

Abstract
Tryptophan-substitution mutagenesis was applied to the first and third transmembrane segments (S1 and S3) of a Shaker-type K+ channel for the purpose of ascertaining whether these sequences are α-helical. Point mutants were examined for significant functional changes, indicated by the voltage-activation curves and gating kinetics. Helical periodicity of functional alteration was observed throughout the entire S1 segment. A similar result was obtained with the first 14 residues of S3, but this periodicity disappeared towards the extracellular side of this transmembrane sequence. In both helical stretches, tryptophan-tolerant positions are clustered on approximately half the α-helix surface, as if the sidechains are exposed to the hydrocarbon region of the lipid bilayer. These results, combined with an analogous study of S2 (Monks, S., D.J. Needleman, and C. Miller. 1999. J. Gen. Physiol. 113:415–423), locate S1, S2, and S3 on the lipid-facing periphery of Kv channels.
Keywords: tryptophan-scanning, α-helix, gating
INTRODUCTION
Voltage-dependent K+ channels are homotetrameric proteins with six transmembrane sequences, S1–S6, in each subunit (Fig. 1). The crystal structure of the bacterial KcsA K+ channel makes it all but certain that the S5 and S6 segments of eukaryotic Kv channels are α-helical and, along with the P-loop, pack close to the fourfold axis to form the K+ conduction pathway (Doyle et al. 1998). The structures of the first four transmembrane segments, which underlie the control of channel gating by voltage (Yellen 1998), are unknown. These transmembrane segments have been generally assumed to fold into α-helices, but the experimental evidence for this is indirect. Analysis of S1–S3 sequences of Kv channels reveals helical periodicity in amino acid variability, a common indicator of lipid-exposed sides of transmembrane helices (Donnelly et al. 1993; Perozo and Rosales 1996; Baldwin et al. 1997). A tryptophan-scanning study of S2 in a Shaker K+ channel indicated that substitution of this bulky residue is tolerated with a helical periodicity (Monks et al. 1999). There is also a striking correspondence of the sequence-variable residues with the Trp-tolerant positions. These facts taken together argue that S2 is indeed α-helical and that ∼50% of its surface is exposed to the fatty acyl chains of bilayer lipids.
Figure 1.

Standard model for membrane topology of Kv channels. The lower cartoon represents an aerial view of the assembled tetramer from the extracellular side. The positions of S5, P-loop, and S6, encircled by heavy curve, are modeled to scale from a slice through the structure of the KcsA channel at the level of the external lipid headgroups. Helices are represented by circles of 11.8 Å diameter. Unlabeled helices represent hypothetical positions of S1–S4.
The present study expands our Trp-scanning analysis of Shaker to S1 and S3, the other two transmembrane segments of Kv channels that are predicted by sequence analysis to make contact with lipid (Monks et al. 1999). Tryptophan is substituted individually at each position in these sequences, and the resulting functional consequences are examined. We find that S1 shows a clear helical pattern of Trp toleration, while S3 gives a similar pattern along only part of its length. These results, together with previous work on S2, identify the lipid-exposed faces of the first three transmembrane stretches of Kv channels.
MATERIALS AND METHODS
In general, materials and procedures used in this study were identical to those described in the previous report (Monks et al. 1999).
Mutagenesis and In Vitro Transcription
The channel used in this study is an inactivation-removed (Δ6–46) Shaker B (Schwarz et al. 1988; Hoshi et al. 1990) carrying one point mutation, F425G, in the external vestibule. This Shaker variant is considered “wild type” in these studies. Tryptophan point mutants were made on this background in pBluescript KS(+) using PCR-based mutagenesis. PCR products were purified by agarose gel electrophoresis, digested, and reintroduced into the Shaker cDNA backbone. All constructs were confirmed by sequencing through the cloning cassette. cDNA plasmid was linearized by NotI digestion. cRNA was made by in vitro transcription from the linearized plasmid using T7 RNA polymerase (Promega Corp.).
Electrophysiology
Xenopus oocytes were prepared and incubated for 70–80 min in collagenase (2 mg/ml; Worthington Biochemical Corp.) in Ca2+-free solution containing (mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, pH 7.5. Defolliculated oocytes were injected with 27 or 50 nl of cRNA (0.03–3 mg/ml), and then stored at 17°C in ND96-gentamicin solution containing (mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 10 mM HEPES, pH 7.6, and 0.1 mg/ml gentamicin. Oocytes were examined 1–3 d after injection using two-electrode voltage clamp (oocyte clamp OC-725B; Warner Instruments) in ND96 solution (containing 0.3 mM CaCl2) to check expression levels and K+ selectivity. Electrodes were filled with 3 M KCl, 5 mM EGTA, and 10 mM HEPES, pH 7.6. Electrodes had resistances of 0.3–1.5 MΩ. Tail currents were recorded in KD98 solution containing (mM): 98 KCl, 0.3 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.6. The standard pulse protocol was a holding potential of −90 mV, test pulses between −60 and +50 mV in 10-mV increments (50-ms duration), followed by a tail pulse to −70 mV (30-ms duration). The interpulse interval was 1 s. The values of test and tail voltages, pulse duration, and interpulse interval were modified according to the kinetic properties of individual mutants. The data were obtained from 6–14 oocytes for each mutant.
