

Abstract
We analyzed the kinetics of interaction between the peptide KIFMK, containing the isoleucine, phen-ylalanine, and methionine (IFM) motif from the inactivation gate, and the brain type IIA sodium channels with a mutation that disrupts inactivation (F1489Q). The on-rate constant was concentration dependent, consistent with a bimolecular reaction with open sodium channels, while the off rates were unaffected by changes in the KIFMK concentration. The apparent K d was ∼33 μM at 0 mV. The on rates were voltage dependent, supporting the hypothesis that one or both of the charges in KIFMK enter the membrane electric field. The voltage dependence of block was consistent with the equivalent movement of ∼0.6 electronic charges across the membrane. In contrast, the off rates were voltage independent. The results are consistent with the hypothesis that the KIFMK peptide enters the pore of the open sodium channel from the intracellular side and blocks it.
Keywords: Voltage-dependent gating, inactivation, sodium channel, peptide
introduction
The time course of macroscopic current through sodium channels during membrane depolarization has a characteristic shape that is determined by the processes of activation and inactivation (Hodgkin and Huxley, 1952; Hille, 1992). While the primary structures of the α subunits of brain sodium channels are known (e.g., Noda et al., 1986; Auld et al., 1988, 1990), the molecular components of the protein involved in channel gating are still under active investigation. The cytoplasmic loop between domains III and IV of the sodium channel α subunit has been shown to have an essential role in inactivation by experiments using site-directed antipeptide antibodies (Vassilev et al., 1988, 1989) and site-directed mutagenesis (Stühmer et al., 1989; West et al., 1992; Patton et al., 1992; Kellenberger et al., 1996). This loop contains 53 amino acids including isoleucine, phenylalanine, and methionine (IFM)1 at positions 1488–1490, respectively. Inactivation is disrupted if these three amino acids together or phenylalanine 1489 alone are converted to hydrophilic residues by site-directed mutagenesis (West et al., 1992). From these results, it has been postulated that the IFM motif acts as the inactivation particle of sodium channels.
Application of an IFM-containing peptide, acetyl-KIFMK-NH2 (KIFMK), to the cytoplasmic side of sodium channels produces both a rapid block, which occurs in milliseconds and resembles inactivation in kinetics and voltage dependence, and a cumulative, use-dependent block, which occurs during repetitive high voltage pulses (Eaholtz et al., 1994). In this study, we focus on the kinetics of peptide block that occurs in a few milliseconds. Our results support a bimolecular reaction scheme in which KIFMK binds to open sodium channels and blocks them on the millisecond time scale.
materials and methods
Expression of F1489Q Sodium Channel in Xenopus Oocytes
Messenger RNAs were transcribed from the cDNA plasmid pCDM8Sal-F1489Q encoding the α subunit of rat brain type IIA sodium channels with disrupted inactivation (F1489Q) as well as pCDNA3-β1, which encodes the β1 subunit (Isom et al., 1992). Stage six oocytes were isolated from Xenopus laevis, enzymatically dissociated using collagenase (Sigma Chemical Co., St. Louis, MO), and injected with 0.2–5 ng of cRNA (Zagotta et al., 1989). Both F1489Q α and β1 subunit cRNA were coinjected (1:1) into each oocyte. Oocytes were bathed in an ND-96 solution containing (mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH 7.6, with added penicillin (100 U/ml), streptomycin (100 μg/ml), and 2.5 mM Na+-pyruvate. They were maintained at 16°C until electrophysiological recordings were made 3–6 d after injection.
Recording from Macropatches of F1489Q Mutant Sodium Channels
Recording solutions.
