

Abstract
Spinophilin (SPL) and neurabin (NRB) are structurally similar scaffolding proteins with several protein binding modules, including actin and PP1 binding motifs and PDZ and coiled-coil domains. SPL also binds regulators of G protein signaling (RGS) proteins and the third intracellular loop (3iL) of G protein-coupled receptors (GPCRs) to reduce the intensity of Ca2+ signaling by GPCRs. The role of NRB in Ca2+ signaling is not known. In the present work, we used biochemical and functional assays in model systems and in SPL−/− and NRB−/− mice to show that SPL and NRB reciprocally regulate Ca2+ signaling by GPCRs. Thus, SPL and NRB bind all members of the R4 subfamily of RGS proteins tested (RGS1, RGS2, RGS4, RGS16) and GAIP. By contract, SPL, but not NRB, binds the 3iL of the GPCRs α1B-adrenergic (α1BAR), dopamine, CCKA, CCKB and the muscarinic M3 receptors. Coexpression of SPL or NRB with the α1BAR in Xenopus oocytes revealed that SPL reduces, whereas NRB increases, the intensity of Ca2+ signaling by α1BAR. Accordingly, deletion of SPL in mice enhanced binding of RGS2 to NRB and Ca2+ signaling by αAR, whereas deletion of NRB enhanced binding of RGS2 to SPL and reduced Ca2+ signaling by αAR. This was due to reciprocal modulation by SPL and NRB of the potency of RGS2 to inhibit Ca2+ signaling by αAR. These findings suggest a novel mechanism of regulation of GPCR-mediated Ca2+ signaling in which SPL/NRB forms a functional pair of opposing regulators that modulates Ca2+ signaling intensity by GPCRs by determining the extent of inhibition by the R4 family of RGS proteins.
Keywords: Ca2+ signaling, pair, regulation, spinophilin/neurabin
Introduction
Ca2+ signaling regulates virtually all cellular functions on timescales from milliseconds to days (Berridge et al, 2003), and in all cellular compartments (Berridge et al, 2003; Kiselyov et al, 2003; Rizzuto and Pozzan, 2006). This requires compartmentalization and extraordinary specificity in Ca2+ signaling. Several mechanisms have been identified to contribute to Ca2+ signaling specificity that affect all steps in the Ca2+ signaling pathway (Kiselyov et al, 2003). One of these mechanisms is generating Ca2+ oscillations of defined amplitude and frequency. Ca2+ oscillation amplitude and, in particular, frequency, are determined by the intensity of the stimulus (Berridge, 1997; Kiselyov et al, 2003). Hence, controlling intensity of the stimulus controls the type of the Ca2+ signal evoked by the receptors.
A ubiquitous mechanism by which stimuli are translated into a [Ca2+]i response is activation of G protein-coupled receptors (GPCRs). GPCRs are the largest protein family in mammals, and decode stimuli as diverse as light, pheromones and peptide hormones. The GPCR complex that generates a Ca2+ signal includes the receptor, a heterotrimeric G protein composed of Gq class α subunit and Gβγ (Gilman, 1987; Freissmuth et al, 1989; Dessauer et al, 1996), PLCβ (Rebecchi and Pentyala, 2000) and regulators of G protein signaling (RGS) proteins (Ross and Wilkie, 2000; Ishii and Kurachi, 2003). At the resting state, Gα is bound with GDP and the Gα·GDP binds Gβγ to form Gαβγ. Activation of GPCRs includes receptor catalyzed GDP–GTP exchange on Gα, leading to dissociation of Gαβγ to form Gα·GTP and Gβγ that are free to interact with and activate their respective effectors.
Termination of the GPCR stimulated state involves hydrolysis of GTP by the intrinsic Gα GTPase activity. The Gα GTPase activity is accelerated by the RGS proteins. The RGS proteins have a conserved GTPase activating protein (GAP) domain and divergent C- and N-termini (Ishii and Kurachi, 2003; Ross and Wilkie, 2000). The function of the C-terminus of most RGS proteins that participates in Ca2+ signaling is not well understood, although in several cases it acts as a scaffold. The N-terminal domain functions in membrane targeting and confers receptor recognition (Zeng et al, 1998) to mediate the receptor-specific action of several RGS proteins (Xu et al, 1999). Regulation by RGS proteins is a primary mechanism of generating receptor-specific signals (Luo et al, 2001; Kiselyov et al, 2003).
In an effort to understand how RGS proteins communicate with GPCRs, we reported that the N-terminal domain of RGS2 binds to the scaffolding protein spinophilin (SPL) and that SPL binds several members of the R4 subfamily of RGS proteins and GAIP. Most notably, SPL also binds the third intracellular loop (3iL) of several GPCRs (Smith et al, 1999; Richman et al, 2001). SPL mediates the inhibition of GPCR signaling by RGS2, and therefore reduces the intensity of the signal conveyed by GPCRs (Wang et al, 2005).
Neurabin (NRB) is an SPL homologue, the function of which in regulation of GPCR signaling is not known. Recent work showed that NRB and SPL have distinct roles in dopamine-mediated neuronal plasticity, whereby deletion of SPL results in the loss of induction of long-term depression, whereas deletion of NRB results in the loss of long-term potentiation (Allen et al, 2006), raising the possibility that SPL and NRB may differentially affect their target proteins. SPL and NRB have multiple protein–protein interacting domains and a protein phosphatase 1 (PP1) binding sequence (Allen et al, 1997; Satoh et al, 1998; Ouimet et al, 2004). The domains in NRB and SPL that bind PP1 and RGS proteins are highly homologous, whereas the SPL domain that binds the 3iL of GPCRs has little homology with the comparable NRB domain (Wang et al, 2005). This raises the possibility that the actions of SPL and NRB are reciprocal, and that SPL and NRB cooperate as a functional pair of opposing regulators to tune the intensity of Ca2+ signaling by GPCRs. We hypothesized that SPL recruits the signaling terminator, that is, the RGS protein, to GPCR signaling complexes to reduce signaling intensity, whereas NRB removes RGS proteins from the complexes to increase signaling intensity. We tested this hypothesis by biochemical and functional assays in model systems, and by using SPL−/− and NRB−/− mice. We report that SPL and NRB bind the same RGS proteins equally well. By contrast, SPL, but not NRB, binds the 3iL of several GPCRs, including the α1BAR. Coexpression of SPL or NRB with the α1BAR revealed that SPL reduces whereas NRB increases the intensity of Ca2+ signaling by α1BAR. Accordingly, deletion of SPL in mice reduces whereas deletion of NRB in mice enhances the potency of the RGS2 to inhibit Ca2+ signaling. Deletion of SPL also increases the binding of RGS2 to NRB. These findings suggest a novel mechanism of regulation of GPCR-mediated Ca2+ signaling in which the SPL/NRB functional pair regulates Ca2+ signaling intensity by determining the extent of inhibition of Ca2+ signaling by RGS proteins.