Data Analysis
Tail-current amplitudes were measured at 2–3 ms after the tail pulse. Voltage-activation curves were calculated using standard tail-current analysis (Liman et al. 1992). Data were fit to a Boltzmann function, I/I max = 1/{1 + exp[−zF(V − Vo)/RT]}, to determine Vo (half-maximal activation voltage) and z (slope factor). The free energy of channel opening at zero voltage (ΔG o, the intrinsic stability of the open conformation with respect to the closed) was calculated according to ΔG o = zFVo.
Activation and deactivation kinetics were compared among the various mutants and wild type. Activation kinetics, which are nonexponential, were quantified by the time required for half-opening, t o, at a test voltage near Vo. The time constants for deactivation, τd, were determined by fitting the tail current with the falling exponential function. The “open-state stabilization energy” is defined as: ΔΔG o = ΔG o mut − ΔG o wt; it should be noted that this convention is opposite from that in the previous report (Monks et al. 1999), where closed-state stabilization energy was reported.
RESULTS
The sequences of S1 (positions 225–246) and S3 (positions 311–332), when examined in BLAST searches of >100 Kv channels, show patterns of sequence variability that hint at the locations of lipid-exposed and protein-packed faces of α-helices. Fig. 2 displays these sequences in Shaker, indicating the variable and conserved residues (circles and squares, respectively). The variable residues in S1, which are nearly all aliphatic, cover about half the surface of a helical wheel, all clustered on the same side; these residues were predicted to define a lipid-exposed surface of an α-helical S1 (Monks et al. 1999). In contrast, S3 contains a larger fraction of conserved residues, with a small, restricted area of variable residues clustered in a corner of the helical wheel diagram, as though this transmembrane stretch is surrounded by other parts of the protein or, perhaps, is not helical at all.
Figure 2.

Sequence characteristics of S1 and S3 segments in Shaker. (A and B) Amino acid sequences of S1 and S3. Boldface underlined residues are those conserved in 120 S1 and 135 S3 K+ channel sequences, using a BLAST search (Altschul et al. 1997) of the nonredundant database. (C and D) α-helical projections of S1 and S3. Conserved residues, inscribed in black squares, are identified by two criteria. First, the most common residue is found in over 65% of instances; second, no more than four alternative substitutes are observed, and these must all be of similar chemical character. Listed in parentheses are alternative amino acids found at equivalent positions in other K+ channels.
To subject these sequence-based conjectures to experimental examination, we employed Trp-scanning mutagenesis of S1 and S3 in Shaker. Each residue in S1 and S3 was replaced individually by tryptophan, and the mutant channels were analyzed by two-electrode voltage clamp after expression in Xenopus oocytes. In particular, we assessed the effects of mutations on the equilibrium voltage-activation curve and on activation and deactivation kinetics.
S1 Segment
Of the 22 tryptophan mutants in S1, 21 expressed voltage-dependent, K+-selective currents, the single exception being I237W. Responses to families of depolarizing pulses are shown in Fig. 3 for wild-type Shaker and two mutants—L238W, which is left-shifted by 21 mV, and I243W, which is right-shifted by 27 mV. The activation and deactivation kinetics of L238W are slowed substantially, while those of I243W are similar to wild type. All S1 mutants were analyzed in this way, and gating parameters are reported in Table .
Figure 3.

Recordings from tryptophan-substituted Shaker channels. (A) Two-electrode voltage-clamp recordings of wild-type channels expressed in Xenopus oocytes. Currents were recorded in KD98 medium with a holding potential of −90 mV, test pulses (30-ms duration) from −60 to +50 mV in 10-mV increments, and a tail potential of −70 mV. (B and C) Similar recordings taken for mutations I243W and L238W. For L238W, test pulses were from −80 to +20 mV with duration of 200 ms and each test pulse was followed by a fixed tail voltage −80 mV. (D) Voltage-activation curves for wild-type, I243W, and L238W mutant channels, calculated from tail-current analysis. Solid curves are Boltzmann fits to the equilibrium activation data. Scale bars in all data panels represent 1 μA, 10 ms.
Table 1.
