation, and that recovery of accessibility of cys1304 is not slowed after long (2–10 s) depolarizations. These results suggest that (a) fast and slow inactivation are structurally distinct processes that are not tightly coupled, (b) fast and slow inactivation are not mutually exclusive processes (i.e., sodium channels may be fast- and slow-inactivated simultaneously), and (c) after long depolarizations, recovery from fast inactivation precedes recovery from slow inactivation.
Keywords: methanethiosulfonate, SkM1, sodium channels, mutagenesis, patch clamp
introduction
Upon repolarization from brief depolarizations (<50 ms), Na+ channels recover from inactivation with a single kinetic phase whose time constant is on the order of a few milliseconds. After long depolarizations (seconds to minutes), recovery from inactivation proceeds through multiple kinetic phases whose time constants range over several orders of magnitude (a few milliseconds to tens of seconds). The most rapid phase of recovery is thought to correspond to fast inactivation (the Hodgkin- Huxley h-gate), which is responsible for limiting the duration of the action potential and regulating the availability of sodium channels for subsequent discharges (Hodgkin and Huxley, 1952a , 1952b ). All other kinetic phases of recovery have been associated with a relatively poorly understood process called slow inactivation, first described in lobster giant axons (Narahashi, 1964) and subsequently found in every Na+ channel isoform studied to date, both in native tissues (Adelman and Palti, 1969; Almers et al., 1983; Quandt, 1987; Ruben et al., 1992; Simoncini and Stühmer, 1987) and in heterologous expression systems (Cummins and Sigworth, 1996; Hayward et al., 1997; Townsend and Horn, 1997). The physiological importance of slow inactivation as an additional regulator of channel availability was not established until recently, when defects in slow inactivation were found to underlie the pathophysiology of some forms of heritable muscle disease (Cannon, 1996; Cummins and Sigworth, 1996; Hayward et al., 1997).
While fast inactivation is thought to occur via intracellular occlusion of the ion-conducting pore by a cluster of hydrophobic amino acids in the III–IV interdomain (West et al., 1992), little is known of the structural basis for slow inactivation. A growing body of evidence supports the hypothesis that fast and slow inactivation are structurally distinct: application of intracellular proteases disrupts fast but not slow inactivation (Rudy, 1978); site-directed mutagenesis in the Na+ channel III–IV interdomain also disrupts fast but not slow inactivation (Cummins and Sigworth, 1996; Featherstone et al., 1996); and extracellular metal cations inhibit slow inactivation but do not affect fast inactivation (Khodorov et al., 1976; Townsend and Horn, 1997). Unfortunately, the few mutations known to affect slow inactivation do not colocalize or clearly implicate a particular region of the protein (Cummins and Sigworth, 1996; Hayward et al., 1997; Wang and Wang, 1997).
There is also very little known of the kinetic relationship between fast and slow inactivation, primarily because transitions between different inactivated states do not involve changes in ionic current and cannot be monitored with standard electrophysiological techniques. Some questions that are still unanswered include the following: Are fast and slow inactivation mutually exclusive? Do the fast and slow inactivation gates compete for the same binding site? Are fast and slow inactivation tightly coupled or are they kinetically independent? After long depolarizations, must recovery from slow inactivation precede recovery from fast inactivation, or vice-versa?
In this work, we have tried to address some of these questions by using a conformational marker for the position of the fast inactivation gate and by monitoring its movement during entry to and recovery from slow inactivation. Kellenberger et al. (1996) have shown that the accessibility of a cysteine substituted for phe1489 in the III–IV loop of the rat brain IIA Na+ channel is reduced when the channel is fast inactivated. We have measured the accessibility of a cysteine substituted at the analogous site, phe1304, in the rat adult skeletal muscle sodium channel (μ1) using modification by [2-(trimethylammonium)ethyl]methanethiosulfonate (MTS-ET).1 This accessibility measurement provided a conformational marker for the fast inactivation gate, allowing us to examine the relationship between fast and slow inactivation. Our results indicate that fast and slow inactivation are not mutually exclusive, nor do they compete for the same binding site. We also found that dissociation of the fast inactivation particle during recovery from inactivation occurs within a few milliseconds, regardless of the position of the slow inactivation gate. This indicates that fast and slow inactivation are not tightly coupled, and that recovery from fast inactivation precedes recovery from slow inactivation after long depolarizations.
materials and methods
Mutagenesis and mRNA Preparation
The rat adult skeletal muscle Na+ channel α subunit cDNA (μ1) containing a silent ClaI site at base 3865 was subcloned into the EcoRI site of the Xenopus oocyte expression vector pGEMHE (Liman et al., 1992). Mutation F1304C was engineered into the construct using polymerase chain reaction overlap extension to generate a 503-bp fragment that was subcloned into a unique ClaI-SacII segment spanning the III–IV interdomain. The mutagenic fragment was ligated into the expression construct and the presence of the mutation was verified by sequencing the cassette and flanking regions. The human sodium channel β1 subunit cDNA (McClatchey et al., 1993) was also subcloned into the EcoRI site of pGEMHE. RNA for μ1, F1304C, and human β1 subunit were all generated by in vitro translation of linearized plasmids (Megascript kit; Ambion Inc., Austin, TX).
Expression of Sodium Channels
Xenopus oocytes were harvested and coinjected with either wild type (WT) μ1 + human β1 RNA or F1304C + human β1 RNA as described in Chen and Cannon (1995). Before electrophysiological recording, oocytes were incubated for 2–3 d at 18°C in ND-96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.6) supplemented with pyruvate (2.5 mM) and gentamicin (50 μg/ml).