The bath contained an intracellular solution composed of (mM): 150 KCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES, pH 7.3. The pipette was filled with an extracellular solution composed of (mM): 150 NaCl, 4 KCl, 2 MgCl2, 10 HEPES, pH 7.4. Patches of oocyte membranes were excised in the inside-out configuration for recording with the patch-clamp technique (Hamill et al., 1981) at room temperature (∼25°C). Electrodes were pulled from borosilicate glass tubes (o.d. 1.5 mm, i.d. 0.86 mm; Sutter Instruments, Co., Novato, CA) and had resistances of 0.5–1.0 MΩ when filled with the pipette solution. Application of solutions used to perfuse the cytoplasmic side of the channels in the membrane patch was controlled by a rapid solution changer (RSC-100; Molecular Kinetics, Pullman, WA). Amino acid analysis (AAA Laboratory, Seattle, WA) of HPLC-purified KIFMK was used to determine the amount of material needed to be dissolved into the intracellular recording solutions to achieve a range of peptide concentrations from 5 to 250 μM.
Recording apparatus.
Sodium currents from macropatches of oocytes membranes were recorded using an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA). Pipette capacitance was compensated by adjusting the amplifier while any remaining capacitance and leakage currents were subtracted using a P/−4 protocol (Armstrong and Bezanilla, 1977). No corrections were made for junction potentials. The amplifier output was low pass filtered at 2 kHz through an eight-pole Bessel filter (Frequency Devices, Inc., Haverhill, MA), and digitized at 50 kHz. Data acquisition was controlled by a Macintosh Quadra 800 (Apple Corp., Cupertino, CA) computer running PULSE software (HEKA Elektronik, Lambrecht, Germany) and equipped with an ITC-16 computer interface (Instrutech Corp., Greatneck, NY). Data were stored on computer disks and analyzed on a Macintosh Quadra 800 computer using IGOR software (WaveMetrics, Lake Oswego, OR).
Data Analysis
Measurement of blocking rates induced by peptides.
A holding voltage of −100 mV was applied to macropatches of oocyte membranes to maintain the channels in the resting state. The blocking and unblocking rates were calculated from the time course of the decaying phase of the macroscopic current after a voltage step as previously described by Murrell-Lagnado and Aldrich (1993a , 1993b ; see also Patton et al., 1993). A steady state level of current remaining after the decay was assumed to be the equilibrium condition between blocking and unblocking of channels by peptide. The currents were fitted with the exponential function:
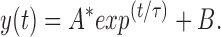 |
1 |
To estimate the fractional steady state current (Iss/Ipeak) in the absence of any peptide, the offset current (B) was divided by the sum of the amplitude of the fast exponential component (A) and offset (Iss/Ipeak = B/[A+B]). This value, along with the time constant of the peptide-induced decay, τ, were used to calculate the blocking and unblocking rates from the following expressions:
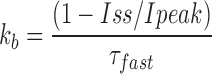 |
2 |
and
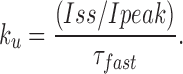 |
3 |
For a bimolecular reaction, the blocking rates were related to the association rate constant by the equation k b = k on*[KIFMK], while the dissociation rate constant was equal to the off rate, k ub = k off. Measurements were performed on currents averaged from three pulses to a specified voltage. All values are shown as means ± SEM unless otherwise indicated.
Estimates of Error and Model Predictions
We estimated the error in our calculated blocking and unblocking rates by analyzing simulated currents from a kinetic model. This kinetic model included four voltage-dependent activation transitions leading to an open state (Hodgkin and Huxley, 1952) and a voltage-independent inactivation transition coupled to channel opening. Peptide block was modeled as a bimolecular reaction between the peptide and open state (open channel block). This model accurately predicted the time course of activation and inactivation over the voltage range of −30 to +60 mV of the F1489Q currents as well as the concentration and voltage dependence of peptide block. The simulated currents were then fitted in the same manner as the data traces and the blocking and unblocking rates were calculated from the decay. From this analysis, we estimate the error in magnitude and voltage dependence of k on at 0 mV to be <10%. In addition, these calculations accurately predicted the lack of voltage dependence in k off but consistently underestimated its magnitude by ∼30%. A similar conclusion was reached after analyzing a model where residual inactivation and peptide block were treated as independent processes. This analysis indicates that the rates of activation and residual inactivation of F1489Q channels do not appreciably affect the conclusions of our analysis of peptide block.