Electrophysiological Properties of S1 Segment Trp Mutant Channels
| Construct | V0 | z | ΔG o | ΔΔG o | t o | τd |
|---|---|---|---|---|---|---|
| mV | kcal/mol | kcal/mol | ms | ms | ||
| WT | −19 ± 3 | 2.6 ± 0.3 | −1.2 ± 0.3 | — | 4.5 ± 0.3 | 1.1 ± 0.2 |
| A225W | −16 ± 2 | 2.5 ± 0.3 | −0.9 ± 0.2 | 0.3 | 4.5 ± 0.3 | 0.8 ± 0.1 |
| A226W | −49 ± 2 | 4.9 ± 0.6 | −5.5 ± 1.0 | −4.3 | 32.8 ± 3.7 | 6.9 ± 0.8 |
| R227W | −1 ± 1 | 2.6 ± 0.3 | −0.0 ± 0.4 | 1.2 | 5.6 ± 0.3 | 1.1 ± 0.1 |
| V228W | −23 ± 3 | 3.3 ± 0.6 | −1.8 ± 0.5 | −0.6 | 6.2 ± 0.6 | 1.9 ± 0.2 |
| V229W | −35 ± 2 | 6.7 ± 0.8 | −5.5 ± 0.8 | −4.3 | 12.1 ± 2.0 | 3.2 ± 0.3 |
| A230W | 50 ± 6 | 1.0 ± 0.1 | 1.2 ± 0.1 | 2.4 | 2.5 ± 0.3 | 1.0 ± 0.1 |
| I231W | −30 ± 5 | 2.1 ± 0.3 | −1.4 ± 0.5 | −0.2 | 3.3 ± 0.5 | 1.5 ± 0.3 |
| I232W | −23 ± 2 | 3.6 ± 0.6 | −1.9 ± 0.6 | −0.7 | 9.5 ± 1.2 | 2.7 ± 1.0 |
| S233W | >80 | * | * | * | * | * |
| V234W | 26 ± 3 | 1.4 ± 0.1 | 0.8 ± 0.1 | 2.0 | 4.7 ± 0.4 | 1.0 ± 0.3 |
| F235W | −5 ± 2 | 3.2 ± 0.4 | −0.4 ± 0.2 | 0.8 | 9.6 ± 1.1 | 1.2 ± 0.1 |
| V236W | −7 ± 2 | 2.2 ± 0.2 | −0.4 ± 0.1 | 0.8 | 3.8 ± 0.4 | 0.8 ± 0.1 |
| I237W | × | × | × | × | × | × |
| L238W | −44 ± 1 | 5.8 ± 0.5 | −5.9 ± 0.7 | −4.7 | 24.0 ± 2.2 | 6.8 ± 0.8 |
| L239W | −11 ± 3 | 2.6 ± 0.3 | −0.7 ± 0.2 | 0.5 | 5.8 ± 0.5 | 1.1 ± 0.1 |
| S240W | 44 ± 2 | 2.3 ± 0.1 | 2.3 ± 0.1 | 3.5 | 44.9 ± 2.2 | 6.9 ± 0.2 |
| I241W | 51 ± 2 | 2.7 ± 0.2 | 3.2 ± 0.2 | 4.4 | 7.6 ± 0.3 | 4.1 ± 0.2 |
| V242W | −10 ± 1 | 2.6 ± 0.1 | −0.6 ± 0.1 | 0.6 | 4.9 ± 0.3 | 1.5 ± 0.1 |
| I243W | 5 ± 1 | 2.5 ± 0.1 | 0.3 ± 0.1 | 1.5 | 4.8 ± 0.4 | 0.8 ± 0.1 |
| F244W | 30 ± 3 | 2.7 ± 0.4 | 1.8 ± 0.3 | 3.0 | 9.1 ± 0.6 | 1.8 ± 0.2 |
| C245W | 23 ± 4 | 1.1 ± 0.1 | 0.6 ± 0.1 | 1.8 | 2.8 ± 0.1 | 0.9 ± 0.2 |
| L246W | −14 ± 3 | 1.9 ± 0.3 | −0.6 ± 0.3 | 0.6 | 4.3 ± 0.4 | 1.5 ± 0.1 |
Data from individual activation curves, obtained from 6–14 oocytes, were fit to a Boltzmann function. Vo, t o, and τd were obtained as described in materials and methods. Values are mean values ± SD. *For S233W, Vo is too high to permit accurate determination of activation curves. × indicates no expression of current.
In Fig. 4, we display graphically the pertinent gating parameters, including the open-state stabilization free energy, ΔΔG o, a nearly model-independent measure of the effect of Trp substitution on the channel's open–closed equilibrium (Monks et al. 1999). In all, nine mutant channels have electrophysiological properties similar to wild type, with |ΔΔG o| < 1 kcal/mol. The remaining thirteen positions (A226, R227, V229, A230, S233, V234, I237, L238, S240, I241, I243, F244, and C245) have significantly altered properties. We refer to these as “low-impact” and “high-impact” residues, respectively; the cutoff of 1 kcal/mol is arbitrary, but it falls naturally out of the free energy data. None of the resides scored as low-impact shows more than a threefold alteration of kinetic properties, whereas all of the kinetically disrupted positions (A226, L238, S240, I241) were also high-impact by the equilibrium criterion. As with S2 (Monks et al. 1999), some of the equilibrium-altered positions showed unaltered kinetics.
Figure 4.
