

Abstract
c-Src is a tightly regulated non-receptor tyrosine kinase. We describe the C-terminus of c-Src as a ligand for a PDZ (postsynaptic density 95, PSD-95; discs large, Dlg; zonula occludens-1, ZO-1) domain. The C-terminal residue Leu of c-Src is essential for binding to a PDZ domain. Mutation of this residue does not affect the intrinsic kinase activity in vitro, but interferes with c-Src regulation in cells. As a candidate PDZ protein, we analysed AF-6, a junctional adhesion protein. The AF-6 PDZ domain restricts the number of c-Src substrates, whereas knockdown of AF-6 has the opposite effect. Binding of c-Src to the AF-6 PDZ domain interferes with phosphorylation of c-Src at Tyr527 by the C-terminal kinase, and reduces c-Src autophosphorylation at Tyr416, resulting in a moderately activated c-Src kinase. Unphosphorylated Tyr527 allows binding of c-Src to AF-6. This can be overcome by overexpression of CSK or strong activation of c-Src. c-Src is recruited by AF-6 to cell–cell contact sites, suggesting that c-Src is regulated by a PDZ protein in special cellular locations. We identified a novel type of c-Src regulation by interaction with a PDZ protein.
Keywords: cell junction, PDZ domain, signal transduction, Src protein kinase
Introduction
c-Src is a non-receptor tyrosine kinase and the prototype of the Src family kinases (SFKs). Three SFKs, Src, Yes and Fyn, are ubiquitously expressed, whereas others such as Lyn, Lck, Hck and Blk are mainly expressed in non-adherent cells of the haematopoietic system. SFKs play key roles in cell proliferation and survival, as well as in cell adhesion, cell morphology and motility (Frame, 2002; Yeatman and Roskoski, 2004). Src knock-out mice die within the first week after birth and show defects in osteoclast development resulting in osteopetrosis (Soriano et al, 1991). Src, Yes and Fyn can partially replace each other, thus demonstrating their close functional relationship. Mice deficient in all three kinases die at early stage of embryonic development and cells derived from these embryos show severe defects in migration and survival (Klinghoffer et al, 1999). Expression of constitutively activated mutants of c-Src promotes cell proliferation and invasive growth of cells. v-Src, the viral homologue of the cellular proto-oncogene c-Src encoded by the avian Rous Sarcoma Virus (RSV), is constitutively activated and causes tumours in chickens (Martin, 2001). Elevated c-Src protein expression level and increased kinase activity have been found in several human cancers and a constitutively activated c-Src mutant was described in human colon carcinoma (Irby and Yeatman, 2000).
The Src family proteins are characterized by four highly conserved Src homology (SH) domains termed SH1 to SH4 (Figure 1B). The SH4 domain includes a myristoyl group involved in membrane targeting. SH3 and SH2 domains are protein–protein interaction domains interacting with proline-rich sequences and phosphotyrosine containing motifs, respectively. A proline-rich linker connects the SH2 domain with the protein kinase domain (SH1 domain). The short C-terminal regulatory tail contains Tyr527, which upon phosphorylation allows intramolecular interaction with the SH2 domain. This interaction and binding of the linker to the SH3 domain stabilize an inactive closed conformation of c-Src. Competitors for the SH2 or SH3 domains and dephosphorylation of Tyr527 by protein tyrosine phosphatases (PTP) result in opening of c-Src and partial activation of its kinase activity (Frame, 2002). Complete activation of c-Src requires, in addition, autophosphorylation of c-Src at Tyr416 located in the activation loop of the catalytic centre. Phosphorylation of Tyr527 by the C-terminal Src kinase, CSK, together with dephosphorylation of Tyr416 by PTP, negatively regulates the c-Src kinase activity (Sun et al, 1998; Schneider et al, 1999; Chong et al, 2005).
Figure 1.
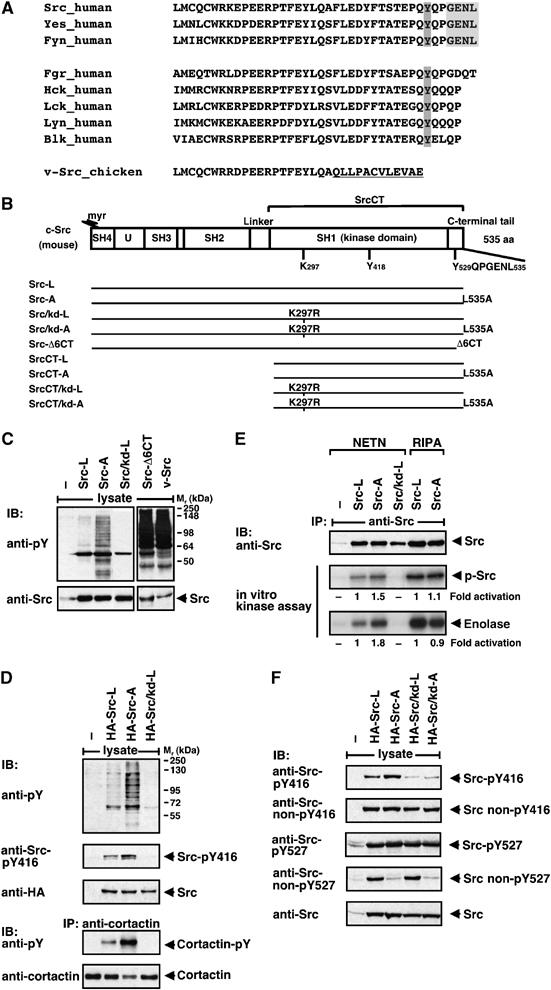
Regulation of c-Src by its very C-terminal sequence. (A) C-terminal sequence alignment of Src-family kinases. The PDZ binding site is depicted in light grey and the regulatory Tyr527 in dark grey. The v-Src-specific C-terminus is underlined. (B) Domain structure of murine c-Src. The molecule is composed of an N-terminal Src homology (SH)4 domain attached with a myristoyl group (myr), a unique region (U), SH3 and SH2 protein–protein interaction domains, a kinase domain (SH1) that contains Lys297 and Tyr418 (Tyr416 in chicken) and a C-terminal regulatory tail that contains Tyr529 (Tyr527 in chicken) and a PDZ binding sequence (GENL). The regions encoded by the different Src constructs are shown. (C–F) Regulation of Src by its C-terminal sequence. HEK 293 cells were transiently transfected with empty vector (−) or constructs, as indicated. Lysates of starved cells were analysed by blotting (IB) with anti-phospho-tyrosine (pY), anti-Src-pY416, anti-Src non-pY416, anti-Src-pY527 and anti-Src non-pY527 antibodies. Src expression was controlled by blotting with anti-Src or anti-HA antibody. (D) For analysis of cortactin phosphorylation, lysates were precipitated (IP) with anti-cortactin antibody and blotted with anti-pY and anti-cortactin antibodies. (E) Immunopurified Src proteins were analysed by in vitro kinase assay for autophosphorylation (p-Src) and for substrate phosphorylation (enolase). Numbers indicate fold activation normalized to Src expression levels.