results
Block of F1489Q Channels by KIFMK
To assess the effects of KIFMK quantitatively, F1489Q sodium channels were expressed in Xenopus oocytes, and recordings were made from excised inside-out patches of membrane to which KIFMK was applied. The advantages of these techniques compared with previous whole-cell voltage-clamp recordings (Eaholtz et al., 1994) were threefold: (a) KIFMK could be applied directly to the cytoplasmic surface of the membrane-containing channels and was not hindered by diffusion through the cytosol or nonspecific interactions with cytosolic proteins, (b) experiments with and without peptide could be done on the same population of channels, and (c) a range of KIFMK concentrations could be applied to a given population of channels so that the concentration dependence of the peptide action could be accurately determined. The F1489Q mutant α subunit and wild-type β1 subunit of the type IIA sodium channels were coexpressed and will be referred to as simply F1489Q channels. The kinetics of activation and inactivation were slow for wild-type IIA channels composed solely of α subunits expressed in Xenopus oocytes. Coexpression of the β1 subunit accelerates the activation and inactivation of type IIA sodium channels and thereby makes the time course of the currents more similar to native brain sodium channels (Isom et al., 1992; Patton et al., 1994).
The cytoplasmic surface of excised patches of membrane was continually perfused throughout these experiments. In peptide-free solution, F1489Q channels activated rapidly in response to depolarizing voltage steps from a holding potential of −100 mV (Fig. 1 A). Currents reached peak amplitudes and then decayed to reduced steady state values during each voltage step. The small decline in current indicates that the F1489Q mutation disrupts fast inactivation incompletely, as shown previously (West et al., 1992; Eaholtz et al., 1994). An exponential fit to the time course of current decay at 0 mV yielded a time constant for this residual inactivation of 4.0 ± 0.2 ms (SEM, n = 6) in the absence of added peptide. We plotted the magnitude of peak sodium current versus test pulse voltage to illustrate the current–voltage (I-V) relationship. The largest inward currents from one example patch were elicited by voltage steps between −10 and −20 mV, and currents reversed direction at ∼+75 mV (Fig. 1 B). Expression levels varied from patch to patch. The magnitude of the currents at the peak of the I-V curves ranged from −50 to −159 pA (n = 6).
Figure 1.

(A) Voltage-clamped currents from F1489Q mutant sodium channels expressed in Xenopus oocytes. Shown are macroscopic currents recorded from excised inside-out patches of membrane expressing the F1489Q α subunits and wild-type β1 subunits. The currents were elicited by voltage steps to −60, −40, −20, 0, 20, 40, 60, 80, and 110 mV from a −100 mV holding potential. (B) Current–voltage relations of F1489Q channels in oocytes. Peak currents were measured, and then plotted versus the voltage of the applied test pulse. For this patch, the largest current (107 pA) observed was elicited by a voltage step from −100 to −20 mV and the current changed direction at +75 mV.
KIFMK was applied to the cytoplasmic side of F1489Q sodium channels in concentrations ranging from 10 to 100 μM. Application of this IFM-containing peptide rapidly blocked F1489Q channels during a voltage pulse to 0 mV (Fig. 2 A). With peptide, the current decay to steady state was faster and the steady state level of current was smaller than controls without peptide (Fig. 2 A). Both the rate and extent of block increased when the KIFMK concentration was increased. A 100-μM concentration of KIFMK blocked >70% of the steady state sodium current elicited by a voltage step to 0 mV.
Figure 2.
