

Abstract
It is well known that the sodium current (INa) and the degree of gap-junctional electrical coupling are the key determinants of action potential (AP) conduction in cardiac tissue. Immunohistochemical studies have shown that sodium channels (NaChs) are preferentially located in intercalated disks (IDs). Using dual immunocytochemical staining, we confirmed the colocalization of NaChs with connexin43 in cultures of neonatal rat ventricular myocytes. In mathematical simulations of conduction using the Luo-Rudy dynamic model of the ventricular AP, we assessed the hypothesis that conduction could be modulated by the preferential localization of NaChs in IDs. Localization of INa at the ID caused a large negative potential in the intercellular cleft, which influenced conduction in two opposing ways, depending on the degree of electrical coupling: (1) for normal and moderately reduced coupling, the negative cleft potential led to a large overshoot of the transmembrane potential resulting in a decreased driving force for INa itself (self-attenuation), which slowed conduction; (2) for greatly reduced coupling (<10%), the negative cleft potential induced by INa in the prejunctional membrane led to suprathreshold depolarization of the postjunctional membrane, which facilitated and accelerated conduction. When cleft potential effects were not incorporated, conduction was not significantly affected by the ID localization of INa. By enhancing conduction through the establishment of cleft potentials, the localization of NaChs in IDs might protect the myocardium from conduction block, very slow conduction, and microreentry under conditions of greatly reduced coupling. Conversely, by supporting moderately slow conduction, this mechanism could also promote arrhythmias
Keywords: action potential conduction, sodium current, gap junctions, intercalated disks, slow conduction
It is well known that the velocity of cardiac conduction is determined by the density and type of voltage-dependent ion channels carrying inward currents and by the degree of intercellular gap-junctional coupling.1-4 In ventricular tissue, the fast inward sodium current (INa) is the major depolarizing current, whereas connexin43 (Cx43) is the major junctional channel-forming protein permitting current flow between adjacent cells. A decrease of INa or a decrease in gap-junctional coupling both lead to conduction slowing, increasing the risk of life-threatening reentrant arrhythmias.3-5
Even under physiological conditions, gap junctions represent discrete resistive barriers for the flow of current. As shown in single cell strands,6-9 these locations of increased resistance result in a discontinuous pattern of conduction at the cellular level: across junctions, conduction is characterized by local conduction delays. The duration of these delays (and thus conduction velocity, θ) is strongly dependent on the degree of gap-junctional coupling.3,4,9
In immunocytochemical studies, it was observed that Nav1.5 sodium channels (NaChs) are preferentially located in intercalated disks (IDs) between neighboring myocytes.10-13 The observation that NaChs are concentrated at intercellular junctions led to the hypothesis that this colocalization is a physiological adaptive mechanism to overcome the local junctional resistive barriers and hence to stabilize conduction.
It was the aim of this study (1) to determine the subcellular distribution of NaChs in cultures of neonatal rat ventricular myocytes and (2) to evaluate the functional consequences of a preferential localization of NaChs in IDs using a mathematical model of conduction. In the cultures, we observed colocalization of NaChs with Cx43. In the model, the presence of a large INa in IDs led to a modified dependence of θ on gap-junctional coupling. This effect was caused by large intercellular cleft currents and potentials induced by the large density of INa. This suggests that the localization of NaChs in IDs leads to extracellular cleft interactions that modulate the transfer of the action potential (AP) from one cell to the next.14
Materials and Methods
Immunocytochemical Staining of Cardiac Cell Cultures
Cultures of neonatal rat ventricular myocytes (Wistar) were prepared according to previously published procedures.3 Five to 6-day-old cultures were washed with PBS, fixed with 2% paraformaldehyde for 5 minutes at 20°C, and incubated for 1 hour at 20°C in blockingbuffer (PBS containing 3% goat serum). Primary antibodies, directed against rat heart NaChs (rH1, polyclonal antibody raised against residues 492 to 510 of the α-subunit of rat cardiac type NaCh; Alomone) and Cx43 (mouse anti-Cx43 monoclonal antibody; Chemicon) were diluted at 1:10 (rH1) and 1:100 (Cx43) in a buffer containing 1% goat serum and 0.15% Triton X-100. Preparations were exposed to these antibodies for 1 hour at 20°C followed by 16 hours at 4°C. They were then washed with PBS and incubated with the secondary antibodies for 2 hours at 37°C (CY3-conjugated goat anti-rabbit IgG for rH1, Jackson ImmunoResearch; Alexa 488 conjugated goat anti-mouse IgG for Cx43, Molecular Probes). Animals were obtained from the central animal facility of the University Hospital of Bern (Bern, Switzerland). They were used according to the ethical principles and guidelines of the Swiss Academy of Medical Sciences.
Mathematical Model of Conduction
The latest version of the Luo-Rudy dynamic (LRd) ventricular cell model15-17 was used to simulate conduction along linear strands of 64 cells. As shown in Figure 1, the membrane of every cell (length L: 100 μm; radius r: 11 μm) was discretized into 10 axial patches (length Lp: 10 μm) and 2 disk (junctional) patches, one at each end of the cell. Every patch had a capacitance proportional to its area (Cax and Cdisk, respectively) and generated transmembrane currents according to the LRd formulation. The intracellular potential (Vi) was computed at the nodes corresponding to the patches. The myoplasmic resistance between adjacent nodes, Rmyo, was ρmyo · Lp/πr2, where ρmyo is the myoplasmic resistivity. The last node within a cell was connected to the first node in the next cell through a gap-junctional resistance Rgap. As done previously,4 a value of 150 Ω · cm was used for ρmyo and a control (normal) value of 395 kΩ (conductance: 2.534 μS) was used for Rgap.