Inactive c-Src is predominantly localized at the perinuclear membrane. Upon stimulation, c-Src is recruited to the cell periphery (Bjorge et al, 2000). At the plasma membrane, c-Src mediates mitogenic signalling, at cytoplasmic endosomes it is involved in trafficking, at focal adhesion plaques it plays a role in cell spreading and migration and at cell–cell contacts it contributes to regulation of cell adhesion. The intracellular localization and the cell type determine the kind of substrates phosphorylated by c-Src. Substrates include focal adhesion proteins, cell adhesion molecules, adaptor proteins and transcription factors.
The basis for this study was our observation that the C-terminus of c-Src, GENL (GlyGluAsnLeu), is a putative ligand for PDZ (postsynaptic density 95, PSD-95; discs large, Dlg; zonula occludens-1, ZO-1) domains (Figure 1A) (Songyang et al, 1997). PDZ domains can be classified according to their specificity for C-terminal peptides of their ligands (Nourry et al, 2003). Class I PDZ domains preferentially recognize the motif S/T–X–V/L/I (Ser/Thr–X–Val/Leu/Ile; X means any residue), class II Φ−X–Φ (Φ means hydrophobic residue), and class III D/E–X–Φ (Asp/Glu–X–Φ). Some PDZ domains belong to more than one class, such as the ones of AF-6 and Erbin (Buchert et al, 1999; Boisguerin et al, 2004; Wiedemann et al, 2004; Ress and Moelling, 2006). The C-terminus of c-Src, GENL, fits to the consensus sequence typical for binding to the rare class III PDZ domains of proteins such as the neuronal nitric oxide synthase (nNOS), a protein recruited to the postsynaptic membrane, and LIN-10, a protein involved in vesicle transport (Nourry et al, 2003).
The binding between a PDZ domain and its ligand is often of low affinity, enabling its regulation by phosphorylation, either of the C-terminal sequence of the ligand or of the PDZ domain protein (Schneider et al, 1999; Nourry et al, 2003; Radziwill et al, 2003). PDZ domain proteins can multimerize and function as scaffolds to organize cell adhesion complexes and to cluster transmembrane proteins. They also align signalling proteins, thereby directing and anchoring them to specific subcellular compartments.
Here, we identified the C-terminus of c-Src as a ligand for the PDZ domain of AF-6, an interaction that regulates c-Src. In addition to a PDZ domain, AF-6 contains Ras binding sites that interact with and regulate the small GTPases Ras and Rap1 (Boettner et al, 2000; Radziwill et al, 2003). AF-6 has an F-actin binding domain to connect the cytoskeleton with transmembrane proteins at cellular junctions. AF-6 plays a role in formation and maintenance of cell junctions (Takai and Nakanishi, 2003; Lorger and Moelling, 2006).
We demonstrate a specific interaction between c-Src and the PDZ domain of AF-6. Exchange of the C-terminal residue Leu of c-Src to Ala reduced its binding to the AF-6 PDZ domain. This mutation did not affect the intrinsic kinase activity in vitro, but released c-Src from a PDZ domain-dependent regulation. c-Src bound to AF-6 was dephosphorylated at Tyr527 and shielded against phosphorylation by CSK at this site. The AF-6 PDZ domain reduced autophosphorylation of c-Src at Tyr416 and allowed an only partially activated kinase. Binding of c-Src to AF-6 was overcome by overexpression of CSK or strong activation of c-Src. c-Src and AF-6 colocalized at cellular junctions, suggesting that c-Src regulation by a PDZ protein is restricted to certain cellular locations.
Results
The C-terminus of c-Src is a recognition site for PDZ domains
The C-terminal sequence, GENL, of c-Src, resembles other ligands for PDZ domains, and was most similar to a type III PDZ domain binding motif (Nourry et al, 2003). Sequence analysis of human SFKs showed that only Src, Yes and Fyn harboured such a sequence, whereas the other Src family members did not (Figure 1A). To study the biological relevance of the putative PDZ ligand sequence, we mutated the C-terminal Leu of c-Src (Src-L) to Ala (Src-A) (Figure 1B). It is known for other PDZ ligands that this single amino-acid exchange interferes with binding to the PDZ domain (Radziwill et al, 2003). Expression of Src-L in human embryonic kidney (HEK) 293 cells resulted in tyrosine phosphorylation of only a few proteins in serum-starved cells. In contrast, expression of the mutant Src-A increased the number of proteins phosphorylated at tyrosines (Figure 1C and D). Quantification of tyrosine phosphorylated proteins in Src-A expressing cells showed an up to six-fold increase compared with Src-L. Src-A expression elevated tyrosine phosphorylation of its known substrate cortactin by four-fold (Figure 1D). The kinase activity of Src-A protein purified from cells, which were lysed under mild conditions (NETN), allowing protein–protein interactions, was slightly increased compared with Src-L (1.5-fold for autophosphorylation and 1.8-fold for enolase phosphorylation; Figure 1E). This correlated with a moderate increase (two-fold) in the autophosphorylation of c-Src at Tyr416 (Figure 1D). Under more stringent lysis conditions (RIPA), no difference in autophosphorylation and enolase phosphorylation was observed between Src-A and Src-L (Figure 1E). Thus, the Leu to Ala mutation in the PDZ ligand sequence of c-Src does not affect the intrinsic c-Src kinase activity, but interferes with c-Src regulation.
The kinase activity of c-Src is positively regulated by autophosphorylation at Tyr416 and negatively regulated by CSK-dependent phosphorylation of Tyr527. Under basal growth conditions, 90–95% of cellular c-Src is kinase-inactive (Roskoski, 2004). Analysis of the phosphorylation state of Src-L and Src-A in serum-starved cells revealed that the majority of both proteins was dephosphorylated at Tyr416 and phosphorylated at Tyr527, indicating inactive c-Src kinase (Figure 1F). Compared with Src-L, Src-A, the mutant that escapes binding to a PDZ domain, showed a larger subpopulation phosphorylated at Tyr416 correlating with increased substrate phosphorylation (Figure 1E and F). A subpopulation of Src-L but not Src-A was dephosphorylated at Tyr527.
Summarized data show that the C-terminal sequence of c-Src is a PDZ domain ligand and mutation of this sequence increased the number of substrates phosphorylated by c-Src, presumably because of loss of negative regulation by a PDZ protein.
We compared Src-A effects with the oncogenic mutant Src-Δ6CT carrying a deletion of the C-terminal six residues and the viral oncoprotein v-Src in which the 19 C-terminal residues of c-Src are substituted by 12 v-Src-specific residues (Figure 1A). Both Src-Δ6CT and v-Src do not contain the PDZ ligand sequence and cannot be regulated by phosphorylated Tyr527. This led to a significantly stronger tyrosine phosphorylation of numerous substrates than Src-A (Figure 1C, right blot).
Effect of the c-Src PDZ domain recognition motif in cellular transformation assays
The mutation of the PDZ ligand sequence in Src-A did not increase the intrinsic kinase activity in vitro but increased substrate phosphorylation in the cellular context. In order to analyse the significance of c-Src GENL as a ligand for PDZ domains, following three biological assays for cellular transformation were performed: growth of cells in soft agar correlating with anchorage-independent growth, focus formation correlating with the loss of contact inhibition and migration through a matrigel matrix correlating with invasive growth. These assays were performed in SYF−/− cells, a mouse fibroblast cell line deficient in expression of Src, Yes and Fyn (Klinghoffer et al, 1999). SYF−/− cells were stably transduced with recombinant retroviruses expressing Src-L or the mutant Src-A under control of a tetracycline-regulatable promoter (Figure 2A). Autophosphorylation at Tyr416 was increased in Src-A compared with a similar amount of Src-L and indicated an elevated activation state of Src-A. Expression of Src-A led to increased colony formation in soft agar, focus formation as well as invasive growth compared with Src-L (Figure 2B). However, these Src-A effects on transformation were weak compared with the highly oncogenic Src-Δ6CT protein, which is far more effective in transformation of cells (data not shown). Thus, the point mutation in the PDZ binding site of c-Src led to moderate cellular transformation compared with the oncogenic deletion mutant.