Electrophysiology
All experiments were performed at room temperature (20– 22°C). Oocytes were placed in a hypertonic solution to facilitate removal of the vitelline membrane before transfer into the recording chamber. Na+ currents were measured in excised inside-out macropatches with an Axopatch 200B amplifier (Axon Instruments Inc., Foster City, CA). The amplifier output was filtered at 5 kHz and digitally sampled at 20 kHz using a Digidata 1200 interface (Axon Instruments Inc.). Data was stored on a 75-mHz Pentium-based computer under control of a custom AxoBasic data acquisition program. Pipette capacitance was compensated using analog circuitry, and correction of leakage conductance was performed on-line through digital scaling and subtraction of passive currents elicited by hyperpolarization from −120 to −145 mV.
Patch electrodes were fabricated from borosilicate capillary tubes (1.65 mm o.d.) with a two-stage puller (Sutter Instrument Co., Novato, CA). The shanks of the pipettes were coated with Sylgard and the tips were heat polished to a final diameter of 0.5– 1.5 μm. The pipette solution contained (mM): 100 NaCl, 10 HEPES, 2 CaCl2, 1 MgCl2, pH 7.6. The bath contained 100 KCl, 10 HEPES, 5 EGTA, 1 MgCl2, pH 7.6 (+20 mM NaCl only when current–voltage curves were recorded). To evaluate the kinetics of solution exchange, a sodium-based bath solution containing (mM) 100 NaCl, 10 HEPES, 5 EGTA, and 1 MgCl2 was used. Stock solutions (1 or 2 mM) of MTS-ET (Toronto Research Chemicals Inc., Downsview, Ontario, Canada) were prepared from the solid in 1 ml of distilled, deionized H2O and placed on ice at the beginning of each recording day. Appropriate amounts were diluted into 10 ml of bath solution (to a final concentration of 1, 2, 4, or 8 μM) after suitable patches were obtained and immediately before use. MTS-ET solutions were never used for more than 10 min after dilution from the stock.
Rapid solution exchange was achieved with a computer-controlled PZS-200 piezoelectric stack (Burleigh Instruments Inc., Fishers, NY), to which a piece of glass capillary theta-tubing (Warner Instrument Co., Hamden, CT) was attached (2.0 mm o.d.). Theta glass was pulled to a diameter of 50–100 μm per lumen with a two-stage puller (Sutter Instrument Co.) and solutions were passed through each lumen by gravity filtration.
Data Analysis
Curve fitting was performed off-line using AxoBasic or SigmaPlot (Jandel Scientific Co., San Rafael, CA). Conductance was calculated as G(V) = I peak(V)/(V − Erev), where the reversal potential, Erev, was measured experimentally for each patch. Steady state fast inactivation, h∞•, and the voltage dependence of the modification rate were fitted to Boltzmann curves with nonzero pedestals, Io, calculated as I/I peak = [1 − I o]/{1 + exp[(V − V1/2)/k]} + I o, where V1/2 is the voltage at half-maximum, and k is the slope factor. All symbols with error bars indicate means ± SEM.
For modification experiments, the fractional persistent current for a given pulse was calculated by averaging the value of the Na+ current between 40 and 42 ms after depolarization to −20 mV and dividing by the average value over the same interval obtained after the modification reaction was complete. Curves of fractional persistent current (F) versus exposure time (t exp) were fit to a monoexponential with a nonzero pedestal and a maximum value normalized to unity by the trace with maximal modification: F = (1 − F o)[1 − exp(−t exp/τmod)] + F o, where τmod is the reciprocal of the reaction rate and F o is the mean value of the persistent current between 40 and 42 ms before any exposure has occurred. Approximately 20% of patches exhibited multicomponent behavior, with as much as 10–30% of the modification reaction occurring much more slowly than normal. For these patches, we fit the faster exponential only, allowing the maximum value to be a free parameter in the fit.
results
Fast Inactivation Is Mildly Disrupted in F1304C
Sodium channel gating was assessed in inside-out patches taken from Xenopus oocytes injected with RNA encoding either adult skeletal muscle sodium channel α subunit (μ1) + human β1 subunit or F1304C + human β1 subunit. Site 1304 lies near the middle of the sodium channel III–IV interdomain, a region that is crucial for normal fast inactivation (Stühmer et al., 1989). When the corresponding residue in the rat brain IIA isoform (F1489) is mutated to hydrophilic residues, fast inactivation is almost completely disrupted, while mutation to hydrophobic residues leaves inactivation more or less intact (Kellenberger et al., 1997; West et al., 1992). For F1304C, we found that inactivation was mildly disrupted, consistent with the intermediate hydrophobicity of cysteine.
Fig. 1 A shows macroscopic current traces in response to a step depolarization from −120 to −20 mV taken from patches expressing wild-type or F1304C channels. The mutant displays a slowed rate of macroscopic current decay (τh) and a greater level of persistent current (measured as the average current between 40 and 42 ms after depolarization divided by the peak current): 9.1 ± 1.2% (n = 10) for F1304C, compared with 0.3 ± 0.07% (n = 6) for WT. This persistent current eventually decays over hundreds of milliseconds, probably because of slow inactivation.
Figure 1.

Mutation F1304C destabilizes fast inactivation. (A) Na+ currents were elicited from excised inside-out macropatches by depolarization to −20 mV from a holding potential of −120 mV. Averages of two current traces each for WT and mutant F1304C are shown normalized to peak amplitude and superimposed. Mutant channels display a slowed current decay and a greater fraction of persistent current. (B) Voltage dependence of steady state fast inactivation, h∞•(V), in response to a 200-ms prepulse, is right shifted for F1304C (V1/2 = −81.6 ± 1.3 mV, slope = 5.5 ± 0.5, n = 9), compared with WT (V1/2 = −98.3 ± 1.9, slope = 5.7 ± 0.3, n = 12). G(V), computed as peak I Na/(V − Erev), is similar for WT (n = 12) and F1304C (n = 9). These data are consistent with a destabilization of fast inactivation caused by substitution of cysteine for phenylalanine at site 1304.