Figure 1.
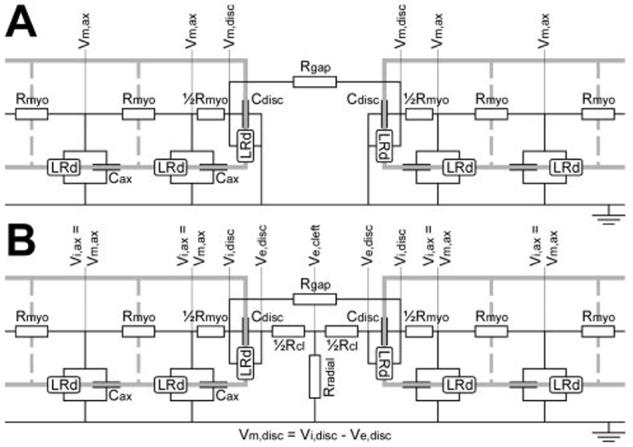
Electric circuit representations of the junction between adjacent cells. A, Noncleft model. The extracellular potential was assumed to be 0 (extensive medium). Adjacent cells interact only through current flowing across the gap-junctional resistance (Rgap). B, Cleft model. The extracellular potential (Ve) was assumed to be 0 except at the junction, where Ve was computed at 3 nodes (pre- and postjunctional Ve,disk, Ve,cleft). In addition to the gap-junctional current, adjacent cells interact through extracellular currents flowing in the extracellular cleft across the T-shaped network of its axial (Rcl) and radial (Rradial) resistances, respectively. See text for details.
In a first approximation, we assumed that the extracellular resistance, including that of the intercellular cleft, is negligible and hence that the extracellular potential is 0 (noncleft model, Figure 1A). Under this assumption, the intracellular potential equals the transmembrane potential, Vm. However, in the very narrow intercellular clefts at IDs, this assumption is likely to be inadequate, especially when resistance in the radial direction (parallel to the cleft) is considered. Thus, extracellular potentials (Ve) in the clefts may be different from 0. Therefore, the circuit of Figure 1A was completed with a T-shaped network of two series axial resistances, each ½Rcl, and a radial cleft resistance Rradial connecting the cleft to the bulk extracellular space (cleft model, Figure 1B). Assuming that the cleft has a cylindrical shape, Rcl was set to ρext · w/πr2, where ρext is the extracellular resistivity and w the cleft width. According to Katz,18 the radial resistance was derived as ρext/8πw. A value of 150 Ω · cm was used for ρext.
To evaluate the consequences of different subcellular distributions of NaChs on conduction, the maximal conductance for INa (gNa,max, representing the number of available NaChs in a given patch) was redistributed within the cells while the total number of NaChs per cell (∑gNa,max) was kept constant. The following distribution patterns were studied: (1) gNa,max was distributed proportionally to patch area (uniform INa distribution); (2) gNa,max was set to 50% ∑gNa,max in each disk patch of the cell and to 0 in the axial patches (100% junctional INa localization); and (3): gNa,max was set to 25% ∑gNa,max in each disk patch and the remaining 50% was distributed uniformly over the axial patches (50% junctional INa localization).
By applying Kirchhoff’s current law at every node and assuming that the LRd currents do not change over an arbitrarily small time interval Δt (ie, between time t and time t+Δt), we obtained a system of coupled first-order linear differential equations with constant coefficients, which was solved for Vm,Vi, and Ve by diagonalization. After solving for potentials at time t+Δt, the LRd currents were recomputed4,15 and the procedure was iterated. A fixed Δt of 0.005 ms was used. Results did not differ with a smaller Δt. Propagating APs were initiated in cell 1; θ was computed by linear regression of the activation times (defined as the time when Vi reached -30 mV) of cells 17 to 48.
Results
Immunocytochemical Staining of Cardiac Cell Cultures
A typical example of the subcellular distribution of rat heart NaChs (rH1) in cardiac cell cultures is shown in Figure 2A. Similar to intact tissue, NaChs were clustered at the sites of cell-to-cell appositions where they colocalized with gap junctions as shown by double-labeling with anti-Cx43 antibodies (Figures 2B and 2C). Control experiments (not shown), where anti-rH1 antibodies were preadsorbed to immunizing peptides, failed to produce a positive signal at the IDs whereas the nuclei still showed an increased level of fluorescence. This suggests that the labeling of nuclei, as evident in Figure 2A, was unspecific.
Figure 2.