Figure 2.

Cellular assays with c-Src and its C-terminal mutant. (A) Immunoblot analysis of SYF−/− stable cell lines inducibly expressing Src-L or Src-A. Src expression was induced by using decreasing levels of tetracycline in the medium. Src autophosphorylation was analysed with anti-pY416-Src antibody and equal loading controlled with anti-ERK2 antibody. (B) Analysis of Src-L expressing SYF−/− cells versus Src-A expressing SYF−/− cells for anchorage-independent growth (upper panel), formation of foci (middle panel) and invasive activity (lower panel). Analyses were performed in duplicates (mean±s.d.), with n=3, P<0.01 (colony formation); n=5, P<0.001 (focus formation); n=3, P<0.05 (matrigel invasion).
c-Src is a ligand for the PDZ domain of AF-6
The exchange of the C-terminal Leu for Ala impaired the PDZ recognition motif of c-Src. This raised the question about the nature of the PDZ protein, which regulates the function of c-Src. It is known that c-Src and its close relatives c-Yes and c-Fyn can localize to cellular junctions and regulate the formation of cell–cell contacts and cell migration by phosphorylating proteins linked to the actin cytoskeleton (Chen et al, 2002; Martinez-Quiles et al, 2004). Therefore, we assumed that a PDZ protein, binding to F-actin and connecting cell adhesion proteins to the cytoskeleton, might have a regulatory effect on c-Src. One potential candidate was AF-6, which had these characteristics (Takai and Nakanishi, 2003; Lorger and Moelling, 2006). Binding between c-Src GENL and the AF-6 PDZ domain was tested in a peptide binding assay using C-terminal Src-related peptides and GST-AF-6 PDZ fusion protein (Figure 3A). Chimeric peptides, where the c-Src-specific PDZ binding sequence GENL was replaced by STEV and GIQV specific for the AF-6 ligands Bcr and EphA7, respectively, were used as controls (Figure 3A) (Hock et al, 1998; Radziwill et al, 2003). Indeed, all three ligands bound to the AF-6 PDZ domain, whereas the control peptides, in which the C-terminal amino acid was replaced by Ala, showed reduced binding (Figure 3A). Calculation of the theoretical dissociation constants (Kd) (Wiedemann et al, 2004) yielded the following values: Src-L, 679 μM; Src-A 1863 μM; Src-STEV 85 μM and Src-GIQV, 36 μM. These values correlate with the data of the peptide binding study, indicating that the C-terminal sequence of c-Src, GENL, is a specific ligand for the PDZ domain of AF-6. Binding of the c-Src peptide to the AF-6 PDZ domain was independent of the phosphorylation state of Tyr527.
Figure 3.
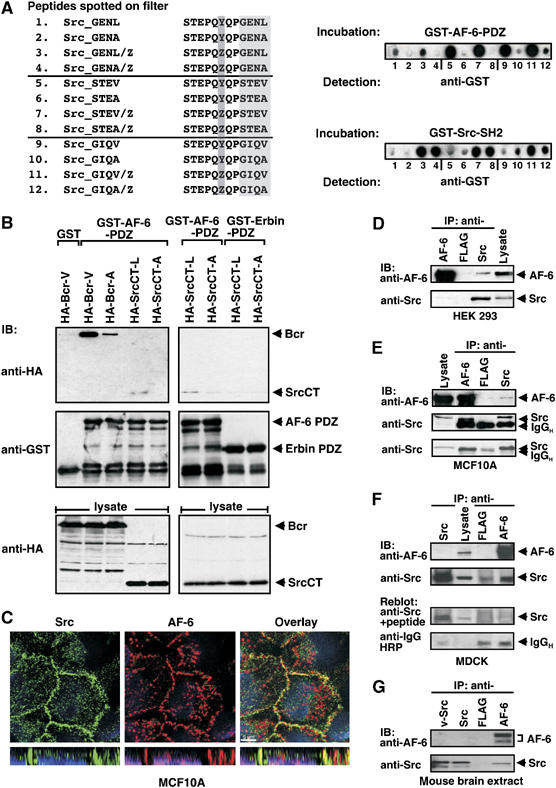
Interaction of c-Src with the AF-6 PDZ domain. (A) Analysis of C-terminal c-Src peptides binding to the AF-6 PDZ domain. GST-AF-6-PDZ bound to peptides immobilized on the membrane was visualized by blotting with anti-GST antibody. GST-Src-SH2 served as control for peptides phosphorylated on Tyr. z indicates pTyr. (B) Interaction of SrcCT with the AF-6 PDZ domain. HEK 293 cells were transiently transfected with expression constructs as indicated. In a pull-down assay, lysates were incubated with GST, GST-AF-6-PDZ or GST-Erbin-PDZ, followed by blotting with anti-HA or anti-GST antibodies. (C) Colocalization of endogenously expressed AF-6 and c-Src in MCF10A. Cells were kept in complete medium, fixed and immunostained with anti-AF-6 (red) and anti-Src (green) antibodies. Single confocal sections in green and red channel and overlay thereof are shown; scale bar is 5 μm. (D–G) Coprecipitation of endogenously expressed AF-6 and c-Src. Cell lysates of HEK 293, MCF10A, MDCK and mouse brain extracts were precipitated with mouse Mab, as indicated, and blotted with mouse anti-AF-6 and rabbit anti-c-Src (D, F, G), or mouse anti-Src GD11 (E). The specificity of the rabbit anti-Src antibody was demonstrated by competition with the appropriate Src peptide antigen and blots with anti-IgG-HRP were used for control of antibody amount; IgGH immunoglobulin G heavy chain (F, lower blots). Exacta Cruz E solutions (Santa Cruz) for detection of native antibody only was used for the lower blot(E).
To support the results of the peptide analysis, we performed a pull-down assay by incubating recombinant GST-AF-6-PDZ with lysates of HEK 293 cells expressing SrcCT-L, comprising the C-terminal part of c-Src (SrcCT; Figure 1B), or SrcCT-A mutated in the PDZ domain ligand sequence. SrcCT-L but not SrcCT-A bound to the PDZ domain of AF-6, indicating that AF-6 might be an interaction partner for c-Src (Figure 3B). As expected, the known AF-6 PDZ domain ligand Bcr-V strongly bound to the PDZ domain of AF-6 compared with the mutant Bcr-A. The PDZ domain of Erbin was used as negative control (Figure 3B). We also excluded binding of Src-L to PDZ domains of ZO-1 and hDlg (data not shown).