Results and discussion
NRB interacts with R4 subfamily RGS proteins to regulate Ca2+ signaling by α1BAR
To study the possible role of NRB in GPCR signaling, we compared the ability of NRB and SPL to bind members of the R4 subfamily of RGS proteins and the 3iL of several GPCRs, including the α1BAR, the binding of which to SPL has been extensively studied (Brady et al, 2003, 2005; Wang et al, 2004a, 2005). In Figure 1A, GST-RGS2 was used to pull down NRB in extracts prepared from human embryonic kidney 293 (HEK) cells expressing NRB and shows that NRB binds RGS2. The N-terminal domain of RGS2 (RGS2N) and RGS2 from which the N-terminal domain was deleted (ΔNRGS2) were used to determine the RGS2 domain that binds NRB. Figure 1A also shows that NRB specifically binds RGS2N, but not ΔNRGS2. Moreover, NRB binds all members of the R4 subfamily tested, RGS1, RGS4, RGS16 and to GAIP (Figure 1C). These findings are identical to those we reported for SPL (Wang et al, 2005).
Figure 1.
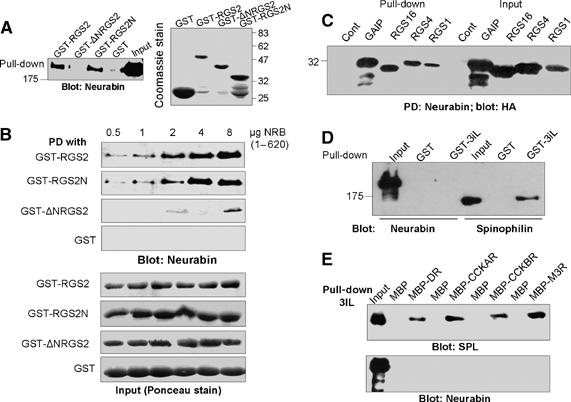
Binding of RGS proteins and GPCRs 3iL to SPL and NRB. Extracts prepared from HEK cells expressing Myc-tagged NRB were used to pull down NRB with GST-RGS2, GST-ΔNRGS2, GST-RGS2N or GST (A). The blots were probed with anti-Myc to detect NRB. The blot on the left is a coomassie stain of the inputs of the purified proteins used for the pull down. (B) Binding of GST-RGS2 constructs to recombinant His-NRB(1–620). The reaction media contained between 0.5–8 μg His-NRB(1–620) and 15 μg of either RGS2, RGS2N, ΔNRGS2 or GST. The complexes were captured on beads and analyzed for NRB with anti-NRB antibodies (upper blots), and for the respective GST constructs (lower blots) by panceau staining. (C) Respective HA-tagged RGS proteins were expressed in HEK cells and extracts prepared from these cells were used for pull down by His-NRB(1–620). (D) Extracts were prepared from HEK cells expressing Myc-NRB or Myc-SPL and the extracts were used for pull down with GST-3iL of the α1BAR. The same blot was used to probe for SPL and NRB since both are Myc tagged. (E) Extracts were prepared from HEK cells transfected with Myc-SPL or Myc-NRB and were incubated with MBP alone (controls) or the MBP-3iL of the dopamine receptor (MBP-DR), the cholecystokinin A receptor (MBP-CCKAR), MBP-CCKBR or the M3 muscarinic receptor (MBP-M3R). The MBP-tagged proteins were pulled down and blotted for SPL (upper blot) or NRB (lower blot).
Binding of purified, recombinant proteins were used to further test the specificity of RGS2 binding to NRB and to determine whether RGS2 directly interacts with NRB. NRB is a 1095-amino acid protein, with four coiled-coil domains that tends to show some nonspecific binding (see Figure 1A). Preliminary experiments showed that His-NRB(1–620) that lacks the coiled-coil domain binds RGS2 as well as full-length (FL)-NRB. Therefore, we prepared purified 6 × His-NRB(1–620) and tested its binding to GST-RGS2, GST-RGS2N, GST-ΔNRGS2 and GST. Figure 1B shows that RGS2 and RGS2N, but not ΔNRGS2 and GST, bind to His-NRB(1–620) and the binding is concentration dependent. These findings indicate that NRB directly binds the N-terminal domain of RGS2.
SPL was shown to bind the 3iL of the α-adrenergic (α1BAR) and dopamine receptors. To determine the binding of the 3iL to NRB, we prepared GST-3iL of the α1BAR and MBP-3iL of the dopamine, CCKA, CCKB and M3 muscarinic receptors. The MBP-3iL of these receptors was used since the GST-3iL did not fold well. The GST-3iL and MBP-3iL were used to pull down SPL and NRB from cell extracts expressing the respective proteins. Notably, Figure 1D shows that SPL, but not NRB, binds the 3iL of the α1BAR. Moreover, SPL but not NRB binds the 3iL of the dopamine, CCKA, CCKB and M3 receptors (Figure 1E).
The results in Figure 1 indicate that SPL and NRB similarly bind the N-terminal domain of RGS2 and bind other members of the R4 subfamily of RGS proteins, whereas, unlike SPL, NRB does not bind the 3iL of GPCRs. To determine the functional significance of these findings, we compared the effect of SPL and NRB on α1BAR-stimulated Ca2+ signaling in Xenopus oocytes. Stimulation of the oocytes with 100 nM epinephrine (Epi) activated a Gq-mediated Ca2+ signaling as revealed by activation of the native oocytes Ca2+-activated Cl− current (see Wang et al, 2005 for details). Figure 2A and C show that SPL, but not NRB, increased the maximal α1BAR-activated current by 2.6-fold. This was likely due to stabilization of α1BAR at the plasma membrane by SPL (Brady et al, 2003; Wang et al, 2004a, 2005), and is consistent with the binding results in Figure 1 in which SPL but not NRB binds to the α1BAR. Despite the increase in total current, SPL reduced the rate of activation of Ca2+ signaling by the α1BAR, with half maximal current attained at 3.9±0.4 s in the absence of SPL and at 14.2±2.2 s in the presence of SPL. By contrast, NRB increased the rate of α1BAR activation by reducing the time for half maximal current activation to 2.6±0.5 s (Figure 2B). Furthermore, measurement of the Epi dose response showed that SPL increased the EC50 for Epi from 17±4 to 193±37 nM and NRB reduced the EC50 to 2.3±1.1 nM (Figure 2D).