The steady state availability curves (Fig. 1 B), measured with 200-ms conditioning pulses, exhibited a 16.7-mV rightward shift in F1304C while G(V) curves were similar for both. Taken together, our data on the gating properties of F1304C suggest a mild, selective defect in fast inactivation, and agree well with the results of a previous study on the analogous mutation in the rat brain IIA sodium channel isoform (Kellenberger et al., 1996).
MTS-ET Dramatically Disrupts Fast Inactivation of F1304C
We examined the effects of substituting a positively charged group at site 1304 by applying MTS-ET to the cytoplasmic face of F1304C macropatches. MTS-ET reacts with free thiol groups, attaching a quaternary-substituted amine through disulfide bond formation. Application of 2 mM MTS-ET to the cytoplasmic faces of macropatches containing WT channels for several minutes had no effect other than a reversible 15% peak current reduction (data not shown). In contrast, modification of F1304C with 4 μM MTS-ET for a cumulative time of 0.5 s (10 exposures of 50 ms) caused a nearly complete disruption of fast inactivation (Fig. 2 A).
Figure 2.

MTS-ET disrupts fast inactivation of F1304C. (A) Selected current traces evoked by depolarization to −20 mV from −120 mV after successive 50-ms exposures to 4 μM MTS-ET are superimposed over a final trace measured after an additional 4 s of exposure (average of two individual records) and a control trace measured before any MTS-ET exposure (average of 10 individual records). MTS-ET application to the cytoplasmic face was accomplished by computer-controlled rapid solution switching (see materials and methods) from bath solution into 4 μM MTS-ET-containing bath solution. In this case, the peak current increased from −129 pA before exposure to −208 pA after the modification reached completion, and the persistent current (measured as the average current between 40 and 42 ms divided by the peak current) increased from 6.1% before exposure to 65.5% after the modification reached completion. (B) After each experiment, the kinetics of solution exchange was evaluated by switching the patch into a solution containing a sodium concentration equal to the pipette solution. The switching was timed to occur for 20 ms at the beginning of a depolarization to 0 from −120 mV. For this patch, there was a 7.4-ms delay from the time of the switching command before the current began to decay, and a monoexponential fit to the current decay gave a time constant for solution exchange of 1.7 ms. (C) The value of the fractional persistent current after each exposure is plotted against the cumulative exposure time. The curve was fit with a monoexponential approaching unity that had nonzero pedestal (solid line): F = (1 − F o)(1 − exp[−t exp/τmod]) + F o, where τmod is the reciprocal of the reaction rate and was 0.279 s (corresponding to a rate of 0.895 μmol−1 s−1), and F o is the value of the fractional persistent current before modification (0.059).
The peak current of F1304C macropatches increased in response to modification by 61 ± 2% (n = 118), a finding consistent with previous studies in which fast inactivation was removed by proteases (Rojas and Armstrong, 1971; Gonoi and Hille, 1987). The increase is thought to reflect a longer mean open time in MTS-ET modified channels, which results in a greater number of superimposed channel openings at any given time after depolarization (Kellenberger et al., 1996; Gonoi and Hille, 1987).
The persistent current relative to the peak current for completely modified patches was 65.3 ± 1.3% (n = 10), a sevenfold increase over the unmodified channels. The MTS-ET-modified current eventually did decay over hundreds of milliseconds, demonstrating that some form of inactivation, presumably slow inactivation, persists after the disruption of fast inactivation. Other studies on sodium channels lacking fast inactivation have also shown that slow inactivation is relatively unaffected by either intracellular protease treatment (Rudy, 1978) or mutagenesis at site 1304 (Cummins and Sigworth, 1996). Brief exposure to 2 mM dithiothreitol completely reversed the effects of MTS-ET modification on F1304C current traces (data not shown).
Measurement of the Reaction Rate of MTS-ET with cys1304
Our next concern was to develop a method for measuring the rate of the chemical reaction between MTS-ET and cys1304. To that end, we constructed a piezoelectric-based rapid solution exchange system capable of millisecond solution switching (see materials and methods). The intracellular face of each F1304C inside-out macropatch was exposed to MTS-ET for brief intervals (20, 50, or 150 ms) and the current was sampled in between each exposure with a 45-ms test pulse from the holding potential (−120 mV) to −20 mV (Fig. 2 A). After a suitable number of pulses (5–50), each patch was exposed for several seconds to MTS-ET to achieve complete modification.
At the end of every modification experiment performed, the fidelity of the solution exchange was confirmed with a 20-ms solution switch during a test pulse to 0 mV into a bath solution containing the same concentration of sodium ions as the electrode solution (Fig. 2 B). This switch zeroed the current, and the timecourse of the disappearance and reappearance of the current was taken as a measure of the speed of our switching system. The kinetics of solution exchange were quantified by fitting the switching-induced current decay with a monoexponential. Only patches whose time constant was <5 ms (for 50-ms exposures) or 3 ms (for 20-ms exposures) were included in the data (∼15–20% of total attempts). The delay between the switching command and the measured commencement of solution exchange was also evaluated (7.1 ± 0.1 ms, n = 54).
To determine the degree of modification after each exposure, the average current between 40 and 42 ms after depolarization was divided by the corresponding value in the traces elicited from completely modified channels. A plot of fraction-modified versus cumulative exposure time was fit with a monoexponential rise that had a nonzero initial value reflecting the presence of persistent current before modification and a time constant whose reciprocal is the reaction rate between MTS-ET and cys1304 (Fig. 2 C). For reactions in 4 μM MTS-ET using 50-ms exposures at −120 mV, the reaction rate was 3.6 ± 0.3 s−1 (n = 10). We repeated this experiment at several different MTS-ET concentrations to test for a linear relationship between MTS-ET concentration and reaction rate, and thereby to confirm the presence of bimolecular kinetics (Fig. 3). A linear regression to this data yielded a slope of 1.03 μmol−1 s−1. Since F1304C has 38 cysteine residues, of which16 are believed to be intracellular, the presence of bimolecular reaction kinetics suggests that other cysteines are not involved in the MTS-ET-induced changes in the macroscopic current. All further experiments were performed either with 4 or 8 μM MTS-ET, both of which are within the linear portion of the concentration–rate curve.