Having demonstrated in an overexpression system that c-Src is a ligand for the PDZ domain of AF-6, we analysed whether endogenous c-Src and AF-6 are able to interact. First, we performed colocalization studies in epithelial MCF10A cells. Indeed, c-Src and AF-6 colocalized exclusively at cell–cell contacts, as shown by confocal microscopy (Figure 3C). Second, we performed coprecipitation assays with different cell lines. Endogenous AF-6 coprecipitated with c-Src in lysates from HEK 293 and MCF10A (Figure 3D and E). In addition, c-Src coprecipitated with AF-6 in lysates from MCF10A and MDCK cells (Figure 3E and F). Finally, in mouse brain extracts, c-Src also coprecipitated with AF-6 (Figure 3G). These experiments imply a physiological relevance of the interaction between c-Src and AF-6 without excluding other PDZ proteins as putative interaction partners of c-Src.
Kinase activity of c-Src influences binding of c-Src to AF-6
We assumed that the interaction between c-Src and AF-6 was regulated and depended on the activation state of the cell or on the kinase activity of c-Src. To test this, we coexpressed Src-L and Src-A with AF-6 in the absence or presence of the activated Ras mutant, RasG12V, in HEK 293 cells. Src-L but not Src-A bound to AF-6, whereas in cells coexpressing activated Ras, neither Src-L nor Src-A bound to AF-6 (Figure 4A). Thus, activation of cells reduced the binding of c-Src to AF-6.
Figure 4.
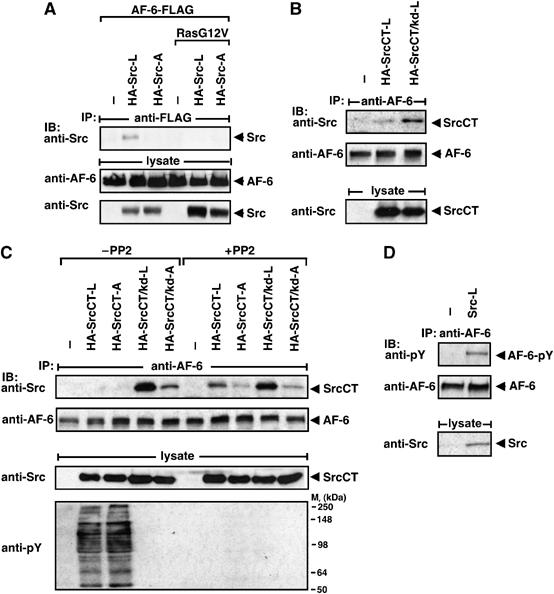
Effect of the kinase activity of c-Src on its binding to AF-6. (A) HEK 293 cells were transiently transfected with empty vector (−) or constructs as indicated. The interaction between AF-6 and Src proteins was detected by precipitation with anti-FLAG and blotting with anti-Src antibodies. Protein expression was controlled by blotting with anti-AF-6 and anti-Src antibodies. (B, C) HEK 293 cells were transiently transfected and pretreated with or without the Src kinase inhibitor PP2 1 h before lysis. The interaction between endogenous AF-6 and SrcCT proteins was detected by precipitation with anti-AF-6 and blotting with anti-Src antibodies. Blotting with anti-pY antibody was included for control of Src activity. (D) Endogenous AF-6 was precipitated from Src-L expressing cells and blotted with anti-pY antibody.
We asked whether a similar effect can be also obtained with activated c-Src. To test this, we used the N-terminal deletion mutant SrcCT (SrcCT-L) (Figure 1B), which is a strong, constitutively active kinase (Figure 4C). SrcCT-L bound only weakly to AF-6, whereas the corresponding kinase-defective protein SrcCT/kd-L bound more strongly (Figure 4B and C). Similarly, inhibition of the kinase activity of SrcCT-L by the Src-specific kinase inhibitor PP2 increased its binding to AF-6 even though slightly less efficiently (Figure 4C). In vitro binding studies showed that the kinase activity did not influence the interaction with the isolated AF-6 PDZ domain (Figure 3B and data not shown). We observed Src-dependent phosphorylation of AF-6 and a weak phosphorylation of the isolated PDZ domain (Figure 4D and data not shown).
Taken together, these data demonstrate that in cells a strong kinase activity of c-Src interferes with its binding to AF-6, and Src-dependent phosphorylation of AF-6 may regulate this interaction.
SrcCT bound to AF-6 is not phosphorylated at Tyr527
We have shown above that the c-Src kinase activity counteracts c-Src interaction with AF-6. Since the kinase activity is regulated by phosphorylation of Tyr527 and Tyr416, we were wondering whether AF-6 affected their phosphorylation. CSK phosphorylates Tyr527, and in concert with dephosphorylation of Tyr416, this inactivates c-Src. Activated c-Src is the substrate for CSK. To prevent an influence of intramolecular interactions between the N-terminal part and the kinase domain, we first analysed SrcCT containing Tyr416 and Tyr527 for phosphorylation.
SrcCT-L showed a stronger phosphorylation on Tyr527 than SrcCT/kd-L (Figure 5A). Thus, increased Tyr527 phosphorylation correlated with decreased binding to AF-6 (Figure 4C). To verify this, we overexpressed AF-6 and found that it markedly reduced phosphorylation of SrcCT-L and SrcCT/kd-L (Figure 5A). SrcCT-A and SrcCT/kd-A, which only showed residual binding to AF-6 (Figure 4C), showed increased phosphorylation of Tyr527, which was not affected by overexpressed AF-6 (Figure 5A).
Figure 5.
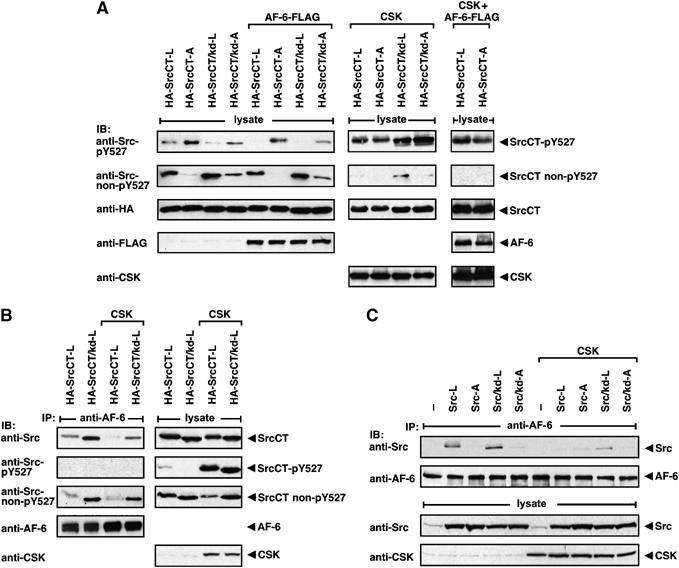
Src phosphorylation on Tyr527 and coprecipitation with AF-6 in the presence of CSK. (A) HEK 293 cells were transiently transfected as indicated. The phosphorylation state of SrcCT on Tyr527 was analysed by blotting using anti-Src-pY527 and anti-Src non-pY527 antibodies. Expression of AF-6, SrcCT and CSK constructs was controlled by immunoblotting. (B, C) HEK 293 cells were transiently transfected as indicated. Cell lysates were incubated with anti-AF-6 antibody and coprecipitating Src proteins were detected by immunoblotting with Src-specific antibodies, as indicated.