Figure 2.
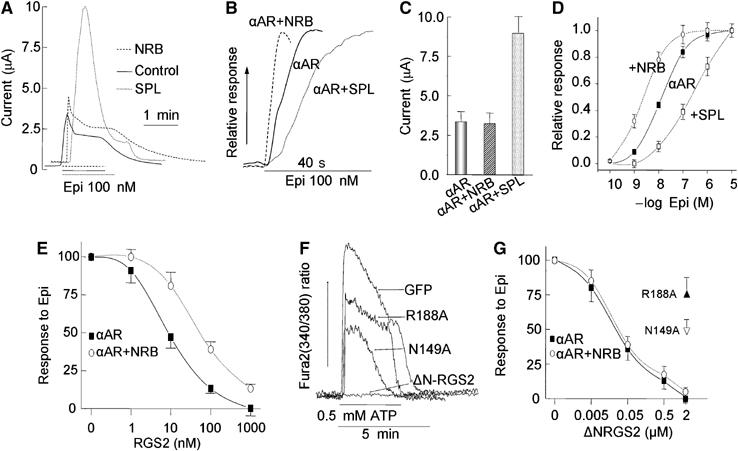
Effect of SPL and NRB on Ca2+ signaling by α1BAR in Xenopus oocytes. (A, B) Oocytes were transfected with α1BAR alone (solid traces), α1BAR+NRB (dashed traces) or α1BAR+SPL (dotted traces) and stimulated with 100 nM Epi while measuring the Ca2+-activated Cl− current. (A) Shows the time course of the overall Ca2+ signal and (B) Shows the initial rate of current increase as an indicator of the rate of cell stimulation. (C) The mean±s.e.m. of the peak current in 10 experiments similar to those in (A). (D) The dose response for Epi in oocytes expressing the α1BAR alone (•),α1BAR+NRB (○) or α1BAR+SPL (□). The results in (D) are the means±s.e.m. of the peak current from four similar experiments. (E) Oocytes expressing α1BAR alone (▪) or α1BAR+NRB (○) were injected with the indicated final concentrations of RGS2 and stimulated with 10 μM Epi. The results are the means±s.e.m. of the peak current from 4–8 experiments. (F) HEK cells grown on coverslips were transfected with 0.5 μg cDNA coding for eGFP alone, ΔNRGS2, ΔNRGS2(N149A) or ΔNRGS2(R188A). The cells were loaded with Fura2 and used to measure the Ca2+ increase in response to stimulation of the native P2Y2 receptors with 0.5 mM ATP. (G) Oocytes were transfected with α1BAR alone (▪) or α1BAR+NRB (○) and injected with 2 μM ΔNRGS2(N149A) or ΔNRGS2(R188A), or the indicated concentrations of ΔNRGS2. The oocytes were used to measure the response to stimulation with 10 μM Epi and the response is plotted as a function of RGS2 constructs concentration. The results are the mean±s.e.m. of the peak current from 4–8 experiments.
The results in Figures 1 and 2A–D are consistent with recruitment of RGS proteins to and away from the GPCR complex by SPL and NRB, respectively. This prediction was tested by measuring the effect of NRB on inhibition of α1BAR-evoked Ca2+ signaling by RGS2. Figure 2E shows that coexpression of NRB with the α1BAR reduces the potency of RGS2 in inhibiting Ca2+ signaling by α1BAR. For control experiments, we tested the effect of ΔNRGS2 GAP mutants ΔNRGS2(R188A) and ΔNRGS2(N149A). These mutants were shown to reduce binding of RGS2 and RGS4 to Giα and reduced acceleration of Giα GTPase activity in vitro (Druey and Kehrl, 1997; Heximer, 2004). Since the effect of these mutants, or of any RGS proteins GAP mutant, on Ca2+ signaling in intact cells was not reported, we first expressed ΔNRGS2 and the mutants in HEK cells and determined their effect of the native P2Y2-evoked Ca2+ signaling. Figure 2F shows that at the expression levels used, ΔNRGS2 completely inhibited Ca2+ signaling, whereas ΔNRGS2(R188A) and ΔNRGS2(N149A) inhibited Ca2+ signaling by only 35 and 50%, respectively (n⩾5). The same constructs were prepared as GST-fusion proteins and injected into oocytes expressing the α1BAR. The experiments in Figure 2G show that ΔNRGS2, which lacks the NRB binding domain (Figure 1A), similarly inhibited Ca2+ signaling by α1BAR in the presence and absence of NRB. Inhibition by ΔNRGS2 is likely mediated by the GAP activity of ΔNRGS2, which is conferred by the RGS domain and is independent of the N-terminal domain (Zeng et al, 1998; Ross and Wilkie, 2000). This was verified by showing that when injected into the oocytes, the GAP mutants ΔNRGS2(R188A) and ΔNRGS2(N149A) only partially inhibited Ca2+ signaling stimulated by α1BAR.
NRB regulates Ca2+ signaling by α1BAR in vivo
Reciprocal regulation of Ca2+ signaling by SPL and NRB requires that they compete for binding of RGS2 in vivo. To test this postulate, we first determined the amount of native SPL and NRB that can be co-immunoprecipitated with native RGS2 from brain extracts of wild-type (WT), SPL−/− and NRB−/− mice. For these, we tested the specificity of several anti-RGS2 antibodies in detecting and immunoprecipitating RGS2. Satisfactory results were obtained only with the chicken anti-RGS2 antibodies from GenWay, which detects recombinant RGS2 and RGS2 expressed in HEK cells. However, the native brain RGS2 runs slightly slower than the expressed RGS2. Nevertheless, the middle blot in Figure 3A shows that the anti-HA and anti-RGS2 antibodies recognize the same expressed protein band. In each of the experiments, a sample of HA–RGS2 was included to positively identify the RGS2.
Figure 3.