Figure 3.

Dose–response curve. Reaction rates for MTS-ET modification of F1304C are shown as a function of MTS-ET concentration used. All exposures were carried out at −120 mV and were either 50 (1 μM, n = 3; 2 μM, n = 6; and 4 μM, n = 10) or 25 (8 μM, n = 5) ms in duration. Rates were determined as described in Fig. 2. The dose–response curve was fit to an unweighted linear regression constrained to pass through the origin, giving a slope of 1.03 μmol−1 s−1.
Voltage Dependence of Site 1304 Accessibility
To test whether the accessibility of cys1304 could serve as a marker for the conformational state of the fast inactivation gate, the voltage dependence of the MTS-ET modification rate was measured. At depolarized voltages, where the channel is nearly completely inactivated (and site 1304 is presumably buried), we expected a slower modification rate than at hyperpolarized voltages, where the channels are predominantly in the resting state (and site 1304 is presumably accessible).
The experimental protocol we used is shown in Fig. 4 A. As in previous experiments, inside-out F1304C macropatches were subjected to a series of 50-ms exposures to 4 μM MTS-ET, with a test pulse between each exposure to determine the extent of modification. In this experiment, however, the exposure occurred during a conditioning pulse to a specific voltage (ranging from −140 to +20 mV). The exposure was timed to occur 200 ms after the beginning of the prepulse to facilitate comparison with the h∞• curve. The prepulse voltage was maintained throughout the exposure and for an additional 100 ms to prevent accidental exposure on repolarization.
Figure 4.

Voltage dependence of F1304C accessibility. (A) The protocol for measuring the accessibility of site 1304 as a function of voltage is shown. 200-ms prepulses were used at each voltage, followed by 50 ms of exposure to 4 μM MTS-ET, and an additional 100 ms at the prepulse voltage to prevent unwanted exposure after repolarization. The time between the prepulse and the test pulse ranged from 3.5 to 18 s, according to the necessary recovery time at −100 mV after 350 ms at the prepulse voltage. Degree of modification was assessed with a 45-ms test pulse to −20 mV. (B) Reaction rates were determined as described in Fig. 2 and are plotted against the voltage at which the exposure occurred (average of n = 6 per voltage; all points at least n = 3). The curve was fit to a Boltzmann (solid line) with a nonzero pedestal: R(V) = (R max − R min)/{1 + exp [(V - V1/2)/k]} + R min, where R max is the maximum rate (0.932 μmol−1 s−1), R min is the minimum rate (0.208 μmol−1 s−1), V1/2 is the voltage at half-maximal rate (−85.1 mV), and k is the slope factor (6.4). For comparison, the measured h∞• curve for F1304C (dotted line) is superimposed scaled to R max and R min (V1/2 = −81.6 ± 1.3 mV and k = 5.5 ± 0.5).
Reaction rates were measured as described above and are plotted as a function of prepulse voltage in Fig. 4 B. As expected, and in agreement with similar studies on the rat brain IIA channel (Kellenberger et al., 1996), the reaction rates were slower when exposures occurred during strongly depolarized prepulses. Furthermore, the voltage dependence was well-fit by a two-state Boltzmann, giving a maximum rate (R max) of 0.932 μmol−1 s−1 (within 10% of the linear regression estimate in Fig. 3) and a minimum rate (R min) of 0.208 μmol−1 s−1. The V1/2 was −85.1 mV and the slope factor was 6.4. The h∞• curve for F1304C (shown above in Fig. 1 B) was scaled and superimposed over the modification data as a dashed line (V1/2[h∞•] = −81.6 ± 1.3 mV; k[h∞•] = 5.5 ± 0.5) and shows good agreement (ΔV1/2 = 3.5 mV; Δk = 0.9).
The correspondence between the two steady state inactivation curves confirms that site 1304 becomes buried during the inactivation process, consistent with the hinged-lid model for fast inactivation, in which a triplet of amino acids, IFM (1303–1305), forms a hydrophobic contact with a site near the inner vestibule of the channel and thereby occludes the ion-conducting pore (West et al., 1992).
Finally, we note the absence of any significant intrinsic voltage dependence associated with the modification reaction. Since MTS-ET has a single positive charge, the modification rate would tend to increase with depolarization and decrease with hyperpolarization if the reaction with cys1304 occurred within the transmembrane electric field. From our data, an upper limit can be estimated for the fractional distance, δ, of cys1304 into the membrane electric field. The data in Fig. 4 B show <15% variation in the reaction rate over the 60-mV range from −40 to +20 mV. Since the reaction rate is predicted to increase an e-fold per ΔV δ/25 mV, an upper limit is established:
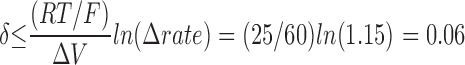 |
Thus, even when cys1304 was buried, it was never further than 6% of the entire distance into the membrane electric field.
In sum, these data on the voltage dependence of the modification rate suggest that cys1304 accessibility is an excellent marker for the conformational state of the fast inactivation gate.