Having demonstrated that binding of the AF-6 PDZ domain to SrcCT-L and SrcCT/kd-L reduced phosphorylation of Tyr527, we attempted to revert this effect by CSK to strengthen the result. Overexpression of CSK strongly increased phosphorylation of Tyr527 even in the presence of AF-6, indicating that CSK overcomes the inhibitory effect of AF-6 on Tyr527 phosphorylation under these conditions (Figure 5A). Therefore, we analysed the effect of overexpressed CSK on the interaction between SrcCT and AF-6 (Figure 5B). Indeed, CSK expression reduced the binding of Src-L to AF-6, correlating with increased phosphorylation of Tyr527. Consistent with this, SrcCT bound to AF-6 was not phosphorylated at Tyr527. The SrcCT/kd-L mutant dephosphorylated at Tyr527 bound more efficiently to AF-6 (Figure 5B), presumably because it lacks the kinase activity that counteracts the binding to AF-6 (Figure 4B). Analysis of Src full-length proteins showed that coprecipitation of Src-L and Src/kd-L with AF-6 was also reduced by overexpression of CSK (Figure 5C). Thus, binding of AF-6 to c-Src prevents the accessibility for CSK and therefore c-Src bound in a complex to AF-6 is not phosphorylated at Tyr527.
The kinase activity of c-Src is reduced by the PDZ domain of AF-6
So far we have shown that binding of the AF-6 PDZ domain to SrcCT diminished phosphorylation on Tyr527. However, c-Src is not only regulated by phosphorylation on Tyr527 but also by autophosphorylation on Tyr416, which correlates with an increased kinase activity and substrate phosphorylation. To test the effect of AF-6 on Tyr416 phosphorylation, we compared the phosphorylation state of Src-L and Src-A in HEK 293 cells stably expressing an AF-6 short hairpin (sh)RNA or a control shRNA (Figure 6A). Endogenously expressed c-Src and overexpressed Src-L showed increased phosphorylation at Tyr416 in AF-6 knockdown cells compared with control cells (Figure 6A, compare lanes 2 and 4 with 1 and 3). As expected, Src-A, with only residual binding to AF-6, is not affected in its phosphorylation at Tyr416 by knockdown of AF-6 (Figure 6A, lanes 5 and 6). Overall phosphorylation of proteins on tyrosine correlated with the level of Tyr416 phosphorylation. The in vitro kinase activity of Src-L is slightly increased in AF-6 knockdown cells compared with control cells, a difference not detectable with Src-L immunopurified from RIPA lysates (Figures 6B and 1E). Recombinant AF-6 PDZ protein was not effective in inhibiting purified Src-L, implying that the PDZ domain of AF-6 per se does not interfere with c-Src kinase activity (data not shown).
Figure 6.
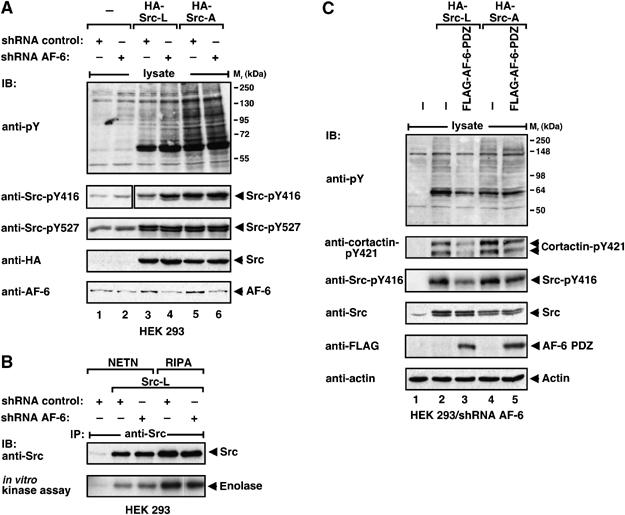
The kinase activity of c-Src is regulated by the AF-6 PDZ domain. (A) HEK 293 cells stably expressing control or AF-6 shRNA were transiently transfected as indicated. Lysates of starved cells were analysed by blotting with anti-pY antibody. For analysis of cortactin and c-Src phosphorylation, lysates were blotted with anti-cortactin-pY421, anti-Src-pY416 or anti-Src-pY527 antibodies. Expression of HA-Src proteins and FLAG-AF-6-PDZ was controlled by blotting with anti-HA or anti-FLAG antibodies, respectively. (B) Immunopurified Src proteins were analysed by in vitro kinase assays with enolase as substrate. (C) HEK 293 cells expressing Src-L or Src-A in the absence or presence of FLAG-AF-6-PDZ were analysed as described in (A).
In AF-6 knockdown cells, the pattern of substrate phosphorylation by Src-L and Src-A were similar, since AF-6 did not restrict substrate phosphorylation by c-Src (Figure 6C, lanes 2 and 4). Expression of the AF-6 PDZ domain reduced the effect of Src-L on tyrosine phosphorylation and phosphorylation of the Src-specific substrate cortactin (Figure 6C, lanes 2 and 3). Reduced substrate phosphorylation correlated with decreased autophosphorylation of Src-L at Tyr416. Expression of the AF-6 PDZ domain did not interfere with Src-A effects (Figure 6C, lanes 4 and 5).
Thus, in cells, the accessibility of c-Src to substrates and their tyrosine phosphorylation and the autophosphorylation of c-Src at Tyr416 are reduced by the presence of the AF-6 PDZ domain.
Cellular localization and substrate phosphorylation of c-Src is regulated by AF-6
Endogenous AF-6 accumulated in cell–cell contacts in MCF10A cells (Figure 3C), where it colocalized with endogenous c-Src. To test whether AF-6 is involved in regulating c-Src in regions of cell–cell contacts, we visualized AF-6 and c-Src in serum-starved or EGF-stimulated MCF10A control or shRNA AF-6 cells by confocal microscopy. c-Src localized to regions of cell–cell contact in starved control cells (arrows; Figure 7A), although staining was less marked than in cells grown in complete growth medium (Figure 3C). However, no accumulation of c-Src staining in cell–cell contacts was detectable in shRNA AF-6 cells (arrowheads; Figure 7A). Upon EGF stimulation, c-Src staining in regions of cell–cell contact increased markedly in control cells but not in shRNA AF-6 cells. These data imply the contribution of AF-6 in maintaining c-Src in regions of cell–cell contact in starved cells and in recruiting c-Src after growth factor stimulation.
Figure 7.