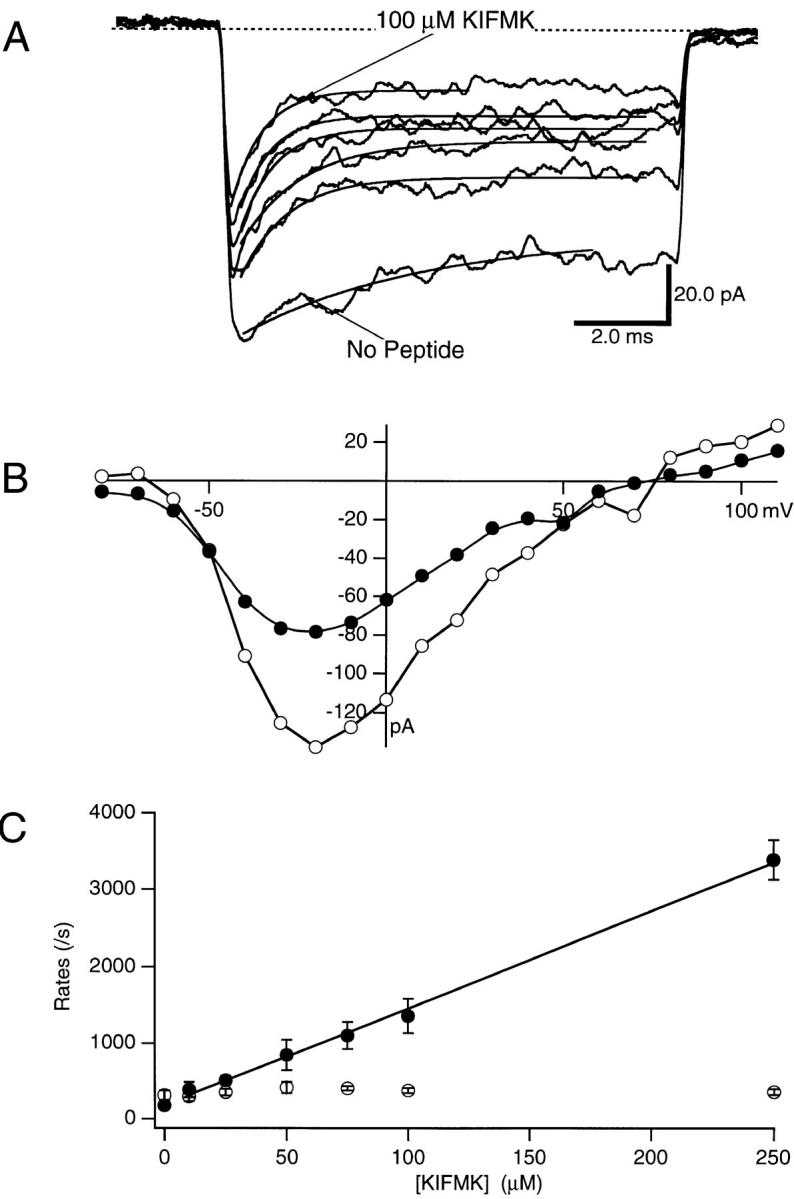
(A) Effects of intracellular KIFMK concentration on F1489Q currents. Macroscopic currents were elicited by voltage steps from −100 to 0 mV, while a solution of KIFMK was applied to the cytoplasmic surface of the inside-out excised patch of membrane. The concentration of KIFMK peptide ranged from 10 to 250 μM. Exponential fits of Eq. 1 (see materials and methods) to determine the blocking and unblocking rates are superimposed on each current trace. (B) Current–voltage relations of F1489Q in the presence of 100 μM KIFMK compared with peptide free. Peak currents were measured for each test voltage ranging between −80 and 110 mV and plotted versus the voltage of the applied test pulse. In this patch, the data from no peptide control currents were plotted as unfilled circles (○) and data from currents in the presence of 100 μM KIFMK were plotted as filled circles (•). (C) Blocking and unblocking rates plotted as a function of KIFMK concentration. The mean blocking (•) and unblocking (○) rates of four patches determined for each concentration are shown (mean ± SEM). The smooth line is a fit to the blocking rates of the equation k b = 1.2 × 107 ± 0.2 M−1s−1 *[KIFMK] + 188. The average k u was 362/s.
The block of sodium current induced by 100 μM KIFMK was observed over a broad voltage range, as illustrated in the I-V curves of Fig. 2 B. There was a negative shift of the I-V relationship when KIFMK was applied as compared with the no peptide control. This shift was expected because KIFMK block was coupled to channel opening.