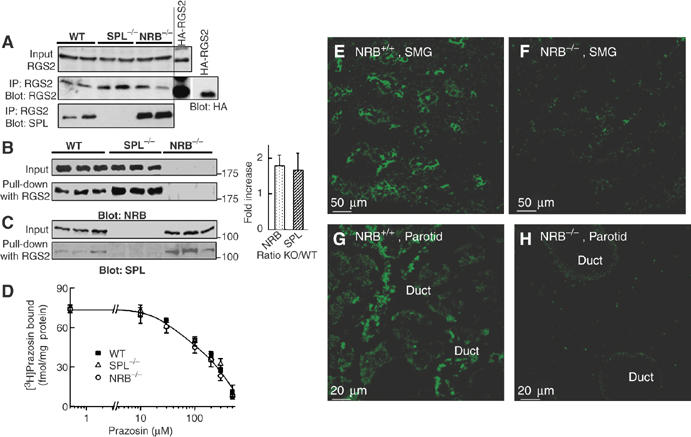
Pull down of native SPL and NRB by RGS2 and localization of NRB in salivary gland ducts. (A) Co-immunoprecipitation of SPL and RGS2. Extracts were prepared from the brains of WT, SPL−/− and NRB−/− mice, or from HEK cells transfected with HA-RGS2. Preliminary experiments showed that the anti-RGS2 antibodies recognized a band that ran slightly slower in brain extract than in transfected HEK cells. The specificity of the anti-RGS2 antibodies was verified by showing that they recognize recombinant RGS2 and the same band was recognized by anti-RGS2 and anti-HA in extract prepared from HEK cells transfected with HA-RGS2 (middle blot in panel A). The anti-RGS2 antibodies were then used to immunoprecipitate RGS2 from the indicated brain extracts and the precipitated proteins were probed for the co-immunoprecipitation of RGS2 and SPL. The upper blot is the RGS2 input, the middle blot is the immunoprecipitation of RGS2 and the bottom blot is the immunoprecipitation of SPL. Interaction of SPL and NRB with RGS2 was probed further by a pull-down assay. (B, C) Extracts prepared from the brains of three each of WT, SPL−/− and NRB−/− mice were used to pull down NRB (B) or SPL (C) with the same amount of GST-RGS2. The pull downs are summarized in the columns as the ratio of WT to that in the knock-out mice (WT/KO). (D) Binding of 3[H]prazosin to brain microsomes prepared from WT (▪), SPL−/− (Δ) and NRB−/− mice (○). (E–H) Localization of NRB in salivary glands was determined by immunolocalization. Frozen sections were prepared from the submandibular (SMG) (D, E) or parotid (F, G) glands of WT (D, F) or NRB−/− mice (panels E, G) and stained with anti-NRB antibodies. Note that NRB is expressed at high levels at the apical pole of all ducts.
The anti-RGS2 antibodies were used to determine the effect of deletion of SPL and NRB on the expression level of RGS2 and to immunoprecipitate RGS2. The upper blot in Figure 3A shows that deletion of SPL and NRB does not affect expression of RGS2. Notably, immunoprecipitation of RGS2 co-immunoprecipitates much more SPL from the NRB−/− than from WT brain extracts. It was not possible to co-immunoprecipitate native NRB and RGS2 from any of the extracts. The reason is not known, but perhaps the RGS2–NRB complex is not as stable as the RGS2–SPL complex in the lysates and does not survive the co-immunoprecipitation procedure. To overcome this problem, we used a complementary assay and determined the amounts of NRB and SPL that are pulled down by recombinant GST-RGS2 from the brain extracts. The blots in Figure 3B confirm the specificity of the anti-NRB antibodies and show that deletion of SPL had no effect of the expression of NRB. On the other hand, deletion of SPL almost doubled the binding of RGS2 to NRB. Moreover, the blots in Figure 3C confirm the specificity of the anti-SPL antibodies and show that deletion of NRB had no effect of the expression of SPL, but deletion of NRB almost doubled the binding of RGS2 to SPL. The results in Figure 3A–C are in agreement with the notion that SPL and NRB compete for binding to RGS2.
To establish the physiological relevance of the findings in Figures 1, 2 and 3 to regulation of Ca2+ signaling in vivo, we determined the effect of deletion of NRB in mice on Ca2+ signaling by αAR and the inhibition of Ca2+ signaling by RGS2. First, we verified that deletion of SPL and NRB does not affect expression level and ligand binding of the αAR, as revealed by measurement of 3[H]prazosin, a ligand for the αAR (Figure 3D). For [Ca2+]i measurements, we used parotid duct cells that robustly respond to stimulation of the αAR with a Ca2+ increase (Xu et al, 1996; Wang et al, 2005). NRB was reported to have restricted expression, with particularly high levels in the brain (Nakanishi et al, 1997). Therefore, it was important to determine expression and localization of NRB in salivary gland cells. Preliminary survey by RT–PCR revealed widespread expression of NRB mRNA (not shown). The immunolocalizations in Figure 3E–H show robust expression of NRB in submandibular (E, F) and parotid (G, H) ducts, and the absence of NRB in NRB−/− cells. Note that NRB was enriched at the apical pole, the site with high expression levels of all Ca2+ signaling proteins, including IP3 receptors, TRPC channels and GPCRs (Lee et al, 1997; Kiselyov et al, 2003; Li et al, 2004; Kim et al, 2006).
[Ca2+]i in parotid duct cells was followed by measuring the Ca2+-activated Cl− current using whole-cell recording to allow infusion of RGS2 and ΔNRGS2 through the patch pipettes (Luo et al, 2004; Wang et al, 2005). Figure 4 shows the response of WT, NRB−/− and SPL−/− parotid duct cells to different concentrations of Epi. The cells were stimulated only with one submaximal (1 or 10 μM) and one maximal (100 μM) Epi concentration to avoid differences due to desensitization. The individual traces and the summary show that SPL and NRB have opposite effects on Ca2+ signaling by αAR. Deletion of NRB reduced whereas deletion of SPL increased the potency of Epi to stimulate Ca2+ signaling, consistent with the effect of the scaffolds on regulation of Ca2+ signaling by RGS proteins.
Figure 4.
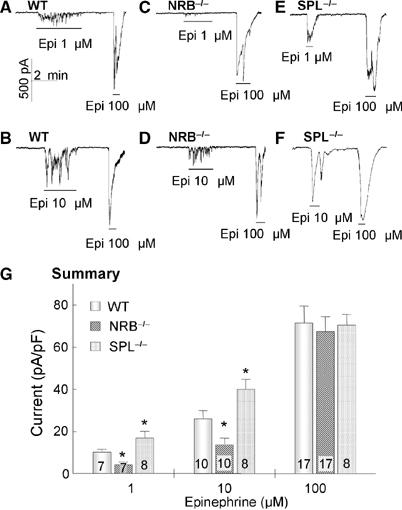
Effect of deletion of NRB and SPL in mice on Ca2+ signaling by αAR in parotid duct cells. The whole-cell Ca2+-activated Cl− current was measured in parotid duct cells prepared from WT (A, B), NRB−/− (C, D) or SPL−/− mice (E, F), and stimulated with 1, 10 or 100 μM Epi, as indicated. (G) The summary of the peak current at each [Epi] was determined and current density was calculated as pA/pF. When the signal was oscillatory, peak current was taken as the highest current measured. The number of cells analyzed at each Epi concentration is listed in the columns. The results are the means±s.e.m. *P<0.05.