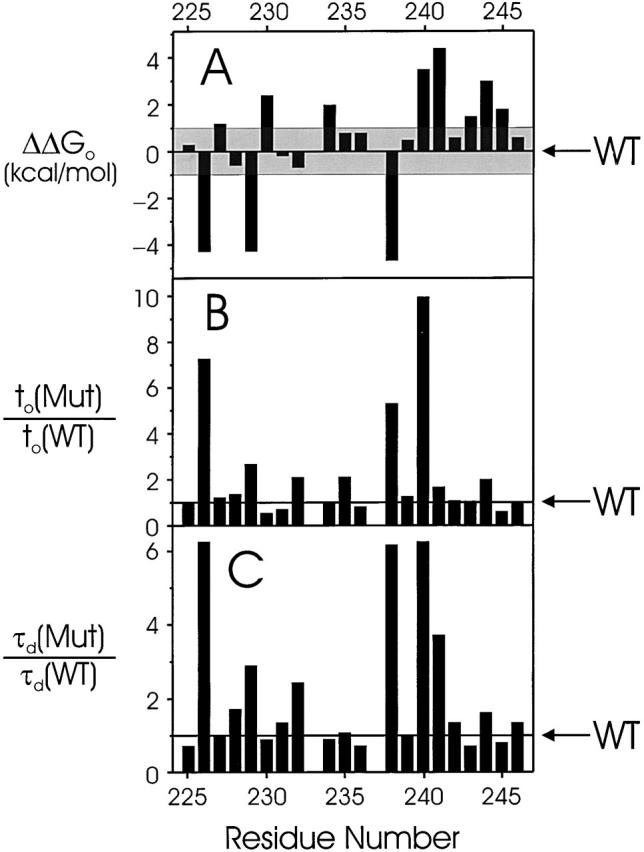
Gating parameters of S1 tryptophan scan. Changes of empirical gating parameters resulting from point Trp mutations are displayed versus residue number. (A) Intrinsic free energy of open-state stabilization, ΔΔG o. The shaded region (|ΔΔG o| < 1 kcal/mol) represents the range of values defining tolerant residues. (B and C) Mutant-to-wild-type ratio of activation times, t o, and deactivation time constant, τd, determined as in materials and methods. Electrophysiological parameters are reported in Table .
S3 Segment
All 22 mutants in S3 segment expressed voltage-dependent, K+-selective currents (Table ). By the above considerations, 11 positions (M312, N313, I315, D316, A319, I325, T326, T329, V330, V331, and A332) were high-impact (Fig. 5), most of them with left-shifted gating, equivalent to a stabilization of the open state by Trp substitution. The expression level of D316W was very low, and both opening and closing kinetics were too fast to quantify accurately. The remaining 11 mutants were low impact, and five of these are contiguous in sequence (I320, I321, P322, Y323, and F324); in contrast to the situation in S1, three of these low-impact substitutions (I321, P322, and F324) show greatly slowed activation kinetics.
Table 2.
Electrophysiological Properties of S2 Segment Trp Mutant Channels
| Construct | Vo | z | ΔG o | ΔΔG o | t o | τd |
|---|---|---|---|---|---|---|
| mv | kcal/mol | kcal/mol | ms | ms | ||
| WT | −19 ± 3 | 2.6 ± 0.3 | −1.2 ± 0.3 | — | 4.5 ± 0.3 | 1.1 ± 0.2 |
| V311W | −9 ± 1 | 1.8 ± 0.1 | −0.4 ± 0.1 | 0.8 | 5.8 ± 0.3 | 1.0 ± 0.1 |
| M312W | 47 ± 13 | 1.2 ± 0.1 | 1.3 ± 0.4 | 2.5 | 2.9 ± 0.4 | 0.7 ± 0.1 |
| N313W | −51 ± 1 | 4.1 ± 0.4 | −4.8 ± 0.6 | −3.6 | 8.0 ± 0.7 | 3.2 ± 0.3 |
| V314W | −14 ± 3 | 1.9 ± 0.2 | −0.6 ± 0.2 | 0.6 | 4.0 ± 0.3 | 1.5 ± 0.2 |
| I315W | −53 ± 1 | 5.2 ± 0.6 | −6.3 ± 0.9 | −5.1 | 23.1 ± 1.7 | 12.1 ± 0.9 |
| D316W | 38 ± 14 | 1.1 ± 0.1 | 1.0 ± 0.3 | 2.2 | <3.0 | <1.0 |
| I317W | −12 ± 2 | 2.3 ± 0.1 | −0.7 ± 0.1 | 0.5 | 6.8 ± 1.0 | 1.5 ± 0.1 |
| I318W | −14 ± 3 | 2.1 ± 0.3 | −0.7 ± 0.2 | 0.5 | 8.2 ± 1.1 | 2.2 ± 0.3 |
| A319W | 1 ± 4 | 2.4 ± 0.2 | 0.1 ± 0.2 | 1.3 | 21.0 ± 1.7 | 3.7 ± 1.0 |