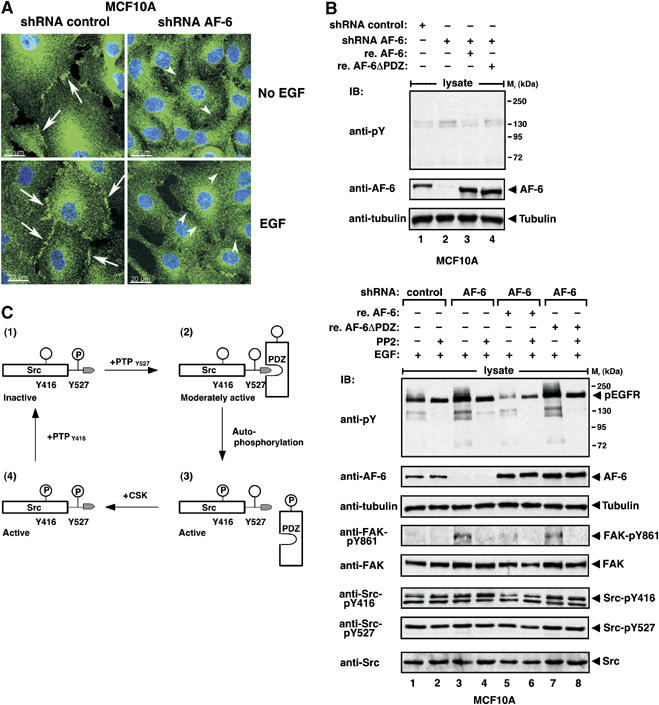
Cellular localization and substrate phosphorylation of c-Src is regulated by AF-6. (A) Cellular localization of c-Src in MCF10A cells stably expressing control or AF-6 shRNA. Cells were starved for 24 h, treated with 100 ng/μl EGF for 10 min as indicated, fixed and immunostained with anti-Src antibody. Knockdown cells show reduced Src localization at cell–cell contacts (arrowheads) compared with control cells (arrows). Maximum-intensity projections of confocal image stacks are shown; scale bar is 20 μm. (B) MCF10A cells stably expressing control or AF-6 shRNA and AF-6 knockdown cells reconstituted (re.) with AF-6 (re. AF-6) or AF-6ΔPDZ (re. AF-6ΔPDZ) were starved for 18 h, pretreated with or without the Src kinase inhibitor PP2 1 h before lysis and stimulated with 100 ng/μl EGF for 10 min, as indicated. Tyrosine phosphorylation of proteins was analysed by blotting using anti-pY antibody. Phosphorylation of Src and FAK was analysed using anti-FAK-pY861, anti-Src-pY416 and anti-Src-pY527 antibodies. Expression of AF-6 and equal loading were controlled by blotting with anti-AF-6 or anti-tubulin antibody, respectively. (C) Model for the regulation of c-Src by PDZ proteins. For details see Discussion.
In order to further analyse the role of AF-6 in c-Src regulation, we tested the phosphorylation state of c-Src- and Src-dependent substrate phosphorylation (Figure 7B). Knockdown of AF-6 in serum-starved MCF10A had slight effects on substrate phosphorylation on tyrosine (Figure 7B, top blot, lanes 1 and 2). This effect was more pronounced in cells stimulated with EGF (Figure 7B, bottom blot, lanes 1 and 3). To determine whether the PDZ domain of AF-6 mediated the regulatory effects on c-Src, we reconstituted AF-6 knockdown cells with AF-6 or AF-6 lacking the PDZ domain (AF-6ΔPDZ). Reconstitution with AF-6 but not with AF-6ΔPDZ reverted the effect and restricted Src-dependent tyrosine phosphorylation, comparable to control cells (Figure 7B, bottom, lanes 1, 5 and 7). FAK, a c-Src substrate that localizes to cell–cell contacts, showed increased phosphorylation at the Src-specific phosphorylation site Tyr861 in AF-6 knockdown cells (Figure 7B, bottom). This effect was reverted by re-expression of AF-6 but not AF-6ΔPDZ, correlating with overall tyrosine phosphorylation of substrates. The Src-specific inhibitors PP2 (Figure 7B, bottom) and SU6656 (data not shown) almost completely prevented Src-dependent substrate phosphorylation apart from a strong phosphorylated protein representing the phosphorylated EGF receptor. Autophosphorylation of c-Src at Tyr416 was slightly increased by AF-6 knockdown and decreased again upon re-expression of AF-6 but not AF-6ΔPDZ. Phosphorylation of c-Src at Tyr527 was not affected under these conditions, presumably since EGF stimulation only affected a subpopulation of c-Src (Figure 7B, bottom). Thus, AF-6 via its PDZ domain reduces Src-dependent substrate phosphorylation in epithelial cell lines.
Discussion
We identified a novel mechanism for c-Src regulation via its extreme C-terminal sequence. We demonstrate that c-Src is restricted in its cellular activity by a PDZ domain (Figures 1 and 2). We attribute this effect to the C-terminal PDZ binding motif of c-Src with a terminal hydrophobic residue Leu and its binding to a PDZ protein. Mutation of this amino-acid residue does not change the intrinsic c-Src kinase activity in vitro (Figure 1E), suggesting that a PDZ domain regulates c-Src properties in cells. Many PDZ proteins play a role at cell–cell junctions. We demonstrate that the PDZ domain of such a junctional protein, AF-6, can bind to c-Src (Figure 3). AF-6 can recruit c-Src to sites of cell–cell contacts (Figures 3C and 7A). SrcCT, which is dephosphorylated at Tyr527, binds to AF-6 and the AF-6 PDZ domain reduces phosphorylation of active SrcCT at Tyr527, the phosphorylation site of CSK (Figure 5A and B). Together with reduced autophosphorylation at Tyr416, this results in a partially activated kinase activity of c-Src in cells (Figures 6 and 7). Strong activation of the c-Src kinase or overexpression of CSK disrupts the c-Src/AF-6 interaction and releases c-Src from restriction in substrate phosphorylation (Figures 4 and 5).
The C-terminal tail of c-Src is an important regulatory region. C-terminal deletions present in the oncogenic proteins v-Src and the colon cancer-specific Src-Δ6CT lead to cellular transformation. The C-terminal PDZ ligand sequence of c-Src, which we characterized here, fine-tunes c-Src more subtly. It allows binding to a PDZ domain and thereby restricts the number of substrates phosphorylated by c-Src and maintains the physiologically required c-Src kinase activity. Since many ligands interact with different PDZ domains, we assume that additional PDZ proteins might bind to c-Src.
The C-terminal sequence of c-Src is a ligand for PDZ domains
The C-terminus of c-Src, GENL, is a typical ligand of class III PDZ domains. So far, the AF-6 PDZ domain has been described to bind ligands typical for class I and class II (Nourry et al, 2003). However, the typical class III domain of nNOS, which binds ligands similar to the C-terminal sequence of c-Src, also interacts with ligands of other PDZ domain classes (Jaffrey et al, 1998).
The affinity between a ligand and its PDZ domain can be regulated by phosphorylation. Phosphorylation of the ligand at position −2 can reduce or prevent its binding to a PDZ domain (Cao et al, 1999; Kuhne et al, 2000). In the case of c-Src, the residue at position −2 is negatively charged, which mimics phosphorylation. This may explain the relatively low affinity of the C-terminus of c-Src for the AF-6 PDZ domain. We show that c-Src kinase activity leads to disruption of the AF-6/c-Src complex, presumably by Src-dependent phosphorylation of AF-6 (Figure 4). Conversely, inhibition of the c-Src kinase activity or use of a kinase-defective c-Src mutant increased the interaction with AF-6. Interaction of two other AF-6 PDZ domain binders, the receptor tyrosine kinase EphA7 and the Ser/Thr kinase Bcr, is known to be regulated by phosphorylation of AF-6 (Hock et al, 1998; Radziwill et al, 2003). However, in these cases, phosphorylation of AF-6 increased its binding to the ligand. The specific responses may depend on phosphorylation at different sites in AF-6 and on the cellular background.
Two other SFKs, c-Yes and c-Fyn, may also be regulated by PDZ domains, as they share with c-Src the identical C-terminal sequence (Figure 1A). Supporting this notion, we could observe that Fyn-L but not the mutant Fyn-A interacted with the PDZ domain of AF-6 (data not shown). c-Src, c-Yes and c-Fyn form a subfamily of ubiquitously expressed SFKs distinct from the other family members, which are mainly present in non-adherent cells such as haematopoietic cells. Our findings indicate that c-Src, c-Yes and c-Fyn are predestined for a role in PDZ domain-dependent regulation of cell–cell contacts.