Further evidence that the effect of SPL and NRB deletion is due to altered inhibition of Ca2+ signaling by RGS proteins is illustrated in Figure 5. Figure 5A–C show that deletion of NRB increased the potency of RGS2 to inhibit αAR-stimulated Ca2+ signaling, as expected from removal of a RGS protein buffer that binds the RGS proteins but not the GPCRs. The control experiments in Figure 5D–F show that deletion of NRB does not affect inhibition of αAR-mediated Ca2+ signaling by ΔNRGS2. The effect of deletion of NRB on inhibition of Ca2+ signaling by RGS2 is exactly opposite to that found with deletion of SPL (Wang et al, 2005).
Figure 5.
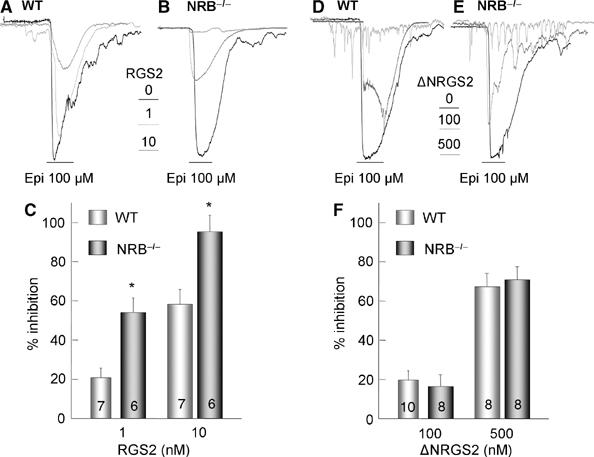
Effect of deletion of NRB in mice on inhibition of αAR-mediated Ca2+ signaling by RGS2 and ΔNRGS2. Parotid duct cells prepared from WT (A, D) and NRB−/− mice (B, E) were infused with the indicated concentrations of GST-tagged RGS2 (panels A, B) or ΔNRGS2 (D, E) and stimulated with 100 μM Epi. The peak currents from the indicated number of experiments were used to calculate current density in pA/pF and the percent inhibition. The summary in (C) is for RGS2 and in (F) is for ΔNRGS2, and are given as the mean±s.e.m. *P<0.05.
Reciprocal effect of NRB and SPL on Ca2+ signaling predicts that SPL should more efficiently impair Ca2+ signaling in NRB−/− than in WT cells. This was tested by comparing the effect of infused His-SPL(1–600) on Ca2+ signaling by αAR in WT and NRB−/− cells. His-SPL(1–600) was used since it is more resistant to degradation than FL-SPL and retains all the effects of FL-SPL on Ca2+ signaling (Wang et al, 2005). Coomassie staining of the purified His-SPL(1–600) showed a single band (not shown). Figure 6 shows that infusing the cells with 100 nM His-SPL(1–600) inhibited Epi-stimulated Ca2+ signaling nearly twice better in NRB−/− than in WT cells.
Figure 6.
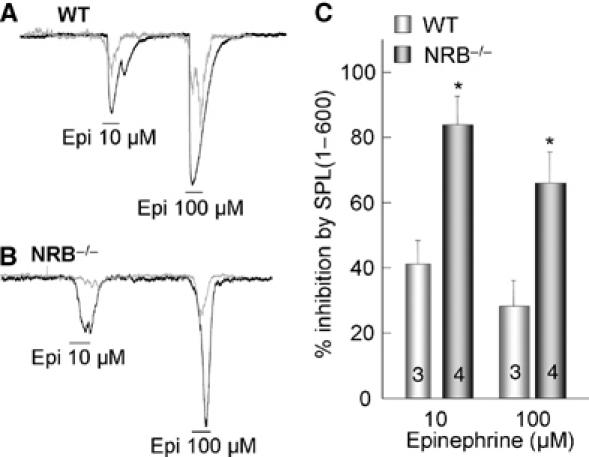
Sensitivity of Ca2+ signaling to inhibition by SPL in WT and NRB−/− cells. Parotid duct cells from WT (A) and NRB−/− mice (B) were infused with 100 nM His-SPL(1–600). The cells were stimulated with 10 and 100 μM Epi and the peak currents were used to calculate current density in terms of pA/pF and to calculate the percent inhibition. The results are summarized in (C) as the mean±s.e.m. of 3–4 experiments. *P<0.05.
Conclusions
The present work shows that both NRB and SPL bind the same members of the R4 subfamily of RGS proteins, but only SPL binds to the 3iL of GPCRs, which includes the α1BAR, dopamine, CCKA, CCKB and M3 receptors. The R4 RGS proteins directly bind to SPL and NRB, and the binding is mediated by the N-terminal domain of RGS2, the domain that confers receptor recognition (Zeng et al, 1998). NRB and SPL appear to compete for binding of RGS2 in vivo, as revealed by the increased co-immunoprecipitation of SPL with RGS2 in NRB−/− cells and the increased pull down of NRB by RGS2 from SPL−/− cells. Consequently, SPL and NRB reciprocally affect regulation of GPCR-evoked Ca2+ signaling by RGS proteins. Multiple lines of evidence support this conclusion: (a) the reciprocal shift by NRB and SPL of the dose–response curve for Epi-stimulated Ca2+ signaling in the Xenopus oocytes model system (Figure 2) and in native cells (Figure 4), (b) the enhance potency of RGS2, but not of ΔNRGS2, to inhibit Ca2+ signaling in NRB−/− cells (Figure 5) and (c) significantly, the enhanced inhibitory effect of SPL in NRB−/− cells. These findings together with those in our previous work (Wang et al, 2005) lead to the model illustrated in Figure 7, in which SPL and NRB form a functional pair of opposing regulators that modulates the intensity of Ca2+ signaling by GPCRs. The binding of R4 subfamily members of RGS proteins to SPL and NRB is dynamic. By binding to R4-RGS proteins and the 3iL of GPCRs, SPL recruits the RGS proteins to the GPCRs signaling complex to reduce the intensity of Ca2+ signaling. On the other hand, NRB binds the same R4-RGS proteins to remove them from the GPCRs signaling complex and enhance the intensity of Ca2+ signaling. The relative binding of R4 RGS proteins to SPL and NRB determines the intensity of Ca2+ signaling, and thus the frequency and amplitude of Ca2+ oscillations.