| I320W | −13 ± 3 | 2.4 ± 0.3 | −0.8 ± 0.2 | 0.4. | 5.7 ± 0.3 | 1.7 ± 0.1 |
| I321W | −31 ± 2 | 2.5 ± 0.2 | −1.8 ± 0.3 | −0.6 | 14.5 ± 2.6 | 2.6 ± 0.2 |
| P322W | −22 ± 2 | 2.4 ± 0.2 | −1.2 ± 0.2 | 0.0 | 51.5 ± 3.2 | 4.7 ± 0.7 |
| Y323W | −15 ± 2 | 1.9 ± 0.2 | −0.7 ± 0.1 | 0.5 | 5.1 ± 3.2 | 4.7 ± 0.7 |
| F324W | −15 ± 2 | 1.9 ± 0.2 | −0.7 ± 0.1 | 0.5 | 5.1 ± 0.5 | 1.6 ± 0.4 |
| I325W | −17 ± 3 | 3.2 ± 0.4 | −1.2 ± 0.3 | 0.0 | 34.2 ± 1.5 | 1.6 ± 0.4 |
| T326W | −25 ± 1 | 4.7 ± 0.2 | −2.7 ± 0.2 | −1.5 | 24.3 ± 1.7 | 4.4 ± 0.6 |
| L327W | −20 ± 3 | 3.6 ± 0.9 | −1.7 ± 0.6 | −0.5 | 13.3 ± 1.7 | 2.0 ± 0.5 |
| A328W | −22 ± 3 | 3.1 ± 0.6 | −1.6 ± 0.5 | −0.4 | 5.4 ± 0.4 | 1.3 ± 0.2 |
| T329W | −58 ± 1 | 5.9 ± 0.6 | −7.9 ± 1.0 | −6.7 | 15.3 ± 2.0 | 6.7 ± 0.8 |
| V330W | −31 ± 2 | 5.4 ± 0.5 | −4.0 ± 0.6 | −2.8 | 11.9 ± 1.0 | 2.9 ± 0.5 |
| V331W | −28 ± 3 | 4.7 ± 0.9 | −3.1 ± 0.9 | −1.9 | 12.2 ± 1.9 | 2.2 ± 0.3 |
| A332W | −33 ± 1 | 4.6 ± 0.3 | −3.5 ± 0.3 | −2.1 | 12.5 ± 2.5 | 2.3 ± 0.2 |
Parameters are as described in Table . The expression level of D316W was very low and the opening and closing kinetics were too fast to measure accurately.
Figure 5.
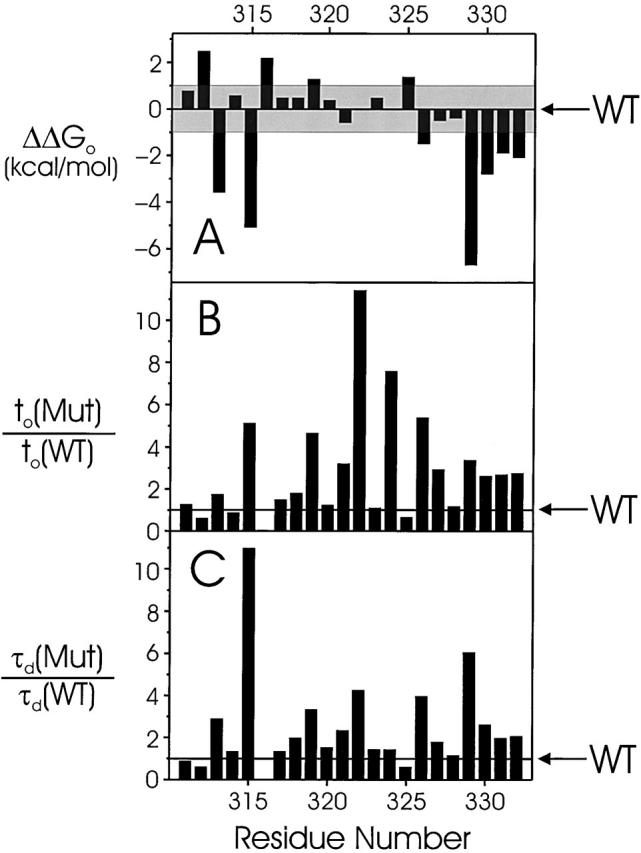
Gating parameters of S3 tryptophan scan. Details are as in Fig. 4. Electrophysiological parameters are reported in Table .
We worried that this uninterrupted string of five low-impact residues might be an artifact arising from the nature of the wild-type residues in this sequence. Since four of these are large or aromatic, it is possible that substitution by Trp is inherently nondisruptive even at a protein-packed interface. Previous work on S2 had shown that in such a case, substitution by Asn, a small, polar residue could be used to buttress an argument for a lipid-exposed position (Monks et al. 1999). We therefore changed the four nonproline residues individually to Asn (Fig. 6, Table ). By this criterion, one of these residues, I321, scores as low impact and one, Y323, as high impact. The two remaining residues, I320 and F324, are ambiguously near the borderline and cannot be reliably scored. Taking these adjustments into account, the S3 scan identifies 2 ambiguous, 8 low-impact, and 12 high-impact positions.