Concentration Dependence of Block by KIFMK
The effects of KIFMK concentration on the blocking and unblocking rates of the sodium channels were determined quantitatively. Blocking (k b) and unblocking (k u) rate constants, in a bimolecular reaction scheme (Eqs. 2 and 3), were determined by fitting each current trace at 0 mV with a single exponential and the resulting rates were plotted as a function of peptide concentration from 10 to 250 μM KIFMK (Fig. 2 C). The unblocking rates were not affected by changes in the peptide concentration (Fig. 2 C, ○) and therefore were averaged for all concentrations to yield k u = 342 ± 21 s−1 (n = 4). In contrast, the blocking rate, k b, increased with increasing peptide. A linear regression was fitted to the blocking rates (k b) and showed a linear dependence of the blocking rates on the KIFMK concentration up to 250 μM with a bimolecular on-rate constant (k on) of 1.1 × 107 ± 0.2 M−1 s−1 (n = 4) (Fig. 2 C, •). From the ratio of the rate constants, the equilibrium dissociation constant (K d = k off/k on) for KIFMK block of the F1489Q channel was determined to be 33 ± 7 μM (n = 4). These results closely fit those expected for a bimolecular reaction scheme where a single peptide would bind within the channel to block current.
Our data showed an apparent “blocking” rate in the absence of KIFMK (Fig. 2 C) due to the remaining intrinsic inactivation of the F1489Q mutant channel. Our analytical procedures measured the time course of the decay of macroscopic currents so that the residual inactivation of F1489Q was included in our values for k b (see the control trace in Fig. 2 A). This value is plotted as the y-intercept, so the regression line does not pass exactly through the origin as expected. We used a kinetic model to assess the effects of activation and residual inactivation of F1489Q channels on the blocking and unblocking rates of KIFMK, as well as the percent error of our measurements, and found that neither of these processes appreciably affected the conclusions of our analysis of peptide block (see materials and methods).
Voltage Dependence of the On and Off Rates
Inactivation of sodium channels is primarily a voltage-independent process that is coupled to activation from which it gains its apparent voltage dependence (Armstrong and Bezanilla, 1977; Aldrich et al., 1983; Aldrich and Stevens, 1987). The time constants for both inactivation of expressed type IIA sodium channels by their intrinsic inactivation gate and for KIFMK block are both steeply voltage dependent at voltages more negative than −30 mV and become only weakly voltage dependent at membrane potentials more positive than −30 mV (Eaholtz et al., 1994). The steep voltage dependence at hyperpolarized potentials is due to the voltage-dependent rate of activation, which is slow and incomplete at these voltages and limits the rate of the reaction of KIFMK with open sodium channels. In contrast, at more positive voltages, where activation is more rapid and complete, much less voltage dependence is observed.
The voltage dependence of KIFMK block of F1489Q sodium channels was measured using excised inside-out macropatches perfused with 100 μM KIFMK intra-cellular solution, while voltage steps from −100 to between −30 and 30 mV were applied to the membrane (Fig. 3 A). The time course of the peptide-induced block was measured by fitting Eq. 1 to the decaying sodium currents elicited at each voltage step. From these exponential fits, the rates for blocking (k b) and unblocking (k u) were determined and plotted as a function of voltage (Fig. 3 B). k b increased exponentially with increasing voltage, even at voltages where sodium channels were completely activated. These results are consistent with a pore-blocking mechanism in which the positively charged peptide enters the electric field applied across the membrane. In contrast, the unbinding rate was not affected by voltage with a mean k u of 336 ± 14 s−1 (n = 4) over the voltage range tested.
Figure 3.

(A) Effects of voltage on KIFMK block of F1489Q currents. Macroscopic currents were elicited from excised inside-out patches of membrane expressing the F1489Q channels in the presence of 100 μM KIFMK by voltage steps from −100 mV to a range between −30 and 30 mV in increments of 10 mV. Also shown are the superimposed single exponential fits of Eq. 1: y(t) = A * exp(t/τ) + B (see materials and methods) to each decaying current trace. (B) Effects of voltage on the rates of KIFMK block of F1489Q currents. Plot shows the on (•) and off (○) rates of 100 μM KIFMK as a function of voltage. The smooth lines represent a fit of the equation k b = 1,390*exp(V/43 mV) to the blocking rates. The unblocking rates showed no voltage dependence (○).