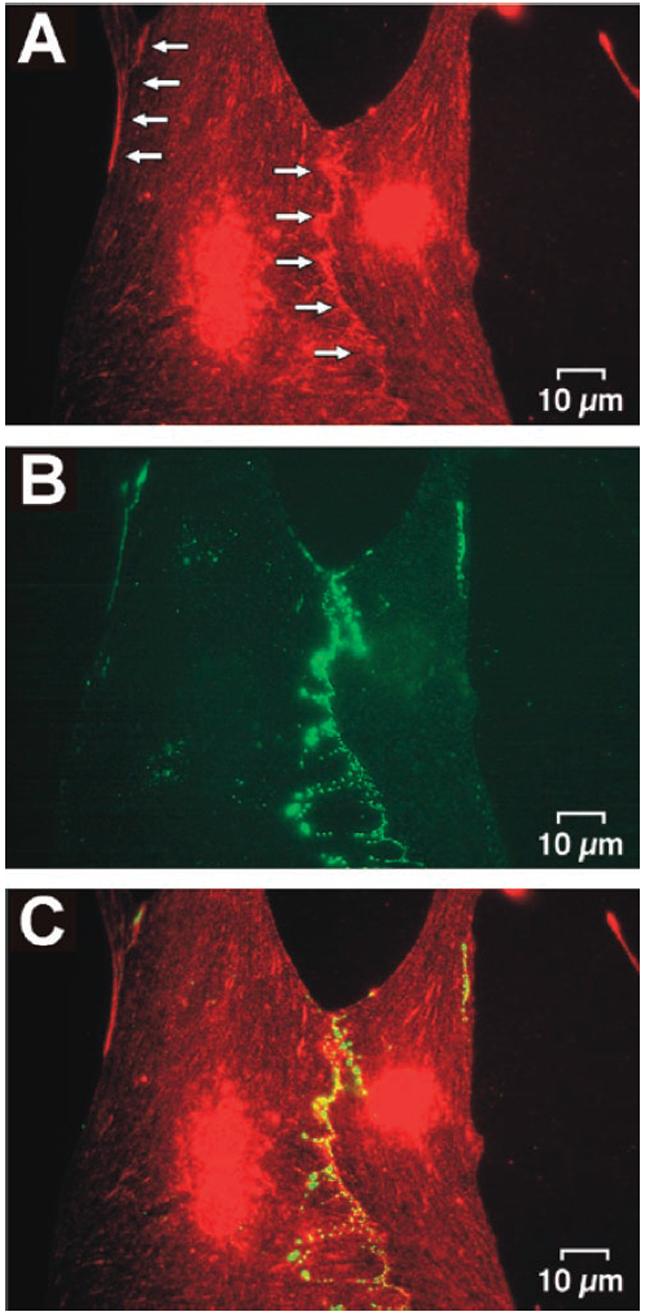
Double-labeling immunocytochemistry for NaChs and Cx43 in a culture of neonatal rat ventricular myocytes. A, NaChs. The increase in fluorescence along the boundaries between cells (arrows) indicates a local clustering of NaChs at the sites of cell-to-cell appositions. The concomitant increase in fluorescence in the region of the nuclei is most likely nonspecific (see text). B, Cx43. The subcellular distribution of Cx43 in the same preparation is highly correlated to that of NaChs suggesting a high degree of colocalization of the two channels as illustrated by superposition of the images (C).
Mathematical Modeling of Conduction
Because θ primarily depends on the degree of gap-junctional coupling,4 we evaluated steady-state θ for gap-junctional conductances ranging from 0 to 2.534 μS (normal value, 100%).
First, we assessed the dependence of θ on coupling under the assumption that NaCh distribution is uniform and that extracellular currents and potentials in the intercellular clefts do not influence conduction (noncleft model). As shown previously,4 θ decreased with decreasing coupling from ≈55 cm/s at 2.534 μS (100% of normal) to <1 cm/s at 0.00634 μS (0.25% of normal). When we assumed that NaChs are localized at cell-cell junctions but that cleft currents and potentials do not influence conduction, we observed only a minimal relative increase of θ (<1.5%). When we assumed that NaChs are distributed uniformly but that extracellular cleft currents and potentials can modulate conduction (cleft model), we observed a very slight reduction of θ (<0.5%) over the entire range of cleft widths investigated (20 to 1000 nm).
Localization of INa in Intercalated Disks Modulates Conduction Through Cleft Currents and Potentials
In contrast to the negligible effects described above, conduction was prominently influenced by cleft currents and potentials under the dual assumption that INa is concentrated at IDs and that cleft currents and potentials are significant (cleft model). In Figure 3, θ was evaluated for 100% junctional INa as a function of cleft width for various degrees of coupling. In wide clefts (>500 nm), there was only a negligible effect on θ when compared with the noncleft model. However, for narrower clefts, the following effects were observed. For higher degrees of coupling (50% to 100%), θ became progressively and monotonically slower when cleft width was narrowed. For medium-range degrees of coupling (3% to 10%), a biphasic behavior was observed: with decreasing cleft width, θ exhibited a progressive increase followed by a decrease with further cleft narrowing. For low degrees of coupling (0.3% to 1%), this initial increase was substantial and occurred abruptly at cleft widths of 40 to 50 nm.
Figure 3.
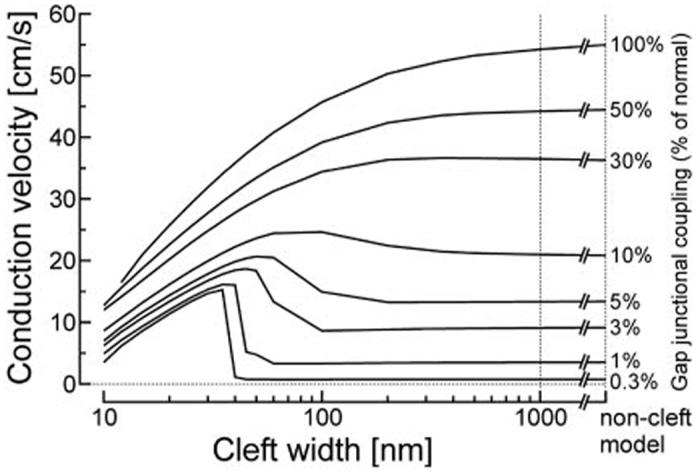
θ as a function of cleft width, evaluated for various degrees of coupling (labels on the right), for 100% junctional INa. The rightmost points of the curves correspond to θ in the non-cleft model with 100% junctional INa.