Figure 6.
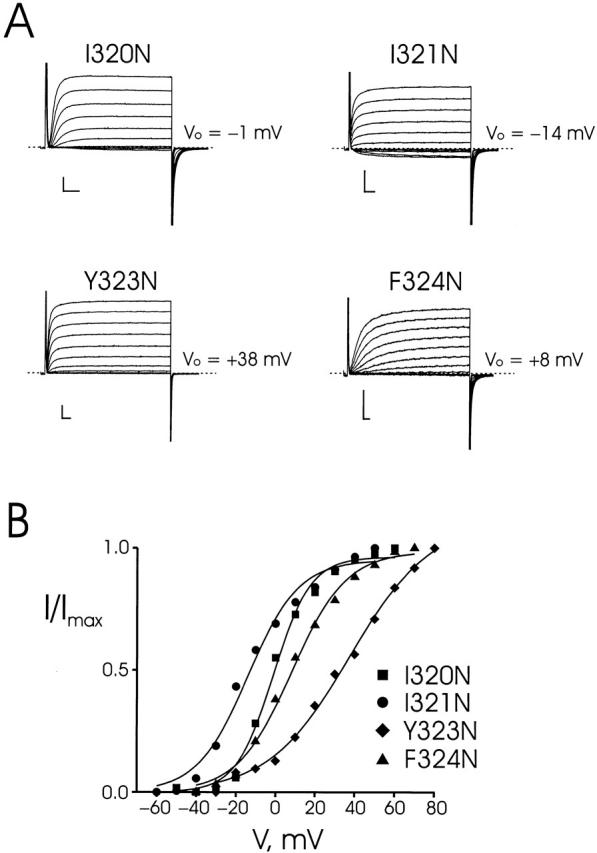
Effects of Asn substitutions at certain Trp-tolerant positions. Two-electrode voltage clamp recordings (A) and voltage-activation curves (B) of the indicated mutants displayed as in Fig. 3. Electrophysiological parameters are reported in Table .
Table 3.
Electrophysiological Properties of S3 Segment Asn Mutant Channels
| Construct | Vo | z | ΔG o | ΔΔG o | t o | τd |
|---|---|---|---|---|---|---|
| mV | kcal/mol | kcal/mol | ms | ms | ||
| WT | −19 ± 3 | 2.6 ± 0.3 | −1.2 ± 0.3 | — | 4.5 ± 0.3 | 1.1 ± 0.2 |
| I320N | −1 ± 1 | 2.7 ± 0.2 | −0.1 ± 0.0 | 1.1 | 21.4 ± 0.8 | 2.1 ± 0.2 |
| I321N | −15 ± 4 | 2.0 ± 0.2 | −0.7 ± 0.2 | 0.5 | 5.6 ± 0.6 | 2.3 ± 0.2 |
| Y323N | 36 ± 3 | 1.3 ± 0.1 | 1.1 ± 0.2 | 2.3 | 5.6 ± 0.3 | 0.9 ± 0.2 |
| F324N | 8 ± 2 | 2.0 ± 0.1 | 0.4 ± 0.1 | 1.6 | 59.6 ± 1.9 | 4.1 ± 0.3 |
Parameters are as described in Table .
DISCUSSION
We have applied tryptophan-scanning mutagenesis to the S1 and S3 segments of a Shaker K+ channel to ask whether these are helical structures, as is widely imagined, and, if so, to locate the lipid-exposed areas of the helices. The method rests on the idea that substitution with Trp, a bulky hydrophobic amino acid, will disrupt or significantly alter channel function if it is substituted for amino acids that contact or directly interact with other parts of the channel protein. In contrast, Trp substitutions of sidechains lipid-exposed in both open and closed conformations should be readily accommodated and should not alter the functional properties of the channel. Though this method has no a priori justification, it has been productively used on several membrane proteins, including S2 of Shaker (Choe et al. 1995; Sharp et al. 1995; Monks et al. 1999). The use of Trp as a probe for helical structures and lipid-exposed regions of membrane proteins has been discussed in detail (Monks et al. 1999), and we will not repeat the same arguments here. As in the previous study on S2, doubling the free energy cutoff that defines high- and low- impact residues does not alter the conclusions reached.
S1 Segment
In Fig. 7, helical wheel and helical net diagrams are shown for S1, with low- and high-impact residues displayed as open and shaded circles, respectively. The two classes of residues segregate to distinct faces of the helix, with only a single exception, I243, a high-impact residue falling on the low-impact face. Each face accounts for approximately half of the α-helical surface. In addition, the pattern of functional segregation agrees well with the expectations based on sequence conservation, also indicated in Fig. 7. The results indicate that the entire S1 segment has an α-helical structure and that half of the helix faces a lipid environment.