Figure 7.
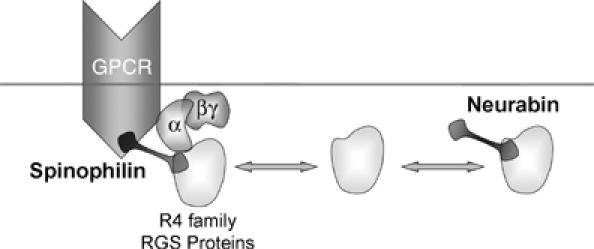
A model illustrating the role of SPL and NRB in regulating GPCR Ca2+ signaling by the R4 subfamily of RGS proteins.
Materials and methods
Plasmid constructs
The 3 × HA-tagged human RGS1, 2, 4, 16, GAIP and the human dopamine type 2, CCKA, CCKB and M3 receptors cDNA clones in pCDNA3.1+ vector were purchased from the UMR cDNA Resource Center. Myc-SPL and 6xHis-tagged SPL(1–600) were prepared as described before (Wang et al, 2005). To obtain His- and Myc-tagged NRB constructs, NRB was cloned from an established plasmid by PCR to include SalI and NotI restriction sites. After digestion with SalI/NotI, the PCR products were subcloned into the Myc-pRK5 vector. The full-length RGS2 construct was used as a template to prepare Myc-tagged ΔNRGS2 by PCR with primers encoding the SaII–KpnI sites and subcloned into a PCMV-Myc vector. GST-RGS2, GST-ΔNRGS2 (aa 79–211) and GST-RGS2N (aa 1–82) were obtained by PCR with primers encoding the XbaI–XhoI sites. The GST-third intracellular loop (3iL) of the α1BAR (aa 225–295) was generated from α1BAR by PCR with primer encoding for the EcoRI–KpnI sites and was inserted in-frame into the pGEX-KG vector. The MBP-3iL of the dopamine 2 (aa 209–373), CCKA (aa 233–315), CCKB (aa 242–332) and M3 (aa 250–493) receptors were prepared by PCR using plasmids coding for the full-length receptors as templates, and with primers encoding for XbaI–HindIII, BamHI–HindIII, XbaI–HindIII, EcoRI–XbaI sites, respectively. After digestion, the fragments were inserted into the pMAL-c2X vector. His-tagged NRB(1–620) was obtained by PCR with primer encoding for the SalI–NotI sites and the fragment was inserted into the pET28A vector. All constructs were confirmed by sequencing.
Generation of SPL−/− and NRB−/− mice
Deletion of SPL (Feng et al, 2000) and NRB (Allen et al, 2006) in mice was accomplished as described before. The mice had free access to standard mouse chow and water and were 5–6 weeks old when used.
Immunolocalization of NRB in salivary glands
The salivary glands of WT and NRB−/− mice were excised and frozen in Tissue-TEK OCT compound. Immunofluorescence localization of NRB in frozen sections (4 mm) was exactly as detailed before (Shin et al, 2003) using the anti-NRB antibodies characterized before (Allen et al, 2006). The sections were fixed and permeabilized with cold MeOH and nonspecific sites were blocked by 1 h incubation at room temperature with PBS containing 5% goat serum, 1% BSA and 0.1% gelatin. The sections were incubated with a 1:250 dilution of anti-NRB antibodies in blocking media, washed and the antibodies were detected with goat anti-rabbit IgG tagged with fluorescein. Images were collected with a confocal microscope (BioRad model MRC 1024).
Protein expression and purification
BL21 (DE3) cells expressing the desired proteins were grown in LB medium with 100 μg/ml ampicillin in 37°C and protein production was induced with 0.5 mM IPTG for 3 h in 37°C. GST and MBP fusion proteins were extracted from the cells by sonication, lysis in 0.5% Triton 100, 50 mM imidazole, 100 mM NaCl, 10 mM EDTA, 0.5 mM DTT, pH 7.4, 20 μg/ml aprotinin, leupeptin 1 μg/ml, pepstatin 1 μg/ml and 1 mM phenylmethylsulfonyl fluoride and centrifugation at 30 000 g for 30 min at 4°C. The supernatant was harvested with glutathione–agarose beads (GST) or amylose resin beads (MBP) by 1 h incubation at 4°C with gentle rotation. GST and GST fusion protein attached to the beads were washed four times with lysis buffer and suspended in PBS containing 0.1% NaN3 for further pull-down assays. The same amount of protein of GST, GST-RGS2, GST-ΔNRGS2, GST-RGS2N and GST-3iL was used for the pull-down assays. For the His-tagged proteins, NTA-Ni2+–agarose and proteins were purified following the manufacturer's instructions. The eluted proteins were dialyzed overnight in 4°C in assay buffer (140 mM KCl, 10 mM HEPES, pH 7.4) and concentrated using centricons for the electrophysiological experiment.
Cell culture, transfection, pull-down and co-immunoprecipitation assays
HEK cells were cultured in DMEM medium containing 10% FBS and 100 U/ml penicillin and streptomycin. Lipofectamine 2000 (Invitrogen) was used as the transfection reagent and cells transfected for 36–48 h were used for pull-down assays or for measurement of [Ca2+]i. Briefly, the cells were extracted in lysis buffer containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 20% glycerol, 1.5 mM MgCl2, 0.5% Triton X-100, 1 mM DTT, 1 mM PMSF and protease inhibitors cocktail, by a 30 min incubation at 0°C. The extracts were collected by centrifugation and incubated with 20–30 μl beads bound with 6xHis-NRB or with GST-RGS2, GST-ΔNRGS2 and GST-RGS2N for 2–4 h at 4°C. The beads were washed three times in lysis buffer and once in PBS, and the proteins were released in SDS loading buffer by a 3 min incubating in a boiling water bath and analyzed by SDS–PAGE.
For RGS2-SPL co-immunoprecipitation, the brains from WT, SPL−/− and NRB−/− mice were collected on ice and each of the brains was immediately homogenized in 1 ml of the lysis buffer specified above. The homogenates were placed on ice for 1 h and then centrifuged at 1000 g for 5 min. The supernatant was collected and centrifuged at 55 000 g for 30 min and was used for immunoprecipitation of RGS2 by incubation of 400 μl extract with 6 μl anti-RGS2 antibodies overnight at 4°C with gentle agitation. The immune complexes were collected with 30 μl of 1:1 goat anti-IgY antibody-conjugated microbead slurry and incubation for an additional 2 h at 4°C. The beads were collected, washed with lysis buffer and the precipitates were probed for RGS2 and SPL.