Regulation of c-Src by the PDZ domain of AF-6
c-Src colocalizes with AF-6 at cell–cell junctions in epithelial cells, and stimulation of cells with EGF increases AF-6-dependent recruitment of c-Src to these sites (Figure 7A). One known Src substrate localized at cellular junctions is cortactin, which links membrane-associated proteins to the underlying cytoskeleton. Cortactin plays a role in cell migration and invasive growth (Lua and Low, 2005). Tyrosine phosphorylation of cortactin increases F-actin turnover to promote actin dynamics, which may support invasive growth. The PDZ domain of AF-6 restricts Src-dependent phosphorylation of cortactin (Figure 6). This may contribute to the maintenance of cellular junctions and is in agreement with our recent observation that knockdown of AF-6 in epithelial cells reduces cell–cell adhesion (Lorger and Moelling, 2006).
For maintenance of cell–cell junctions, a role of c-Yes, a close relative to c-Src, has been described. (Chen et al, 2002). c-Yes phosphorylates the junctional protein occludin to promote the maintenance of an epithelial cell layer. Inhibition of c-Yes basal kinase activity loosens cell–cell contacts. Strongly activated or oncogenic c-Src has the same effect, leading to disruption of cell–cell contacts (Behrens et al, 1993). We observed that highly activated c-Src or Ras disrupts the binding of c-Src to AF-6, allowing additional substrates to be phosphorylated, whereas a moderately activated c-Src is bound to AF-6 and restricted in the number of substrates perhaps by spatial limitations. We suggest that the restriction of the c-Src activity by AF-6 may promote the formation and maintenance of cell–cell contacts depending on moderate c-Src kinase activity.
Model of the molecular mechanism for regulation of c-Src by PDZ proteins
Based on the results of this study, we propose the model for regulation of c-Src shown in Figure 7C. For simplification, we only considered regulation of c-Src by phosphorylation of Tyr416 and Tyr527 located at its C-terminal part (Roskoski, 2004; Yeatman and Roskoski, 2004). Inactive c-Src is not phosphorylated on Tyr416 but on Tyr527 (Figure 7C, 1), which allows an intramolecular interaction with the SH2 domain, leading to inhibition of the kinase activity. Dephosphorylation of Tyr527 leads to partial activation of c-Src, after which c-Src interacts with the PDZ protein AF-6 (Figures 5 and 6 and 7C, 2). This interaction functionally and locally restricts c-Src. Full activation of c-Src kinase activity requires Tyr416 phosphorylation, which decreases PDZ domain binding (Figures 4C and 7C, 3), presumably due to Src-dependent phosphorylation of AF-6 (Figure 4D). Release of c-Src from AF-6 increases accessibility of c-Src for CSK, which could explain the increased phosphorylation on Tyr527 (Figures 5 and 7C, 4). After CSK-mediated phosphorylation of Tyr527, phosphatases specific for Tyr416 inactive c-Src (Figure 7C, 1). We propose that a PDZ protein maintains c-Src in an intermediate moderately active state. This may be physiologically relevant for certain biological processes such as regulation of cellular junctions.
The crystal structure of an unphosphorylated Src mutant, carrying a small truncation in its N-terminus, shows the C-terminal residue Leu bound to a hydrophobic pocket located in the catalytic domain (Cowan-Jacob et al, 2005). Interestingly, this Src mutant has a conformation typical for an active kinase, with the intramolecular interaction preventing phosphorylation on Tyr527. We demonstrate that the C-terminal residue Leu binds to the hydrophobic pocket of a PDZ domain. Whether inter- and intramolecular interactions compete will require further studies. However, inter- and intramolecular binding of the C-terminal sequence of c-Src to hydrophobic pockets shield c-Src against Tyr527 phosphorylation and prevent its inactivation as well as its aberrant activation. The function of AF-6 in our model is to recruit c-Src to specific subcellular regions in the cellular periphery and to maintain it there in a partially activated state. Thus, AF-6 allows c-Src to phosphorylate only a limited or specific set of substrates. Other c-Src binding PDZ proteins may exist and may have effects similar as AF-6. Ligands with low binding affinities for the PDZ domain may be competed out by ligands with stronger binding affinity. In the cellular context, the theoretically calculated values may not apply, because the ligand/PDZ domain interaction may be favoured by compartmentation and regulated by modifications, for example, phosphorylation. Furthermore, PDZ proteins composed of other domains may impose additional roles on c-Src. This study provides evidence for a role of PDZ proteins on regulation of c-Src, which may be of general importance.
Materials and methods
Plasmids
cDNAs for mouse Src-L and Src/kd-L (K296R) were purchased from Upstate. Src-Δ6CT was generated using standard PCR procedures. Src-A and Src/kd-A mutants were generated by using the QuickChange site-directed mutagenesis kit (Stratagene). The coding sequence for the HA tag was inserted by PCR 3′ to the Src myristoylation sequence (residues 1–16 in c-Src). HA-SrcCT constructs were generated by inserting the HA tag coding sequence 5′ to the cDNAs corresponding to residues 254–535. For inducible expression, Src cDNAs were subcloned into pRTP (Heinrich et al, 2000). AF-6 and RasG12V plasmids were described elsewhere (Radziwill et al, 2003). The SH2 domain of Src (residues 144–249) was cloned into the vector pGEX-6P2 (Amersham Pharmacia Biotech). pCMV-Ad-CSK was a gift from Dr Robert Schneider.
Antibodies
The following antibodies were used: anti-FLAG-M2 (Sigma); anti-HA (12CA5) (Roche); anti-AF-6 (clone 35) (Transduction Laboratories); anti-β-tubulin (Babco); anti-phosphotyrosine 4G10, anti-Src GD11 (Upstate); anti-c-Src (Src2), anti-cortactin (H-191), anti-FAK (C-20), anti-actin (I-19), anti-ERK2 (C-14) and anti-CSK (C-20) (Santa Cruz); anti-Src-pY416, anti-Src non-pY416, anti-Src-pY527-Src, anti-Src non-pY527 and anti-Src (32G6) (Cell Signaling Technology); anti-GST (Amersham Pharmacia Biotech); anti-pY421-cortactin and anti-pY861-FAK (Biosource).
Cell culture, transfection and retroviral transduction
HEK 293 cells, SYF−/− cells (Klinghoffer et al, 1999), MDCK and MCF10A cells were obtained from the American Tissue Culture Cooperation and cultured as described (Lorger and Moelling, 2006). Inducible SYF−/− cells additionally were supplemented with 100 ng/ml tetracyclin.
Transfection was performed by the jetPEI (Polyplus-Transfection) or the Lipofectamine 2000 (Invitrogen) method. For starvation of cells, the medium was replaced by medium without FCS 24 h after transfection and cells were incubated for additional 18 h.
HEK 293 cells stably expressing shRNAs against AF-6 or firefly luciferase (GL2), respectively, and SYF−/− cells inducibly expressing Src constructs, were generated by retroviral transduction as described previously (Ziogas et al, 2005). MCF10A cells stably expressing shRNA against AF-6 or GL2, and AF-6 knockdown cells reconstituted for AF-6 and AF-6ΔPDZ are described (Lorger and Moelling, 2006). For inhibition of Src kinase activity, cells were incubated with the Src inhibitor PP2 or SU6656 (Calbiochem) 1 h before lysis.