Figure 7.

Results of Trp-scanning mutagenesis of S1. An α-helical wheel (A) and a net (B) diagram of S1 are shown with residues scored as either not expressed (diamonds), high impact (shaded circles), or tolerant (open circles), according to criteria described in text. Black squares represent conserved residues as in Fig. 2.
S3 Segment
In stark distinction to S1, S3 substitutions fail to produce an orderly pattern of functional segregation on a helical wheel diagram (Fig. 8 A). The only trend apparent is a tendency for low-impact residues to cluster in the upper right-hand quadrant of the diagram, in rough correspondence with the location of the sequence-variable residues. The preponderance of the wheel's perimeter is festooned by high-impact residues, but several low-impact positions are scattered among these. An additional source of confusion is that three functionally tolerant positions (P322, F324, and L327) are tightly conserved among S3 segments, while five high-impact residues (M312, T326, V330, V331, and A332) show a remarkable sequence variability among polar, nonpolar, large, and small sidechains (Fig. 2 D). It is tempting to conclude from the results that S3 is largely surrounded by protein and is poorly exposed to lipid. However, this conclusion would be logically flawed, since the Trp-scanning strategy is useful only if an unforced functional periodicity emerges from the data, as with S1 and S2. The absence of a pattern is uninterpretable; in such a case, the scanned sequence might not have a periodic secondary structure at all, or it might be a helix (or any other structure) specifically packed on all sides against other parts of the protein.
Figure 8.

Results of Trp and Asn mutagenesis of S3. An α-helical wheel (A) and a net (B) diagram of S3 are shown as in Fig. 7. *Residues that were tested by Asn substitution. Striped circles represent ambiguous residues on the basis of Asn mutants, as discussed in text. (C) A restricted helical wheel diagram limited to the first 14 residues of S3.
Nevertheless, a helical pattern is discernible in the S3 scan when the results are displayed on a helical net diagram (Fig. 8 B), which avoids a problem of the wheel representation: that the projection of the entire sequence is being displayed. The net diagram makes it clear that the first 14 residues of S3 form an unconfused pattern of functional periodicity encompassing four helical turns. It is the last eight residues beginning at I325, which lie towards the external side of the membrane, that abruptly depart from helical periodicity and are wholly responsible for obscuring the pattern on the wheel diagram. It is also in this part of S3 that most of the discrepancies between functional impact and sequence conservation are found. On a helical wheel restricted to the first 14 residues, the low- and high-impact positions are well segregated, each type to its own half of the helical surface (Fig. 8 C). We therefore propose that the first two thirds of S3 is indeed helical, with about half its surface exposed to lipid, as with S1 and S2. Any discussion about what happens in the middle of the bilayer towards the external side, where the pattern disappears, would be idly speculative.
Sequence–Structure Mapping of S5
The transmembrane helices S5 and S6 of Shaker are confidently expected to be structurally similar to the two transmembrane helices in KcsA (Doyle et al. 1998). In the bacterial channel, both of these helices are partly lipid-exposed, but some or perhaps all of the corresponding surfaces in Kv channels will be packed against protein rather than lipid. In our analysis of S1, S2, and S3, we were struck by the faithful correspondence between functional response to tryptophan substitution and sequence variability. We were therefore motivated to examine S4, S5, and S6 sequences, to try to identify regions of these helices that remain lipid-exposed in the six-transmembrane Kv channels. Comparisons (not shown) of over 120 S4 and S6 sequences revealed high sequence conservation throughout these segments, with no indication of variability patterns that would indicate lipid exposure. Comparisons of S5 sequences, however, produced a different result. Of the 22 positions in S5 (396–417), six are variable by the criteria used for S1–S3 (G397, F401, F404, V408, A413, and V414). These six positions do not segregate to one side of a helical wheel diagram of S5. However, the first transmembrane segment of KcsA is a continuous α-helix only for its first 16 residues (positions 29–45). At position 45, it kinks slightly, and then returns to helical structure. Fig. 9 shows two views of these 16 helical residues of KcsA. The black atoms mark those residues known from the crystal structure to be packed against other parts of the channel protein. The remaining surface in the bacterial channel is all lipid-exposed. Restricting our analysis to the first 16 residues of S5, the four sequence-variable positions are confined to only a “stripe” in the KcsA lipid-exposed surface (grey atoms). On the basis of this pattern, we propose that the S5 helix in Kv channels is not fully surrounded by protein, but rather that a narrow region of S5 is exposed to membrane lipid.
Figure 9.

Structural interpretation of sequence variability in S5. The first 16 residues in the outer helix of KcsA (positions 29–45), equivalent to S5 in Shaker (396–412), are represented in two views. (Left) View down the helix axis from the intracellular end; (right) longitudinal view, with intracellular end at bottom. (Black atoms) α and β carbons of sidechains packed directly against protein in KcsA; (grey atoms) α and β carbons of positions equivalent to variable residues in S5.