NRB and SPL pull down
Brains from three WT, SPL−/− and NRB−/− mice were collected on ice and each of brain was immediately homogenized in 1 ml lysis buffer. The homogenates were placed on ice for 1 h and then centrifuged at 1000 g for 5 min. The supernatant was collected and centrifuged at 60 000 g for 30 min, and was used for pull-down and Western blot assays. Equal amount of protein from each extract was incubated with the same amount of GST-RGS2 coupled to beads at 4°C for 4–5 h. The beads were collected by centrifugation, washed twice with PBS, and the proteins were eluted by incubating with SDS sample buffer and boiling for 3 min. NRB and SPL were analyzed by Western blot using the anti-NRB and anti-SPL antibodies characterized before (Allen et al, 2006).
NRB and RGS2 binding assay
Purified 6 × His-NRB(1–620) in 25 mM HEPES, pH 8.0, 5 mM EDTA, 2 mM DTT, 0.5 M NaCl and 10% glycerol was diluted into 250 μl binding medium, which is the same as the lysis buffer, for a final concentration between 0.5–8 μg. Then 15 μg GST-RGS2, GST-RGS2N, GST-ΔNRGS2 or GST coupled to glutathione beads were added and the mixtures were incubated overnight at 4°C with continued rotation. The NRB-RGS2 construct complexes were collected by centrifugation and washed once with binding buffer and once with PBS.
Ligand binding
Brains of two WT, two SPL−/− and two NRB−/− mice were transferred to ice-cold homogenization solution composed of 50 mM Tris–HCl, pH 8, 0.5 mM EGTA and 0.5 mM EDTA at a ratio of about 50 volumes of homogenization buffer per brain. The brained were homogenized by 20 strokes in a tightly fitted teflon–glass homogenizer and microsomes were collected by centrifugation at 20 000 g for 20 min. The pellet was resuspended in homogenization buffer containing a mixture of protease inhibitors (Rosh tablets) and stored at −80°C until use. 3[H]prazosin (78 Ci/mmol, Perkin Elmer) binding was measured at 23°C by incubating about 0.5 mg microsomes for 45 min in a binding media composed of homogenization buffer, 3[H]prazosin and the indicated concentrations of unlabeled prazosin in a final volume of 0.45 ml. At the end of the incubation, the microsomes were collected by rapid filtration using GF/C filters, washed three times with 5 ml homogenization buffer and counted.
Ca2+ measurements in HEK cells
HEK cells grown on glass coverslips were transfected with eGFP alone or eGFP and ΔNRGS2, or the ΔNRGS2 box mutants. eGFP fluorescence was used to identify the transfected cells. The cells were used 24–48 h post-transfection. [Ca2+]i was measured by loading the cells with Fura2 and measuring the Fura2 fluorescence at excitation wavelengths of 350 and 380 nm, as detailed before (Wang et al, 2004b). Results are presented as the change in the 350/380 ratio. The cells were continuously perfused with warm (37°C) standard bath solution containing (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES (pH 7.4 with NaOH) and 10 glucose. The P2Y2 receptors were stimulated by including 0.5 mM ATP in the perfusion media.
Preparation, injection and current measurements in Xenopus oocytes
Xenopus oocytes in stages V–VI were used for injection of 2–10 ng cRNA coding for the proteins of interest in a final volume of 30 nl, and kept at 18°C in ND96 solution containing (in mM) 96 NaCl 1 MgCl2, 1 CaCl2 and 5 HEPES (pH 7.6 with NaOH). Current measurement was accomplished 4896 h post-injection with the two-electrode voltage-clamp procedure using an OC-725C amplifier. In some experiments, recombinant RGS proteins were injected into the oocytes for at least 1 h before current measurement. Current injection and voltage recording electrodes were both filled with 3 M KCl and had resistances of 0.5–1 and 0.52 MΩ, respectively. To measure the Ca2+-activated Cl− current, membrane potential was maintained at −60 mV and stepped to+50 mV for 200 ms at 0.2 Hz. Current was digitized and analyzed using DIGI-data 1322A A/D converter and pClamp 8.1 software. The peak of the outward current as a function of time was plotted to obtain the time course at each Epi concentration. Epi dose–response relationships were obtained by plotting the peak of the outward current at each Epi concentration.
Current measurement in parotid duct cells
Single parotid duct cells were prepared from WT and NRB−/− mice by the trypsin and collagenase digestion procedure described previously (Wang et al, 2005). Duct cells were perfused with bath solution containing (in mM) 140 NaCI, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES (pH 7.4 with NaOH) and 10 mM glucose. The patch pipette resistance was 25 MΩ when it was filled with an internal solution containing (in mM) 140 KCl, 1 MgCl2, 0.2 EGTA, 1 Na2ATP and 10 HEPES (pH 7.3 with KOH). After obtaining giga-Ohm seals (>5 GΩ), the holding potential was changed from 0 to 60 mV. The whole-cell configuration was established by gentle suction. The whole-cell Ca2+-activated Cl− current at −60 mV was continuously recorded as a reporter of [Ca2+]i. The current output from the patch-clamp amplifier (Axopatch-200B, Axon Instruments) was filtered at 1 KHz and stored online on a hard disk with a Digi-Data 1200 interface and pclamp6.1 software (Axon Instruments). Infusion of recombinant RGS proteins into the cells was accomplished by including them in the pipette solution and at least 7 min of cell dialysis before the first stimulation. The experiments were carried out at room temperature.
Acknowledgments
This work was supported by a grant from the American Heart Association Inc. Texas Affiliate BGIA 0665192Y to WZ and by NIH grants DE12309 and DK38938 to SM.