This behavior indicates that the dynamic interactions between INa and cleft potentials cause two opposing phenomena: at normal to moderately reduced gap-junctional coupling, these interactions result in an impairment of conduction, whereas, under conditions of substantially reduced gap-junctional conductance, they result in an enhancement of conduction.
Impairment of Conduction at Normal to Moderately Reduced Coupling
For 100% junctional INa localization and normal gap-junctional coupling, θ was 55.0 cm/s in the noncleft model. When 45-nm clefts were introduced, θ decreased to 37.3 cm/s. This slowing phenomenon was specifically investigated as illustrated in Figure 4. Conduction was first simulated in the 64-cell strand using the noncleft model. Then, a single 45-nm cleft was introduced at the junction between the two central cells (32 and 33) while all other junctions were maintained with the noncleft formulation. Before introduction of the cleft at the central junction (Figure 4A, top), steady-state conduction was, as expected, discontinuous at the cellular level and characterized by local conduction delays across the junctions. The high INa density at the junctional membranes (peak: 3850 μA/μF, an order of magnitude greater than for uniform INa distribution) reflects the greatly elevated number of locally available NaChs. However, peak INa integrated over a given cell was not different between uniform and 100% junctional INa repartitions.
Figure 4.
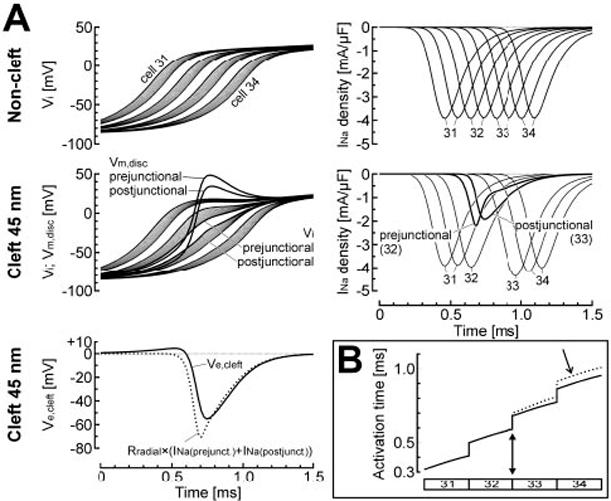
Inhibitory effects of cleft potentials on conduction, for normal gap-junctional coupling and 100% junctional INa. A, top, AP upstrokes (left) and INa density (right) in the 4 central cells (31 to 34) when the noncleft model was used for all junctions. Middle and bottom left, Same simulation, except that a 45-nm cleft was introduced at the junction between cells 32 and 33. As a result of the large negative extracellular cleft potential at this junction (Ve,cleft, solid line in bottom left panel), the transmembrane potential of the pre- and postjunctional disk membranes (middle left panel, Vm,disk) exhibited a large overshoot that led to a reduction of the driving force for INa and consequently to a large reduction of INa itself (middle right panel). The dotted curve in the bottom left panel indicates the potential that would have been induced across the radial cleft resistance by a current equal to INa (prejunctional)+INa (postjunctional). B, Activation profiles of cells 31 to 34 for the two simulations shown in panel A (solid curve: noncleft model; dotted curve: 45-nm cleft between cells 32 and 33, filled arrowhead). Note the conduction delay (arrow) resulting from the reduction of INa at the central cleft.
When the 45-nm cleft was introduced at the central junction (Figure 4A, middle and bottom), INa was associated with a current flowing from the bulk extracellular space through Rradial. This current is necessary to maintain electro-neutrality of the cleft space and satisfies Kirchhoff’s current law in the model. It induced a large negative extracellular cleft potential (Ve,cleft) that peaked at -55 mV. Comparison between Ve,cleft and the potential difference that would have been induced across Rradial by a current equal to INa summed over the pre- and postjunctional membranes (Figure 4A, bottom) confirms that INa was the major determinant of Ve,cleft. This large negative Ve,cleft resulted in depolarization of both pre- and postjunctional transmembrane potentials (Vm) that largely overshot Vm of adjacent patches. This prominent overshoot brought Vm close to the sodium reversal potential, ENa (≈+66 mV). As a consequence, the driving force for INa (ENa-Vm) was greatly reduced, attenuating INa even before voltage-dependent inactivation had started. Therefore, the large Ve,cleft resulted in a ≈50% reduction of INa in the junctional membranes (Figure 4A, middle right).
In cell 33, this “self-attenuation” of INa resulted in a reduction of the current source, and, consequently, in slower upstrokes and delayed distal activation of that cell. As shown in Figure 4B, conduction beyond the central cleft progressed with a delay compared with the conduction pattern observed when the noncleft model was used for all junctions. Thus, under conditions of normal coupling and 100% junctional INa, conduction is impaired in the cleft model due to self-attenuation of INa.