Relationship between Fast and Slow Inactivation
To determine the appropriate protocol for measuring the effects of slow inactivation on the conformation of the fast inactivation gate, we measured recovery from inactivation after various length prepulses in F1304C (Fig. 5). We used a two-pulse protocol with a test pulse placed at varying intervals after a fixed conditioning pulse. With a 20-ms conditioning pulse, channel recovery was nearly complete after just 10 ms. As the conditioning pulse length was increased, the channels recovered much more slowly, reflecting entry into slow inactivated states (the degree of slow inactivation is often estimated as the fraction of channels that do not recover within 20 ms of repolarization; Hayward et al., 1997). After a 10-s conditioning pulse, recovery at −120 mV was slow and incomplete even after 10 s. Relatively little current (<10%) recovered within 20 ms, indicating nearly complete slow inactivation.
Figure 5.

Recovery from inactivation. The two-pulse protocol (top) was used to assess recovery from inactivation in F1304C in response to varying length conditioning pulses. The time axis is displayed logarithmically because recovery rates vary over several orders of magnitude. Symbols indicate means ± SEM (n = 5–7 for each curve). Using this data, experimental protocols (diagrammed in Fig. 6 A) were designed to measure the accessibility of cys1304 when channels are slow inactivated. The shaded area indicates the duration of MTS-ET exposure during the protocol shown in Fig. 6 A (bottom).
The rationale for our next experiment was to determine the conformational state of the fast inactivation gate at the tail end of varying length conditioning pulses to 0 mV. The protocol used is shown in Fig. 6 A (top). As in previous experiments, 4 μM MTS-ET was applied for a fixed duration (either 50 or 150 ms) with test pulses between each exposure. Before each exposure, there was a prepulse to 0 mV of either 0.007, 0.20, 3.0, or 10.0 s, with depolarization maintained during the exposure and shortly after. Reaction rates were determined as described above and are shown in Fig. 6 B (○). The reaction rates for each prepulse used were similar to each other and close to R min, demonstrating that cys1304 is buried within 7 ms of depolarization and remains buried despite the onset of slow inactivation.
Figure 6.

Accessibility of site 1304 recovers rapidly for fast- and slow-inactivated channels. (A) Two protocols were used to measure accessibility of site 1304 in response to variable length conditioning pulses to 0 mV from a holding potential of −120 mV. In the experiment shown on top, the MTS-ET exposure was placed near the end of the conditioning pulse to verify that site 1304 remains buried during long depolarizations and to determine how quickly it becomes buried for short depolarizations. Depolarization was maintained throughout the exposure and shortly after. (Tpre = 0.007, 0.2, 2.0, or 10.0 s; MTS-ET exposure was 20 or 50 ms; [MTS-ET] = 4 or 8 μM; n = 3 for all). In the bottom protocol, the exposure was placed 5 ms after repolarization: 4 μM, 50 ms (average n = 5 per Tcond, except Tcond = 10 s) or 8 μM, 20 ms (average n = 8 per Tcond, all points at least n = 4). The total time between the end of the conditioning pulse and the beginning of the exposure includes a short delay due to lag of the switching apparatus, which we measured as described above (Fig. 2) to be 7.1 ± 0.1 ms, giving a preexposure recovery time of 12.1 ± 0.1 ms. Therefore, for the 20-ms case, exposure occurred from 12.1 to 32.1 ms after repolarization (depicted as the shaded area in the graph in Fig. 5). For both protocols, enough time after the conditioning pulse was allowed for complete recovery (from 5 s for Tcond = 0.02 s to 45 s for Tcond = 10 s) before the 45-ms test pulse was administered. (B) Reaction rates were determined as described above (Fig. 2) for the two protocols described in A. Dotted lines indicate R max (0.932 μmol−1 s−1) and R min (0.208 μmol−1 s−1). The reaction rates for the first protocol, in which the exposures occurred during the conditioning pulse (○) were slow, consistent with site 1304 burial. For the experiments in which the exposure occurred shortly after the end of the conditioning pulse (•), the measured rate was consistent with nearly complete accessibility (experiments without any conditioning pulse are shown for comparison as Tcond = 0). This result suggests that the fast inactivation gate recovers rapidly despite the fact that >90% of the channels remain slow inactivated.
The protocol to assess recovery of accessibility of cys1304 is shown in Fig. 6 A (bottom). Exposures were timed to occur shortly after (12.1 ± 0.1 ms) varying length conditioning pulses (Tcond = 0.02, 0.2, 2.0, or 10 s) to ascertain the position of the fast inactivation gate during recovery from slow inactivation. The exposures were either 20 or 50 ms. There was no significant difference in rates for these two cases. The timing of the exposure in relation to the degree of recovery present for each conditioning pulse is shown as the shaded area of Fig. 5. By confining our exposure to this shaded area, we were able to determine the position of the fast inactivation gate while channels were predominantly slow inactivated. Reaction rates are shown in Fig. 6 B (•). As expected, recovery of cys1304 accessibility occurs rapidly after a 20-ms conditioning pulse, during which channels were not depolarized long enough to slow inactivation appreciably (Fig. 5). More significantly, the modification rates continued to be close to R max (full accessibility) for experiments in which exposures were preceded by long conditioning pulses as well. Even after the 10-s conditioning pulse, when only 5–15% of the current was available, the modification rate was close to R max, suggesting that the fast inactivation gate had almost completely recovered.
discussion
Accessibility of Site 1304 and Fast Inactivation
The major result of this work is that the behavior of the Na+ channel fast inactivation gate is independent of the state of the slow inactivation gate. These observations were only possible because the ability to monitor the conformational rearrangement accompanying fast inactivation allowed us to observe electrophysiologically silent transitions between channel states. Thus, the first set of experiments we performed was directed at characterizing our putative conformational marker for the fast inactivation gate.