References
- Allen PB, Ouimet CC, Greengard P (1997) Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA 94: 9956–9961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen PB, Zachariou V, Svenningsson P, Lepore AC, Centonze D, Costa C, Rossi S, Bender G, Chen G, Feng J, Snyder GL, Bernardi G, Nestler EJ, Yan Z, Calabresi P, Greengard P (2006) Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience 140: 897–911 [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1997) Elementary and global aspects of calcium signalling. J Physiol 499 (Part 2): 291–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529 [DOI] [PubMed] [Google Scholar]
- Brady AE, Wang Q, Allen PB, Rizzo M, Greengard P, Limbird LE (2005) Alpha 2-adrenergic agonist enrichment of spinophilin at the cell surface involves beta gamma subunits of Gi proteins and is preferentially induced by the alpha 2A-subtype. Mol Pharmacol 67: 1690–1696 [DOI] [PubMed] [Google Scholar]
- Brady AE, Wang Q, Colbran RJ, Allen PB, Greengard P, Limbird LE (2003) Spinophilin stabilizes cell surface expression of alpha 2B-adrenergic receptors. J Biol Chem 278: 32405–32412 [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Posner BA, Gilman AG (1996) Visualizing signal transduction: receptors, G-proteins, and adenylate cyclases. Clin Sci (London) 91: 527–537 [DOI] [PubMed] [Google Scholar]
- Druey KM, Kehrl JH (1997) Inhibition of regulator of G protein signaling function by two mutant RGS4 proteins. Proc Natl Acad Sci USA 94: 12851–12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P (2000) Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci USA 97: 9287–9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freissmuth M, Casey PJ, Gilman AG (1989) G proteins control diverse pathways of transmembrane signaling. FASEB J 3: 2125–2131 [PubMed] [Google Scholar]
- Gilman AG (1987) G proteins: transducers of receptor-generated signals. Annu Rev Biochem 56: 615–649 [DOI] [PubMed] [Google Scholar]
- Heximer SP (2004) RGS2-mediated regulation of Gqalpha. Methods Enzymol 390: 65–82 [DOI] [PubMed] [Google Scholar]
- Ishii M, Kurachi Y (2003) Physiological actions of regulators of G-protein signaling (RGS) proteins. Life Sci 74: 163–171 [DOI] [PubMed] [Google Scholar]
- Kim JY, Zeng W, Kiselyov K, Yuan JP, Dehoff MH, Mikoshiba K, Worely PF, Muallem S (2006) Homer 1 mediates store- and IP3Rs-dependent translocation and retrieval of TRPC3 to the plasma membrane. J Biol Chem 281: 32540–32549 [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Shin DM, Muallem S (2003) Signalling specificity in GPCR-dependent Ca2+ signalling. Cell Signal 15: 243–253 [DOI] [PubMed] [Google Scholar]
- Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJ, Kuo TH, Wuytack F, Racymaekers L, Muallem S (1997) Polarized expression of Ca2+ channels in pancreatic and salivary gland cells. Correlation with initiation and propagation of [Ca2+]i waves. J Biol Chem 272: 15765–15770 [DOI] [PubMed] [Google Scholar]
- Li Q, Luo X, Muallem S (2004) Functional mapping of Ca2+ signaling complexes in plasma membrane microdomains of polarized cells. J Biol Chem 279: 27837–27840 [DOI] [PubMed] [Google Scholar]
- Luo X, Ahn W, Muallem S, Zeng W (2004) Analyses of RGS protein control of agonist-evoked Ca2+ signaling. Methods Enzymol 389: 119–130 [DOI] [PubMed] [Google Scholar]
- Luo X, Popov S, Bera AK, Wilkie TM, Muallem S (2001) RGS proteins provide biochemical control of agonist-evoked [Ca2+]i oscillations. Mol Cell 7: 651–660 [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Obaishi H, Satoh A, Wada M, Mandai K, Satoh K, Nishioka H, Matsuura Y, Mizoguchi A, Takai Y (1997) Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol 139: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet CC, Katona I, Allen P, Freund TF, Greengard P (2004) Cellular and subcellular distribution of spinophilin, a PP1 regulatory protein that bundles F-actin in dendritic spines. J Comp Neurol 479: 374–388 [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN (2000) Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev 80: 1291–133511015615 [Google Scholar]
- Richman JG, Brady AE, Wang Q, Hensel JL, Colbran RJ, Limbird LE (2001) Agonist-regulated interaction between alpha2-adrenergic receptors and spinophilin. J Biol Chem 276: 15003–15008 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86: 369–408 [DOI] [PubMed] [Google Scholar]
- Ross EM, Wilkie TM (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 69: 795–827 [DOI] [PubMed] [Google Scholar]
- Satoh A, Nakanishi H, Obaishi H, Wada M, Takahashi K, Satoh K, Hirao K, Nishioka H, Hata Y, Mizoguchi A, Takai Y (1998) Neurabin-II/spinophilin. An actin filament-binding protein with one pdz domain localized at cadherin-based cell–cell adhesion sites. J Biol Chem 273: 3470–3475 [DOI] [PubMed] [Google Scholar]
- Shin DM, Dehoff M, Luo X, Kang SH, Tu J, Nayak SK, Ross EM, Worley PF, Muallem S (2003) Homer 2 tunes G protein-coupled receptors stimulus intensity by regulating RGS proteins and PLCbeta GAP activities. J Cell Biol 162: 293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Oxford GS, Milgram SL (1999) Association of the D2 dopamine receptor third cytoplasmic loop with spinophilin, a protein phosphatase-1-interacting protein. J Biol Chem 274: 19894–19900 [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhao J, Brady AE, Feng J, Allen PB, Lefkowitz RJ, Greengard P, Limbird LE (2004a) Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science 304: 1940–1944 [DOI] [PubMed] [Google Scholar]
- Wang X, Huang G, Luo X, Penninger JM, Muallem S (2004b) Role of regulator of G protein signaling 2 (RGS2) in Ca(2+) oscillations and adaptation of Ca(2+) signaling to reduce excitability of RGS2−/− cells. J Biol Chem 279: 41642–41649 [DOI] [PubMed] [Google Scholar]
- Wang X, Zeng W, Soyombo AA, Tang W, Ross EM, Barnes AP, Milgram SL, Penninger JM, Allen PB, Greengard P, Muallem S (2005) Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol 7: 405–411 [DOI] [PubMed] [Google Scholar]
- Xu X, Diaz J, Zhao H, Muallem S (1996) Characterization, localization and axial distribution of Ca2+ signalling receptors in the rat submandibular salivary gland ducts. J Physiol 491 (Part 3): 647–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zeng W, Popov S, Berman DM, Davignon I, Yu K, Yowe D, Offermanns S, Muallem S, Wilkie TM (1999) RGS proteins determine signaling specificity of Gq-coupled receptors. J Biol Chem 274: 3549–3556 [DOI] [PubMed] [Google Scholar]
- Zeng W, Xu X, Popov S, Mukhopadhyay S, Chidiac P, Swistok J, Danho W, Yagaloff KA, Fisher SL, Ross EM, Muallem S, Wilkie TM (1998) The N-terminal domain of RGS4 confers receptor-selective inhibition of G protein signaling. J Biol Chem 273: 34687–34690 [DOI] [PubMed] [Google Scholar]