Enhancement of Conduction at Greatly Reduced Gap-Junctional Coupling
The same approach (modeling a 45-nm cleft at the central junction in the 64-cell strand while using the noncleft model for the other junctions) was used in Figure 5 to investigate the acceleration of conduction when coupling was 3% of normal. In the noncleft model, θ was slow (9.1 cm/s); θ increased to 18.7 cm/s when 45-nm clefts were introduced at all junctions. The top panels of Figure 5A illustrate conduction in the noncleft model. Because of the low gap-junctional conductance, charge generated by transmembrane currents in a given cell remained essentially confined within that cell. This resulted in an almost simultaneous activation of the entire cell. Furthermore, slow conduction was characterized by long intercellular conduction delays (>1 ms). Peak INa was high in the junctional patches (3850 μA/μF) and was not different from peak INa observed during normal coupling (see Figure 4A, top).
Figure 5.
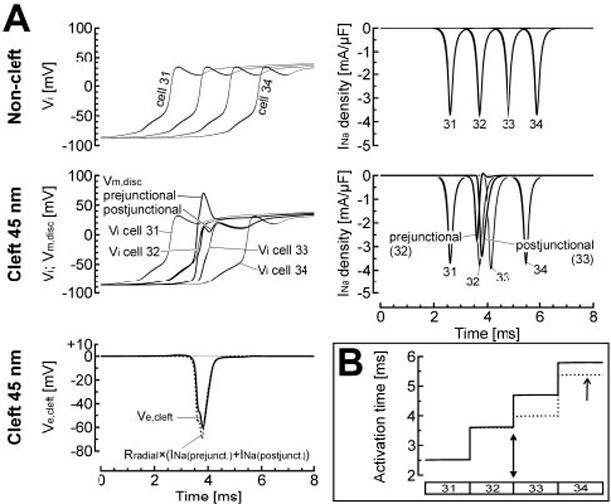
Potentiating effects of cleft potentials on conduction, for reduced gap-junctional coupling (3% of normal) and 100% junctional INa. A, top, AP upstrokes (left) and INa density (right) in the 4 central cells (31 to 34) when the noncleft model was used for all junctions. Middle and bottom left, Same simulation, except that a 45-nm cleft was introduced at the junction between cells 32 and 33. The large negative extracellular cleft potential at this junction (Ve,cleft, solid line in bottom left panel), induced by INa at the prejunctional membrane, resulted in a net depolarization of the postjunctional membrane (Vm,disk postjunctional) and thus in suprathreshold activation of INa in the postjunctional membrane (postjunctional, middle right panel). This activation led to earlier depolarization of cell 33. The dotted curve in the bottom left panel indicates the potential that would have been induced across the radial cleft resistance by a current equal to INa (prejunctional)+INa (postjunctional). B, Activation profiles of cells 31 to 34 for the two simulations shown in panel A (solid curve: noncleft model; dotted curve: 45-nm cleft between cells 32 and 33, filled arrowhead). Note the acceleration of conduction (arrow) at the junction between cells 32 and 33 when the 45-nm cleft was introduced.
When a 45-nm cleft was introduced between cells 32 and 33 only (Figure 5A, middle and bottom), INa was again associated with a current flowing from the bulk extracellular space through Rradial. Because of the junctional INa localization, this current was very prominent and induced a large negative Ve,cleft that peaked at -60 mV. Again, as shown in Figure 5A (bottom), comparison between Ve,cleft and the potential difference that would have been induced across Rradial by a current equal to INa summed over the pre- and postjunctional membranes confirms that INa was the major determinant of Ve,cleft. Also, the negative Ve,cleft led to a large overshoot of Vm of the prejunctional patch. Through the same mechanism as described previously (self-attenuation due to a reduction of the driving force), INa in the prejunctional membrane was reduced by ≈30%.
Importantly, the negative Ve,cleft also led to a synchronous and suprathreshold depolarization of Vm in the postjunctional membrane, where it resulted in voltage-gated activation of INa. Compared with the noncleft model, this activation occurred substantially earlier. As shown in Figure 5B, the early activation of postjunctional INa resulted in earlier activation of cell 33 and of more distal cells. Therefore, intercellular interactions through Ve,cleft were characterized by both self-attenuation of INa and early activation of postjunctional INa. Whereas reduction of INa alone would have resulted in conduction slowing through a reduction of source current, the effect of early activation prevailed and resulted in net acceleration of conduction.
The interaction between these two mechanisms, depending on coupling and cleft width, explains the biphasic behavior of θ depicted in Figure 3. Reducing cleft width increased the amplitude of Ve,cleft and thus promoted self-attenuation, thereby slowing conduction. However, the triggering of an AP in the postjunctional membrane by Ve,cleft explains the abrupt increase of θ at low degrees of coupling and narrow cleft widths.
Dependence of Velocity on Gap-Junctional Coupling Is Attenuated by the Preferential Localization of Sodium Channels at Intercalated Disks
Figure 6 summarizes the dependence of θ on the degree of coupling. Velocities obtained with 100% junctional INa in the noncleft model are indicated by solid curves. Dotted curves indicate θ for 100% junctional INa when 35-nm clefts were incorporated in the entire strand. Although recent work13 strongly suggests that most of INa is conducted through the Nav1.5 channel isoform, which is located in IDs, we nevertheless examined the possibility that a fraction of INa enters the cell through the nonjunctional membrane as well. Therefore, simulations were also performed with only 50% junctional INa localization. The results for this 50% localization and using 10-nm clefts are indicated by dashed curves.