We selected F1304 as a candidate site because previous studies had identified it as an important residue for fast inactivation (West et al., 1992), and because there was some evidence that site 1304 becomes less solvent-accessible during inactivation (Vassilev et al., 1988). Kellenberger at al. (1996) recently examined the effects of different MTS reagents and Ag+ on the analogous mutation (F1489C) in rat brain IIA channels. A primary aim of that study was to examine the effects of substituting groups of different charge and size at position 1489 by determining on- and off-rates for the inactivation particle using single channel recording. The effect of cysteine substitution and MTS-ET modification were similar to what we measured in the muscle channel: the rate of MTS-ET modification of site 1489 in the brain IIA channel, 0.786 μmol−1 s−1, agrees well with the rate we measured for site 1304 in μ1, 0.932 μmol−1 s−1. Kellenberger et al. (1996) were also able to demonstrate that site 1489 in the brain IIA channel exhibits voltage-dependent accessibility that corresponds to the degree of inactivation present and that the modification rate is not intrinsically voltage dependent. Without rapid solution exchange, however, they could not use a defined prepulse duration before exposure for precise comparison with the h∞• curve, nor were they able to examine the effects of slow inactivation on site 1489 accessibility.
While much of the present paper confirms the findings in Kellenberger et al. (1996), our use of a fast solution exchange system allowed us to control the duration of MTS-ET exposure down to the 20-ms level, permitting a more precise comparison between the h∞• curve and the voltage dependence of the modification rate. When we compared the accessibility of cys1304 after 200-ms prepulses to the h∞• curve (also measured with 200-ms prepulses), we found that the two different measures of inactivation agree quite well. In addition to establishing the correlation between cys1304 accessibility and fast inactivation, this result also provides evidence that the inactivation from closed states that occurs during mildly depolarized prepulses involves burial of site 1304 just as inactivation from the open state does. The relatively small differences in V1/2 and k (ΔV1/2 = 3.5; Δk = 0.9) between h∞• and the fit to the modification data might reflect gating shifts that occur in macropatches over time since modification data were, on average, recorded slightly later than h∞• data and took longer to acquire. It is also possible, though the resolution of the data does not permit us to draw such a conclusion, that the small left shift in the fit to the modification data could reflect an additional conformational transition required for channel inactivation after site 1304 burial.
The fit of our modification data to a Boltzmann yielded modification rates R max and R min of 0.932 and 0.208 μmol−1 s−1 for hyperpolarized and depolarized potentials, respectively. Even if cys1304 is completely inaccessible for fast-inactivated channels, R min has a predicted lower limit because fast inactivation is incomplete for F1304C. The ratio of R min to R max, 0.22, should (in theory) be greater than or equal to the fraction of channels that fails to inactivate at strongly depolarized voltages.
Precise estimation of the fraction of channels that are not fast inactivated at equilibrium is complicated by the fact that these channels can become slow inactivated. Close examination of h∞• for F1304C reveals a steep descent that ends at ∼−65 mV, followed by a much shallower descent (Fig. 1 B). This second shallow component is absent from the WT h∞• curve, where fast inactivation is nearly complete. We interpret the shallow descent as the superimposition of slow inactivation onto the fraction of non–fast-inactivating channels. The 200-ms prepulses used to measure h∞• allows some slow inactivation to occur at these voltages, but equilibrium is not reached, so the apparent voltage dependence is mild. The value of h at the plateau (i.e., before the onset of the shallow descent) gives a rough estimate of the fraction of channels that are not fast inactivated at equilibrium, ∼15%. The two-pulse recovery data in Fig. 5 are also consistent with this estimate. After a 20-ms conditioning pulse, 10–15% of the current recovers within 0.1 ms, suggesting that this fraction of channels did not fast inactivate for F1304C. Using this estimate, the theoretical minimum for R min/R max is 0.15, which is below our measured value of 0.22. This difference suggests that burial of cys1304 does not reduce the reaction rate with MTS-ET to zero, although the imprecision in our estimate diminishes the certainty of this interpretation.
Despite the uncertainty in our estimate, we can nevertheless calculate approximate values for the rate of modification for buried channels (R b) and the rate for completely accessible channels (R a). The measured apparent rate at any given voltage (R app) is an average of R a and R b weighted by the equilibrium value of the h parameter at that voltage: R app = (h)R a + (1 − h)R b. At −120 mV, h is nearly one, and R app = R max. Thus, R a = R max = 0.932 μmol−1 s−1. For depolarized voltages, we estimated h at 0.15, and R app = R min (0.208 μmol−1 s−1). Thus, R min = 0.208 = (0.85)R a + (0.15)R b. Substitution of 0.932 for R a gives R b = 0.08 μmol−1 s−1; and therefore R b/R a = 0.09.
According to this calculation, the reaction rate when cys1304 is buried is ∼10-fold slower than when it is accessible. The fact that R b is nonzero implies that there is some degree of accessibility to MTS-ET modification, even when cys1304 is buried. This may be an artifact of the cysteine substitution for a normally inaccessible phenylalanine, or it may reflect an unexpected degree of exposure for site 1304 in the inactivated conformation; our data alone cannot distinguish between these possibilities.
Kellenberger et al. (1996) obtained a ratio of R min to R max for F1489Q in brain IIA channels modified with MTS-ES (negatively charged and smaller than MTS-ET) of 0.04 as compared with our value of 0.22 for F1304C in μ1 modified with MTS-ET. Part of this discrepancy is probably attributable to the fact that fast inactivation is more complete in the brain IIA F1489C than in μ1 F1304C. Additional contributing factors might include other isoform differences between brain IIA and μ1 (such as the nature of the buried conformation), or the use of a different modification reagent. Since the data given in Kellenberger et al. (1996) do not permit an estimation of the equilibrium value of h at depolarized voltages, a precise comparison with our data is not possible.