The smooth line through the data (Fig. 3 B) represents a fit to the blocking rates of the equation:
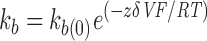 |
4 |
where k b(0) is the rate constant for binding at 0 mV, z is the valence of blocker, δ is the apparent extent of the electric field affecting the binding interaction, V is the membrane voltage, T is absolute temperature, F is Faraday's constant, and R is the ideal gas constant (Woodhull, 1973). From this exponential fit to the blocking rates versus voltage plot, the mean k b(0) for four patches was 1,386 ± 96 s−1. The mean value of zδ (the product of the net charge [z] and the extent of the electric field affecting KIFMK block [δ]) was 0.6 ± 0.08 (n = 4) equivalent electronic charges.
In Woodhull's (1973) analysis, the blocking species is an ion acting as a point charge in the electric field. In the case of the peptide KIFMK, which has a positive charge on the ε-amino group of the side chain of each lysine, we do not know how the charges on the peptide are situated relative to the binding site of the IFM motif. If both charges enter the electric field to an equivalent extent, our results indicate that they traverse 30% of the electric field as the peptide blocks the channel. Our results are consistent with the IFM motif acting similarly to an inactivation particle whose binding in the channel pore blocks ion conductance in the inactivated state.
discussion
KIFMK Blocks Sodium Channels According to a Bimolecular Reaction Scheme
Analysis of the concentration dependence, voltage dependence, and kinetics of channel block showed that KIFMK binds to the mutant sodium channel F1489Q via a bimolecular reaction with the open channel. This reaction has an apparent K d for peptide binding to the open channel of 33 μM at 0 mV. Coupling between channel activation and open channel block can be described by the kinetic Scheme 1.
Where k off is the off-rate constant of peptide block (340 s−1), and k on is the peptide association rate constant (1.1 × 107*e(V/43 mV) M−1s−1), C is the closed state, O is the open state, O-IFM is the blocked state, and IFM denotes a diffusible peptide or the intrinsic IFM of the wild-type sodium channel. Fast (<1 ms) inactivation of sodium channels occurs primarily after channel opening, and the inactivation process derives its voltage dependence from this coupling to activation. Block by KIFMK may be coupled to activation by a similar mechanism since KIFMK appears to preferentially block open channels (Eaholtz et al., 1994).
In addition to the concentration dependence, the on rates of KIFMK block show a significant, exponential voltage dependence. The voltage dependence of block is equivalent to moving 0.6 electronic charges across the membrane. The voltage dependence of k on is consistent with KIFMK entering the membrane electric field to bind with its receptor site, suggesting entry of the peptide into the intracellular mouth of the pore during the blocking reaction. However, the voltage dependence measures the position of the charged amino groups of the lysine residues of KIFMK rather than the IFM motif, so how far into the pore the IFM motif binds remains unknown.
Rapid Block by KIFMK Mimics Inactivation
Open channel block by KIFMK mimics normal inactivation in at least three ways. First, the peptide block and inactivation show the same sequence specificity for IFM (West et al., 1992; Eaholtz et al., 1994). IFM must be intact and a change in any of these amino acids disrupts the function for both the peptide and inactivation. Second, both inactivation and binding of KIFMK are coupled to activation and are both steeply voltage dependent over the voltage range where sodium channels activate (Eaholtz et al., 1994). Finally, the voltage dependence of block and inactivation are small at membrane potentials where sodium channels are fully open (Eaholtz et al., 1994). These results are consistent with the hypothesis that the KIFMK peptide acts like the intrinsic inactivation particle of sodium channels.