Figure 6.
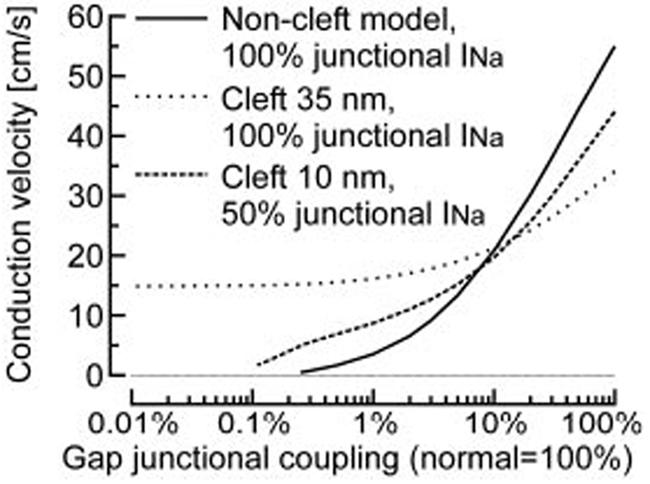
θ as a function of gap-junctional coupling. Comparison of the noncleft model with 100% junctional INa, the model with 35-nm clefts and 100% junctional INa, and the model with 10-nm clefts and 50% junctional INa. Conduction was supported at lower degrees of coupling when the cleft model was used. For 100% junctional INa and 35-nm clefts, conduction plateaus at ≈15 cm/s as coupling is reduced below 1% and is sustained even in the absence of coupling.
For the cleft model and for both 100% and 50% junctional INa, the slope of the curve relating θ to gap-junctional coupling was smaller compared with the curve characterizing the noncleft model. This indicates that the dependence of θ on coupling was attenuated: a given reduction of coupling led to a smaller reduction of θ when clefts were incorporated. Compared with the noncleft model, when the degree of coupling was >10%, self-attenuation in the cleft model resulted in slower conduction velocities for both 100% and 50% junctional INa. However, when coupling was reduced below ≈10%, conduction was faster, revealing the early activation of INa mediated by cleft potentials.
Is Conduction Possible Without Gap-Junctional Coupling?
In Figure 6, the plateau reached by θ at very low degrees of coupling for 100% junctional INa and 35-nm clefts suggests that under these conditions, conduction might be possible during complete uncoupling (0%). This possibility was confirmed in an additional simulation (not shown) where θ was 14.8 cm/s. However, conduction without coupling was only possible when the fraction of junctional INa was close to 100%. Nevertheless, when this fraction was 50%, conduction was still possible at very low degrees of coupling, for which propagation failed in the noncleft model. Whereas the critical degree of coupling before conduction failure was 0.25% of normal in the noncleft model (critical θ: 0.46 cm/s), this value decreased to 0.11% when 10-nm clefts were incorporated (critical θ: 1.67 cm/s). This therefore suggests that under conditions of very low coupling, interactions through Ve,cleft may be crucial for the success of conduction.
Discussion
Irrespective of the spatial distribution of INa at the subcellular level, our study confirms the dominant role of electrical coupling through connexons in cardiac conduction. However, the results indicate that the preferential localization of NaChs in IDs, which was shown in previous studies10-13 and confirmed here in cell cultures, may modulate the dependence of θ on gap-junctional coupling. This modulation is due to cleft potentials based on large cleft currents. These findings provide a functional explanation and suggest a physiological role for the localization of NaChs in IDs.
Mechanisms of Cardiac Conduction
It is widely accepted that connexons are the structures forming the electrical connections between the cytoplasmic compartments of adjacent cardiomyocytes, thus permitting the flow of local circuit current and the propagation of the AP. This concept was challenged by Sperelakis and colleagues, who proposed that cardiac conduction could occur without the need for low-resistance connections between cells through a mechanism that they called “electric field mechanism.”19,20 In a recent review,21 Sperelakis and McConnell pointed out that the increased density of NaChs in IDs would facilitate this mechanism and suggested the possibility that cardiac conduction could be based on multiple mechanisms acting concurrently (electric field, current through gap junctions, accumulation of K+ in the cleft). The principle of the electric field mechanism corresponds to the early activation of postjunctional INa described in the present study: the negative cleft potential during firing of the prejunctional membrane acts to depolarize the postjunctional membrane to threshold. In the model studies of Sperelakis and colleagues, θ ranged up to 32 cm/s in the absence of gap-junctional coupling under conditions where the excitability of the junctional membranes was increased.20 In the present study, where a realistic model of the ionic currents was used, a maximal θ of 14.8 cm/s was observed in the absence of coupling and required 100% INa localization at the IDs. Yet, both velocities are too low compared with normal velocities observed in ventricular tissue (>50 cm/s). The electric field mechanism alone therefore cannot account for normal cardiac conduction.
The possibility of impulse transmission between two cardiac cells in the absence of resistive coupling was also investigated by Hogues et al.22 In their model study, where only a uniform distribution of ionic currents was investigated, they observed changes in the cleft potential that were too small to permit activation of the downstream cell under normal conditions and they estimated the contribution of electric field transmission to be ≈30 to 130 times smaller than transmission through gap-junctional resistive coupling. They nevertheless discussed the possibility that, under conditions of reduced gap-junctional coupling, the electric field mechanism could provide an important contribution to transmission.