Our R max was much faster than MTS-ET modification rates reported for other sites on the sodium channel: 1,000-fold faster than modification of the site of the outermost S4 charge in domain IV (Yang and Horn, 1995), and 3,000-fold faster than the site of the third domain IV S4 charge (Yang et al., 1996). Our rate is also ∼1,000× faster than modification at sites of S4 charges in Shaker K+ channels (Larsson et al., 1996), and ∼50× faster than a site at the outer mouth of the Shaker K+ channel pore (Liu et al., 1996). The relatively high sensitivity of cys1304 to MTS-ET suggests a great degree of solvent accessibility of the site at −120 mV, an interpretation consistent with its putative role as a relatively mobile inactivation particle.
Finally, we were also able to evaluate the time dependence of the accessibility change, though the temporal resolution of our method prohibited precise comparison with rates of entry to and recovery from fast inactivation. Cys1304 is buried after 7 ms at 0 mV, and cys1304 accessibility recovers within 12 ms of repolarization after a 20-ms conditioning pulse to 0 mV. Although these times are substantially longer than the rates for entry to and recovery from fast inactivation, the data do allow us to constrain the rates of burial and recovery of accessibility of cys1304 to within an order of magnitude of the fast inactivation rates measured for ionic currents. This result, along with the close agreement of the voltage dependence of cys1304 accessibility with the h∞• curve, allows us to conclude that cys1304 is indeed an excellent marker for the position of the fast inactivation gate.
Relationship between Fast and Slow Inactivation
In the first experiment designed to test the relationship between fast and slow inactivation, the position of the fast inactivation gate was measured at the tail end of different length depolarizations. If fast and slow inactivation are mutually exclusive, directly competing processes (considered in Featherstone et al., 1996), then cys1304 would be accessible at the tail end of long conditioning pulses when most channels are slow inactivated. If, on the other hand, channels can exist in a state that is fast and slow inactivated simultaneously (suggested in Bezanilla et al., 1982), then cys1304 would remain buried regardless of the length of the conditioning pulse. Our data demonstrate that cys1304 does remain buried regardless of depolarization length, proving that fast and slow inactivation are not mutually exclusive processes; that is, that they can occur simultaneously in the same channel.
These data also show that accessibility does not progressively decline during prolonged depolarizations. The fact that the modification rate does not decrease with longer conditioning pulses suggests that a certain fraction of channels in F1304C remain non–fast inactivated regardless of the length of the conditioning pulse used. This is consistent with the interpretation that the decay of the slow component in the F1304C current trace (Fig. 1 A) is due to slow inactivation and not to fast inactivation, and supports the generally accepted hypothesis that fast and slow inactivation are structurally distinct. In other words, the equilibrium between fast and non–fast inactivated channels is achieved within a few milliseconds at 0 mV and is not measurably biased by the closure of the slow inactivation gate.
In the next experiment, the MTS exposure was placed shortly after the end of variable length conditioning pulses, allowing us to determine whether recovery from fast inactivation could precede recovery from slow inactivation. We found that the accessibility of cys1304 almost completely returned within 12–32 ms, regardless of the length of the conditioning pulse used. For the longest conditioning pulse used, 10 s, the interval between 12 and 32 s corresponds to just 5–15% availability (Fig. 5, shaded area). This result allows us to draw several conclusions. First, fast and slow inactivation must be structurally independent processes, because the fast gate can be almost completely open while the slow gate is nearly completely closed. Second, it rules out a sequential model for the development of and recovery from slow inactivation, according to which sodium channels could only recover from fast inactivation after recovering from slow inactivation. Third, it indicates that in the normal process of recovery from slow inactivation, a state is populated that is slow inactivated but not fast inactivated. It also suggests that fast and slow inactivation are coupled only weakly, if at all, since neither the equilibrium of fast- and non–fast-inactivated channels nor the kinetics of recovery from fast inactivation are altered by the closure of the slow gate. The data from both experiments would support a model in which fast and slow inactivation are independent processes with relatively little interaction.
Relation to Previous Work on Slow Inactivation of Na+ Channels
The relationship between fast and slow inactivation has been difficult to probe, primarily because transitions that occur between different inactivated states are electrophysiologically silent. One line of investigation on the subject has focused on the long-term immobilization of gating charge. For short depolarizations, recovery from inactivation closely parallels recovery of gating charge (Armstrong and Bezanilla, 1977), lending support to the hypothesis that sodium channels must deactivate to recover from inactivation (Kuo and Bean, 1994). In response to prolonged depolarizations, however, some groups have found that available gating charge recovers on the order of hundreds of milliseconds, but still well before recovery from slow inactivation (Bezanilla et al., 1982; Ruben et al., 1992); while another group reports that gating charge recovers with the same time course as slow inactivation (Meves and Vogel, 1977). Our results demonstrate that the accessibility of the fast inactivation gate recovers much more rapidly than this, implying that some other process (perhaps slow inactivation) must be responsible for long-term immobilization of gating charge. Further experiments are needed to determine how recovery from fast inactivation and recovery of gating charge are decoupled during recovery from long depolarizations, and whether this process requires slow inactivation.
Additional data on the relationship between fast and slow inactivation has come from studies on the subtle changes in slow inactivation brought about by disruption of fast inactivation. Several groups have found that slow inactivation is more complete and its onset more rapid after fast inactivation has been removed or disrupted (Featherstone et al., 1996; Hayward et al., 1996; Rudy, 1978). We also found this to be the case for F1304C compared with WT (data not shown). This finding implies either direct competition between the two inactivation gates, or an indirect coupling effect in which the rate of slow inactivation is faster from the open state than from fast-inactivated states (Featherstone et al., 1996; Hayward et al., 1997; Rudy, 1978). Our results rule out direct competition for the same binding site because fast and slow inactivation gates can be closed simultaneously; however, they do not rule out a more subtle coupling between the two forms of inactivation.