Summary
Voltage-dependent K+ channels may be crudely but validly viewed as constructed from two basic units: a pore module, S5–S6, and a gating module that controls the conformation of the pore, S1–S4. The high-resolution structure of KcsA, when combined with compelling functional analogies to Kv channels (Doyle et al. 1998; Moczydlowski 1998; Yellen 1998), shows definitively that S5 and S6 are helical, and that these are packed within ∼20 Å of the channel's central axis. The four remaining segments must therefore be positioned around this central core (Fig. 1), and some of this transmembrane mass must necessarily contact the hydrocarbon region of the lipid bilayer. The present Trp-scanning study, taken together with that reported previously (Monks et al. 1999), locates the major part of the protein–lipid interface in a Shaker channel, and, by implication, in the entire family of Kv channels. The S1 segment is helical, with a well-defined lipid-exposed surface running along its entire length across the membrane. S3 begins on the cytoplasmic side in a helical structure, but shortly after crossing the center of the membrane it becomes lost to our perception. Nevertheless, its helical region, which is ∼20 Å in length, is also exposed to lipid on half its surface. It is worth remembering that the external end of S2 also departs from helical periodicity (Monks et al. 1999).
These results confirm that S1, S2, and S3 are helices lying on the outer periphery of Kv channels, in contact with bilayer lipid. Sequence variability in S5 suggests that a narrow band of lipid-exposed surface also runs along most of this transmembrane segment. We have not attempted to subject S4, the voltage sensor of Kv channels, to a mutagenic scan, since any pattern emerging from such a study might be an artifact of the periodicity intrinsic to the unusual sequence of this highly charged transmembrane segment. In light of the pore-facing location of S6 and the lipid-facing positions of S1–S3, we imagine that both S4 and S6 are surrounded by protein, dwelling somewhere between the central pore and the lipid-exposed edge of the channel. Unfortunately, the results here do not even hint at the details of helix–helix packing. It is notable, though, that E283 and E293 in S2 and D316 in S3, the three anionic residues proposed to form electrostatic contacts with positive sidechains in S4 (Papazian et al. 1995; Tiwari-Woodruff et al. 1997), all lie firmly within the high-impact faces of these helices, far from the proposed lipid-facing areas.
Acknowledgments
We are grateful to Rob Blaustein, Bill Kobertz, and Merritt Maduke for critiques on this manuscript and to Chris Mercogliano for spending a graduate rotation constructing several of the mutants used here. We are also grateful to Kenton Swartz for sharing results before publication on parallel helix-scanning studies coming to very similar conclusions.
Supported in part by National Institutes of Health grant GM-31768.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D. Gapped BLAST and PSI-BLASTa new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin J.M., Schertler G.F.X., Unger V.M. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein–coupled receptors. J. Mol. Biol. 1997;272:144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- Choe S., Stevens C.F., Sullivan J.M. Three distinct structural environments of a transmembrane domain in the inwardly rectifying K+ channel ROMK1 defined by perturbation. Proc. Natl. Acad. Sci. USA. 1995;92:12046–12049. doi: 10.1073/pnas.92.26.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly D., Overington J.P., Ruffle S.V., Nugent J.H., Blundell T.L. Modeling alpha-helical transmembrane domainsthe calculation and use of substitution tables for lipid-facing residues. Protein Sci. 1993;2:55–70. doi: 10.1002/pro.5560020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the K+ channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker K+ channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Liman E.R., Tytgat J., Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Moczydlowski E. Chemical basis for alkali cation selectivity in potassium-channel proteins. Chem. Biol. 1998;5:R291–R301. doi: 10.1016/s1074-5521(98)90288-5. [DOI] [PubMed] [Google Scholar]
- Monks S., Needleman D.J., Miller C. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 1999;113:415–423. doi: 10.1085/jgp.113.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazian D.M., Shao X.M., Seoh S.A., Mock A.F., Huang Y., Wainstock D.H. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Perozo E., Rosales R. Detection and orientation and topoligical arrangement of α-helices in voltage-dependent K+ channels from Fourier analysis of sequence variability Biophys. J. 70 1996. A12(Abstr.) [Google Scholar]
- Schwarz T.L., Tempel B.L., Papazian D.M., Jan Y.N., Jan L.Y. Multiple K+ channel components are produced by alternative splicing at the Shaker locus in Drosophila. Nature. 1988;331:137–142. doi: 10.1038/331137a0. [DOI] [PubMed] [Google Scholar]
- Sharp L.L., Zhou J., Blair D.F. Features of MotA proton channel structure revealed by tryptophan-scanning mutagenesis. Proc. Natl. Acad. Sci. USA. 1995;92:7946–7950. doi: 10.1073/pnas.92.17.7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff S.K., Schulteis C.T., Mock A.F., Papazian D.M. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]