To our knowledge, the present study is the first to simulate cardiac conduction in a model where both resistive current through gap junctions and the electric field mechanism were incorporated. Moreover, motivated by the immunocytochemical findings of other researchers and our own, we investigated the effects of uniform versus nonuniform INa distribution. We conclude that the degree of gap-junctional coupling is the principal determinant of conduction, but that in the presence of electric field interactions through cleft potentials, preferential localization of INa in IDs modulates the dependence of θ on coupling. Obviously, the latter statement will await experimental support. However, because of the technical difficulty of measuring the extracellular potential in very narrow intercellular clefts, experimental approaches appear very challenging.
Study Limitations
The principal limitation of the present study is the simplification that was made concerning cleft shape and the fact that the cleft potential was discretized only along the fiber axis. The shape of the cleft is known to be tortuous and irregular.23 Increased tortuosity of the cleft is expected to be associated with an increased radial resistance Rradial. In Figures 3 and 6, this would translate into a leftward shift of the curves without changing their general behavior. The results of Hogues et al,22 who discretized cleft potential at several nodes in the radial direction, suggest that tortuosity could account for an increase in Rradial by a factor of ≈2.
A major unknown is obviously the cleft width, w. This uncertainty motivated us to use different values of w spanning a range from 20 to 1000 nm. Clefts are certainly not wider than 1000 nm (such clefts would be visible using optical microscopy). In gap-junction plaques, connexons determine an intermembrane distance of ≈5 nm,24 a value that would define the absolute minimal value for w. These plaques, however, occupy only a small fraction of the ID area (in the range of 1%). Between them, the cleft is wider.23 In our study, the most prominent effects of electric field interactions were observed in the range of w=20 to 50 nm when gap-junctional coupling was reduced; however, these effects fell dramatically for w>50 nm. Therefore, to better evaluate the possibility that these interactions play a role in vivo, it would be of greatest value to obtain detailed morphological information about cleft morphology and tortuosity and to model these microstructural details in a future study.
Another issue that arises is the question of a possible depletion of Na+ in the cleft, which would decrease the driving force for INa. Assuming that no ionic exchange occurs between the cleft and the bulk extracellular space, then all Na+ ions leave the cleft through junctional NaChs. Under this assumption, Na+ depletion can be estimated by integration of junctional INa. In our model, it would amount to ≈12 mmol/L in a 20-nm cleft and decrease the Na+ Nernst potential by only ≈3 mV. This value could however be larger if the complicated cleft microstructure gave rise to microdomains between which Na+ diffusion would be restricted. Modeling microstructural compartments and ionic exchanges between them would provide valuable information about ion concentrations in the cleft.
Finally, an uncertainty remains concerning the fraction of functional NaChs that are located in IDs. Although recent work13 suggests that this fraction might be substantial, more research will be necessary to identify not only the location of different NaCh isoforms but also to quantify their respective functional contribution to total INa.
Conduction in Cx43-Deficient Animals
Cx43 knockout mice are used to study the relationship between the degree of coupling and conduction characteristics. Although some research groups reported a θ ≈30% slower in Cx43 knockout heterozygous myocardium compared with wild-type tissue,25,26 others did not observe any significant differences.27,28 More intriguing is the observation that in cardiac-restricted Cx43 knockout mice, where Cx43 expression was reduced by 95%, θ was slowed by only ≈50%.29
In cardiac fiber, total axial resistance (which determines θ) is the sum of its myoplasmic and gap-junctional contributions. In the LRd model, the myoplasmic contribution is 50% (for normal coupling). Because murine myocytes have a smaller diameter compared with the LRd model cell, it is possible that the myoplasmic contribution is higher and that the contribution of Cx43 is relatively lower. This could explain the relatively small effects of reduced Cx43 expression on conduction in some studies. However, the modulation of θ by the electric field mechanism might provide an additional explanation.
Clinical and Physiological Implications
Gap-junctional uncoupling is a hallmark of short- and long-term ischemia, and it is well known that slow conduction favors the occurrence of reentrant arrhythmias. Early INa activation by cleft potentials might act to protect the myocardium against very slow conduction (<1 cm/s) and microreentry. However, it could also make moderately slow conduction (a few centimeters per second) more robust, which would then promote arrhythmias.
Under conditions of normal coupling, the physiological role of the self-attenuation of INa in IDs is yet unclear, because it would act to depress conduction. Intuitively, one would think that the function and distribution of NaChs would be such as to maximize θ. In this study, we only investigated conduction in the direction of fiber orientation. In the direction transverse to myocardial fibers, where the pattern of cell-to-cell coupling is different, conduction is slower. To which extent the cleft-potential mechanisms are involved in transverse propagation and, ultimately, in determining the ratio of longitudinal to transverse conduction velocities needs to be further investigated. Although beyond the scope of this study, this question should be evaluated in two- or three-dimensional models of cardiac tissue.
Acknowledgments
This work was supported by the Swiss National Science Foundation (to J.P.K. and grant 31-50516.97 to S.R.), the Swiss Foundation for Stipends in Biology and Medicine (to J.P.K.), and the NIH National Heart, Lung, and Blood Institute (grants R01-HL49054 and R37-HL33343 to Y.R.).