Shaker K+ channels, for example, exhibit coupling between two structurally independent forms of inactivation (rapid intracellularly mediated N-type inactivation, and slower extracellularly mediated C-type inactivation) that occurs indirectly through effects of the N-type inactivation particle on activation gating and on occupancy of an ion-binding site near the outer mouth of the pore (Baukrowitz and Yellen, 1995). One recent study on the human heart Na+ channel mutant F1485Q found that increasing extracellular Na+ inhibits slow inactivation, while lowering of extracellular Na+ enhances it (Townsend and Horn, 1997). Previous studies have noted effects of external Ca2+ on slow inactivation of Na+ channels in frog nerve (Khodorov et al., 1976). These results have been taken as evidence that binding of a Na+ ion to a site near the outer mouth of the pore inhibits closing of the slow inactivation gate (Townsend and Horn, 1997). If closure of the fast inactivation gate increased the dwell time of an ion at that site, it would explain why fast inactivated channels are slightly more resistant to slow inactivation than open channels.
The results of our study do not directly speak to the possibility of this kind of weak coupling of fast and slow inactivation; however, they do exclude the two extreme cases, that fast and slow inactivation are tightly linked in a sequential gating scheme (employed in Townsend and Horn, 1997), and that fast and slow inactivation are mutually exclusive processes (considered in Featherstone et al., 1996). In addition, our data restrict the number of possible recovery pathways from slow inactivated states: recovery from fast inactivation precedes recovery from slow inactivation.
Acknowledgments
We thank Adriana Pechanova for assistance with site-directed mutagenesis, mRNA preparation, oocyte harvesting, and oocyte injection. We also thank Gary Yellen, Yi Liu, Miguel Holmgren, and Stuart Forman for helpful discussions, and Jim Morrill for critical reading of the manuscript.
This work was supported by the National Institutes of Health (R01-AR42703), the Harvard Mahoney Neuroscience Institute (V. Vedantham), and the Esther A. and Joseph Klingenstein Fund, Inc. (S.C. Cannon).
Footnotes
Abbreviations used in this paper: MTS-ET, [2-(trimethylammonium)- ethyl]methanethiosulfonate; WT, wild type.
references
- Adelman W, Palti Y. The effect of external potassium and long duration voltage conditioning in the amplitude of sodium currents in the giant axon of the squid Loligo Pealei. . J Gen Physiol. 1969;54:589–606. doi: 10.1085/jgp.54.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almers W, Stanfield PR, Stühmer W. Slow changes in currents through sodium channels in frog muscle membrane. J Physiol (Camb) 1983;339:253–271. doi: 10.1113/jphysiol.1983.sp014715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Taylor RE, Fernandez JM. Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J Gen Physiol. 1982;79:21–40. doi: 10.1085/jgp.79.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC. Slow inactivation of sodium channels: more than just a laboratory curiosity. Biophys J. 1996;71:5–7. doi: 10.1016/S0006-3495(96)79203-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CF, Cannon SC. Modulation of Na+ channel inactivation by the β1subunit: a deletion analysis. Pflügers Arch. 1995;431:186–195. doi: 10.1007/BF00410190. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophys J. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in SkM1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonoi T, Hille B. Inactivation modifiers discriminate among gating models. J Gen Physiol. 1987;89:253–274. doi: 10.1085/jgp.89.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium channel III–IV linker. J Gen Physiol. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys J. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. . J Physiol (Camb) 1952a;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Camb) 1952b;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Scheuer T, Catterall WA. Movement of the Na+channel inactivation gate during inactivation. J Biol Chem. 1996;271:30971–30979. doi: 10.1074/jbc.271.48.30971. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, West JW, Scheuer T, Catterall WA. Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997;109:589–605. doi: 10.1085/jgp.109.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodorov B, Shishkova L, Peganov E, Revenko S. Inhibition of sodium currents in frog ranvier node treated with local anesthetics. Role of slow sodium inactivation. Biochim Biophys Acta. 1976;433:409–435. [Google Scholar]
- Kuo C, Bean BP. Sodium channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the Shaker K+Channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, Cannon SC, Slaugenhaupt SA, Gusella JF. The cloning and expression of a sodium channel beta 1-subunit cDNA from human brain. Hum Mol Genet. 1993;2:745–749. doi: 10.1093/hmg/2.6.745. [DOI] [PubMed] [Google Scholar]
- Meves H, Vogel W. Slow recovery of sodium current and ‘gating current' from inactivation. J Physiol (Camb) 1977;267:395–410. doi: 10.1113/jphysiol.1977.sp011819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narahashi T. Restoration of action potential by anodal polarization in lobster giant axons. J Cell Comp Physiol. 1964;64:73–96. doi: 10.1002/jcp.1030640108. [DOI] [PubMed] [Google Scholar]
- Quandt FN. Burst kinetics of sodium channels which lack fast inactivation in mouse neuroblastoma cells. J Physiol (Camb) 1987;392:563–585. doi: 10.1113/jphysiol.1987.sp016797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas E, Armstrong CM. Sodium conductance activation without inactivation in pronase-perfused axons. Nat New Biol. 1971;229:177–178. doi: 10.1038/newbio229177a0. [DOI] [PubMed] [Google Scholar]
- Ruben PC, Starkus JG, Rayner MD. Steady-state availability of sodium channels. Interactions between activation and slow inactivation. Biophys J. 1992;61:941–955. doi: 10.1016/S0006-3495(92)81901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J Physiol (Camb) 1978;283:1–21. doi: 10.1113/jphysiol.1978.sp012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini L, Stühmer W. Slow sodium channel inactivation in rat fast-twitch muscle. J Physiol (Camb) 1987;383:327–337. doi: 10.1113/jphysiol.1987.sp016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Townsend C, Horn R. Effect of alkali metal cations on slow inactivation of cardiac Na+channels. J Gen Physiol. 1997;110:23–33. doi: 10.1085/jgp.110.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Wang S, Wang GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys J. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, George AL, Jr, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]