Unlike inactivation, KIFMK produces two types of block: fast block described here and slow block that occurs at voltages more depolarized than 0 mV (Eaholtz et al., 1994). Recovery from KIFMK block occurs through the open state when the membrane voltage is returned to more hyperpolarizing potentials (Eaholtz et al., 1994). Recovery from inactivation does not occur through the open state (Kuo and Bean, 1994). These differences may reflect the properties of a freely diffusible, unconstrained IFM peptide in contrast to IFM constrained in the sodium channel sequence. The integrity of the cytoplasmic loop between transmembrane domains III and IV is important to the inactivation process because cutting the loop resulted in a loss of inactivation (Stühmer et al., 1989). Evidently, the structure of the loop contributes significantly to the positioning and stability of binding of IFM to the channel during inactivation.
In studies of a cardiac sodium channel with mutations in M1651 and M1652 in the S4/S5 linker of domain IV, Tang et al. (1996) showed that KIFMK blocked mutant sodium channels when the peptide was introduced via the whole-cell recording configuration and outward sodium currents were recorded. A similar K d value of ∼30 μM was estimated from the ratio of the on and off rates (Tang et al., 1996). However, the on and off rate constants they measured were two- to threefold lower than we observed, no voltage dependence of block rate was detected, and the dependence of the on rate on peptide concentration deviated significantly from linearity (Tang et al., 1996), in contrast to our data. The differences in the on and off rates, voltage dependence, and linearity of concentration dependence may reflect in part uncertainties in the true concentration of peptide that diffused into the cells from the whole-cell patch pipette in the experiments of Tang et al. (1996). Our experiments using excised inside-out patches containing brain sodium channels allowed us to study the effects of precisely known concentrations of KIFMK and to vary the concentration to determine its effects on the on and off rates in the same patch of membrane. The close correspondence between the K d values derived from these two studies suggests that the cardiac and brain sodium channels bind the IFM motif in their inactivation gates similarly, but the differences in on and off rates and voltage dependence of KIFMK block may reveal subtype-specific differences in the access of the pentapeptide to these open sodium channels or different effects of the mutations used to slow inactivation in the two sets of experiments.
Comparison to Inactivation of Potassium Channels
Unlike sodium channels, potassium channels have four NH2-terminal inactivation particles that bind to the open channel to produce the inactivated state (Hoshi et al., 1990; Zagotta et al., 1990). Murrell-Lagnado and Aldrich (1993a) reported that block of ShBΔ6-46 potassium channels has a linear dependence on the ShB “ball” peptide concentration, suggesting that Shaker B channels are blocked by a bimolecular reaction mechanism. In spite of the multiple inactivation particles, MacKinnon et al. (1993) showed that a single amino-terminal domain was both necessary and sufficient to inactivate Shaker B channels. The on-rate constant (kon) for KIFMK (∼1.1 × 107 M−1 s−1) is two- to threefold larger than reported for the ShB peptide on potassium channels. Zagotta et al. (1990) and Murrell-Lagnado and Aldrich (1993a) measured blocking rates for peptides binding to open Shaker B potassium channels of 4.3 and 4.8 × 106 M−1 s−1, respectively. The K d value of ∼33 μM is also higher than the value 2.9 μM reported by Murrell-Lagnado and Aldrich (1993a) for potassium channels and reflects the much faster off rate of the KIFMK peptide, 340 s−1 compared with 13.8 s−1 for the Shaker B peptide. The rapid binding and unbinding of the IFM motif may contribute to the more rapid entry into and recovery from the inactivated state of sodium channels relative to potassium channels. It is interesting that the mechanism of inactivation of sodium and potassium channels is so similar when there is no primary structure similarity between the inactivation gates of these two channels. They are also located in different parts of the channel structure, and they are tethered to the pore-forming region in different ways. The experimental approach described here now allows quantitative analysis of the structure–function relationships of IFM analogs in fast open-channel block of sodium channels.
Scheme I.

Acknowledgments
We thank Lorie Devlin and Kevin Black for expert technical assistance and Dr. Todd Scheuer for valuable discussions.