References
- 1.Cranefield PF. The Conduction of the Cardiac Impulse. Futura Publishing Company; New York, NY: 1975. [Google Scholar]
- 2.Cole WC, Picone JB, Sperelakis N. Gap junction uncoupling and discontinuous propagation in the heart: a comparison of experimental data with computer simulations. Biophys J. 1988;53:809–818. doi: 10.1016/S0006-3495(88)83160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohr S, Kucera JP, Kléber AG. Slow conduction in cardiac tissue, I: effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circ Res. 1998;83:781–794. doi: 10.1161/01.res.83.8.781. [DOI] [PubMed] [Google Scholar]
- 4.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue: roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 5.Quan W, Rudy Y. Unidirectional block and reentry of cardiac excitation: a model study. Circ Res. 1990;66:367–382. doi: 10.1161/01.res.66.2.367. [DOI] [PubMed] [Google Scholar]
- 6.Rohr S, Salzberg BM. Discontinuities in action potential propagation along chains of single ventricular myocytes in culture: multiple site optical recording of transmembrane voltage (MSORTV) suggests propagation delays at the junctional sites between cells. Biol Bull Mar Biol Lab. 1992;183:342–343. doi: 10.1086/BBLv183n2p342. [DOI] [PubMed] [Google Scholar]
- 7.Fast VG, Kléber AG. Microscopic conduction in cultured strands of neonatal rat heart cells measured with voltage-sensitive dyes. Circ Res. 1993;73:914–925. doi: 10.1161/01.res.73.5.914. [DOI] [PubMed] [Google Scholar]
- 8.Joyner RW. Effects of the discrete pattern of electrical coupling on propagation through an electrical syncytium. Circ Res. 1982;50:192–200. doi: 10.1161/01.res.50.2.192. [DOI] [PubMed] [Google Scholar]
- 9.Rudy Y, Quan W. A model study of the effects of the discrete cellular structure on electrical propagation in cardiac tissue. Circ Res. 1987;61:815–823. doi: 10.1161/01.res.61.6.815. [DOI] [PubMed] [Google Scholar]
- 10.Cohen SA, Levitt LK. Partial characterization of the rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circ Res. 1993;73:735–742. doi: 10.1161/01.res.73.4.735. [DOI] [PubMed] [Google Scholar]
- 11.Cohen SA. Immunocytochemical localization of rH1 sodium channel in adult rat heart atria and ventricle. Presence in terminal intercalated discs. Circulation. 1994;94:3083–3086. doi: 10.1161/01.cir.94.12.3083. [DOI] [PubMed] [Google Scholar]
- 12.Rohr S, Flückiger R, Cohen SA. Immunocytochemical localization of sodium and calcium channels in cultured neonatal rat ventricular cardio-myocytes. Biophys J. 1999;76:A366. Abstract. [Google Scholar]
- 13.Maier SKG, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–4078. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in inter-calated discs modulates cardiac conduction. Circulation. 2002;106(suppl II):II-88. doi: 10.1161/01.res.0000046237.54156.0a. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential, I: simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 16.Faber GM, Rudy Y. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faber GM. Cardiac Bioelectricity Research and Training Center (CBRTC) The Luo-Rudy dynamic (LRd) model of the mammalian ventricular action potential. Available at: http://www.cwru.edu/med/CBRTC/LRdOnline/. Accessed October 2002.
- 18.Katz B. Nerve, Muscle and Synapse. McGraw-Hill; New York, NY: 1966. [Google Scholar]
- 19.Sperelakis N, Mann JE. Evaluation of electric field changes in the cleft between excitable cells. J Theor Biol. 1977;64:71–96. doi: 10.1016/0022-5193(77)90114-x. [DOI] [PubMed] [Google Scholar]
- 20.Mann JE, Sperelakis N, Ruffner JA. Alteration in sodium channel gate kinetics of the Hodgkin-Huxley equations applied to an electric field model for interaction between excitable cells. IEEE Trans Biomed Eng. 1981;28:655–661. doi: 10.1109/TBME.1981.324756. [DOI] [PubMed] [Google Scholar]
- 21.Sperelakis N, McConnell K. Electric field interactions between closely abutting excitable cells. IEEE Eng Med Biol Mag. 2002;21:77–89. doi: 10.1109/51.993199. [DOI] [PubMed] [Google Scholar]
- 22.Hogues H, Leon LJ, Roberge FA. A model study of electric field interactions between cardiac myocytes. IEEE Trans Biomed Eng. 1992;39:1232–1243. doi: 10.1109/10.184699. [DOI] [PubMed] [Google Scholar]
- 23.Forbes MS, Sperelakis N. Intercalated discs of mammalian heart: a review of structure and function. Tissue Cell. 1985;17:605–648. doi: 10.1016/0040-8166(85)90001-1. [DOI] [PubMed] [Google Scholar]
- 24.Yeager M. Structure of cardiac gap junction intercellular channels. J Struct Biol. 1998;121:231–245. doi: 10.1006/jsbi.1998.3972. [DOI] [PubMed] [Google Scholar]
- 25.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99:1991–1998. doi: 10.1172/JCI119367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eloff BC, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE, Rosenbaum DS. High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc Res. 2001;51:681–690. doi: 10.1016/s0008-6363(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 27.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–1375. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 28.Vaidya D, Tamaddon HS, Lo CW, Taffet SM, Delmar M, Morley GE, Jalife J. Null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of mouse embryonic development. Circ Res. 2001;88:1196–1202. doi: 10.1161/hh1101.091107. [DOI] [PubMed] [Google Scholar]
- 29.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]