This work was supported by research grant NS-15751 from the National Institutes of Health and a research grant from Parke-Davis Division of Warner-Lambert Co. (W.A. Catterall) and by the Howard Hughes Medical Institute (W.N. Zagotta).
Footnotes
Portions of this work were previously published in abstract form (Eaholtz, G., W.N. Zagotta, and W.A. Catterall. 1995. Biophys. J. 68: A159).
Dr. Eaholtz's present address is Howard Hughes Medical Institute, University of Washington, Seattle, WA 98195-7370.
Abbreviations used in this paper: IFM, isoleucine, phenylalanine, and methionine; I-V, current–voltage.
references
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Aldrich RW, Stevens CF. Voltage-dependent gating of single sodium channels from mammalian neuroblastoma cells. J Neurosci. 1987;7:418–431. doi: 10.1523/JNEUROSCI.07-02-00418.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla FM. Inactivation of the sodium channel. II. Gating currents. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Goldin AL, Krafte DS, Catterall WA, Lester HA, Davidson N, Dunn RJ. A neutral amino acid change in segment IIS4 dramatically alters the gating properties of the voltage-dependent sodium channel. Proc Natl Acad Sci USA. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Goldin AL, Krafte DS, Marshall J, Catterall WA, Lester HA, Davidson N, Dunn RJ. A rat brain Na+channel alpha subunit with novel gating properties. Neuron. 1988;1:449–461. doi: 10.1016/0896-6273(88)90176-6. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall WA. Restoration and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041–1048. doi: 10.1016/0896-6273(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Zagotta WN, Catterall WA. Kinetic analysis of time-dependent open channel block by an inactivation gate peptide of non-inactivating type IIa sodium channels. Biophys J. 1995;68:A159. [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic currents of excitable membranes. 2nd edition. Sinauer Assoc., Inc., Sunderland, MA.
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker K+channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Isom LL, DeJongh K, Reber BFX, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, Scheuer T, Catterall WA. Movement of the Na+channel inactivation gate during inactivation. J Biol Chem. 1996;271:30971–30979. doi: 10.1074/jbc.271.48.30971. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+channels must deactivate to recover from inactivation. Neuron. 1994;12:19–29. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Aldrich RW, Lee A. Functional stoichiometry of Shaker K channel inactivation. Science. 1993;262:757–759. doi: 10.1126/science.7694359. [DOI] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Interactions of amino terminal domains of ShakerK channels with a pore blocking site studied with synthetic peptides. J Gen Physiol. 1993a;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Energetics of ShakerK channels block by inactivation peptides. J Gen Physiol. 1993b;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Kurasaki M, Takahashi H, Numa S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature. 1986;320:188–192. doi: 10.1038/320188a0. [DOI] [PubMed] [Google Scholar]
- Patton DE, West JW, Catterall WA, Goldin AL. Amino acid residues required for fast sodium channel inactivation. Charge neutralizations and deletions in the III–IV linker. Proc Natl Acad Sci USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DE, West JW, Catterall WA, Goldin AL. A peptide segment critical for sodium channel inactivation functions as an inactivation gate in a potassium channel. Neuron. 1993;11:967–974. doi: 10.1016/0896-6273(93)90125-b. [DOI] [PubMed] [Google Scholar]
- Patton DE, Isom LL, Catterall WA, Goldin AL. The adult rat brain β1 subunit modifies activation and inactivation gating of multiple sodium channel α subunits. J Biol Chem. 1994;269:17649–17655. [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tang L, Kallen RG, Horn R. Role of an S4-S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol. 1996;108:89–104. doi: 10.1085/jgp.108.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci USA. 1989;86:8147–8151. doi: 10.1073/pnas.86.20.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic residues required for fast sodium channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shakerpotassium channels by a peptide derived from ShB. Science. 1990;250:29–60. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with ShakermRNA. Proc Natl Acad Sci USA. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar, J.H. 1984. Biostatistical Analysis. 2nd edition. Prentice-Hall, Inc., Englewood Cliffs, N.J.