Immunoprecipitations and immunoblots
Cells were lysed in NETN buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40 supplemented with 40 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulphonyl fluoride, Complete Mini (Roche) protease inhibitor). For immunoprecipitation, equal protein amounts from lysates were incubated with antibodies for 2 h, followed by addition of protein G–Sepharose beads for 1 h, and immunoblotted (Radziwill et al, 2003).
In vitro Src kinase assay
Src proteins immunopurified from NETN or RIPA (NETN supplemented with 0.5%. NP-40, 0.5% DOC and 0.1% SDS) lysates were assayed in kinase buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl2, 5 mM MnCl2, 1 mM dithiothreitol, 0.1 mM Na3VO4). containing 5 μCi γ-32P ATP (Redivue; Amersham Pharmacia Biotech) and 1 μg of acetic-denatured recombinant enolase (Roche) as substrate for 15 min at 37°C (Ziogas et al, 2005).
Preparation of GST fusion proteins and pull-down assay
Glutathione-S-transferase fusion proteins were purified from Escherichia coli BL21plus (Stratagene) as described previously (Radziwill et al, 2003). For pull-down assays, GSH–sepharose beads carrying 3 μg of GST fusion protein were used.
PDZ domain binding studies
Peptides corresponding to residues 524–535 of Src and derivatives were synthesized according to standard Fmoc-chemistry on a multiple peptide synthesizer (Abimed GmbH, Langenfeld, Germany) using TentaGel S PHB-aa Fmoc resin (0.25 mmol/g; Rapp Polymere, Tübingen, Germany). Peptides were synthesized with an N-terminal cysteine for the coupling onto maleimide-functionalized Whatman 50 cellulose membrane as described previously (Otte et al, 2003). Membranes were blocked for 1 h with NETG (0.25% gelatine in NET: 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.05% Triton-X 100), incubated with GST-AF-6-PDZ (0.1 μg/ml) or GST-Src-SH2 (0.2 μg/ml) for overnight at 4°C and washed, followed by an incubation with anti-GST antibody for overnight at 4°C. Membranes were washed with NET before being incubated with secondary antibody coupled to horseradish peroxidase. Bound proteins were visualized by ECL (Amersham).
Colony formation (soft agar), matrigel invasion and focus formation assays
Expression of Src proteins in SYF−/−/Src-L, SYF−/−/Src-A or SYF−/−/Src-Δ6CT was induced by removing tetracycline for 24 h. For the soft agar assay, cells were seeded in 0.7% top agar in six-well plates (5 × 104 cells/well) coated with 1% agar, and incubated for 10–14 days until colonies were formed. For the matrigel invasion assay, 1 × 105 cells were seeded into matrigel chambers (BD Bioscience) in serum- and tetracycline-free medium, with full medium in the well below, and assayed according to the manufacturer's protocol. For the focus formation assay, 1 × 105 cells were seeded on 60-mm plates and expression was induced by removing tetracycline for 24 h. Cells then were incubated in tetracycline-free media with 3% FCS for 10–14 days until foci were formed, which were stained with crystal violet and counted.
For statistical analysis, Student's t-test was used to compare data between two groups. Values are expressed as mean±s.d. of duplicate samples. P<0.05 was considered as statistically significant.
Immunofluorescence microscopy
Cells were fixed and permeabilized in ice-cold 100% methanol for 2 min at −20°C and after three washes in PBS blocked with 10% FBS in PBS for 15 min. After fixation and blocking, cells were incubated for 1 h with primary anti-AF-6 (clone 35) and anti-Src GD11 antibodies at 1:200 dilution in 5% FBS in PBS. Secondary anti-mouse Ig subclass-specific antibodies (Jackson Immuno Research) coupled to FITC (fluorescein isothiocyanate) or Rhodamine Red-X were used for detection at 1:100 dilution in 5% FBS in PBS. Confocal image stacks were recorded with an inverted Leica SP5 microscope and corresponding Leica software. Image analysis was performed on Imaris and images are shown either as single confocal sections or as maximum-intensity projections of confocal sections.
Acknowledgments
We thank Drs Rüdiger Erdmann, Günter Dollenmeier, Gabriela Burkard and Mihaela Lorger for their support in the initial phase of this study. We are grateful to Julia Dennler and Denise Strasser for their excellent technical assistance. This work was supported in part by the Swiss National Fonds and the Cancer League Zurich
References
- Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol 120: 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorge JD, Jakymiw A, Fujita DJ (2000) Selected glimpses into the activation and function of Src kinase. Oncogene 19: 5620–5635 [DOI] [PubMed] [Google Scholar]
- Boettner B, Govek EE, Cross J, Van Aelst L (2000) The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc Natl Acad Sci USA 97: 9064–9069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisguerin P, Leben R, Ay B, Radziwill G, Moelling K, Dong L, Volkmer-Engert R (2004) An improved method for the synthesis of cellulose membrane-bound peptides with free C termini is useful for PDZ domain binding studies. Chem Biol 11: 449–459 [DOI] [PubMed] [Google Scholar]
- Buchert M, Schneider S, Meskenaite V, Adams MT, Canaani E, Baechi T, Moelling K, Hovens CM (1999) The junction-associated protein AF-6 interacts and clusters with specific Eph receptor tyrosine kinases at specialized sites of cell–cell contact in the brain. J Cell Biol 144: 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M (1999) A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature 401: 286–290 [DOI] [PubMed] [Google Scholar]
- Chen YH, Lu Q, Goodenough DA, Jeansonne B (2002) Nonreceptor tyrosine kinase c-Yes interacts with occludin during tight junction formation in canine kidney epithelial cells. Mol Biol Cell 13: 1227–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YP, Ia KK, Mulhern TD, Cheng HC (2005) Endogenous and synthetic inhibitors of the Src-family protein tyrosine kinases. Biochim Biophys Acta 1754: 210–220 [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T (2005) The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 13: 861–871 [DOI] [PubMed] [Google Scholar]
- Frame MC (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 1602: 114–130 [DOI] [PubMed] [Google Scholar]
- Heinrich J, Bosse M, Eickhoff H, Nietfeld W, Reinhardt R, Lehrach H, Moelling K (2000) Induction of putative tumor-suppressing genes in Rat-1 fibroblasts by oncogenic Raf-1 as evidenced by robot-assisted complex hybridization. J Mol Med 78: 380–388 [DOI] [PubMed] [Google Scholar]
- Hock B, Bohme B, Karn T, Yamamoto T, Kaibuchi K, Holtrich U, Holland S, Pawson T, Rubsamen-Waigmann H, Strebhardt K (1998) PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proc Natl Acad Sci USA 95: 9779–9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irby RB, Yeatman TJ (2000) Role of Src expression and activation in human cancer. Oncogene 19: 5636–5642 [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH (1998) CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron 20: 115–124 [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J 18: 2459–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhne C, Gardiol D, Guarnaccia C, Amenitsch H, Banks L (2000) Differential regulation of human papillomavirus E6 by protein kinase A: conditional degradation of human discs large protein by oncogenic E6. Oncogene 19: 5884–5891 [DOI] [PubMed] [Google Scholar]
- Lorger M, Moelling K (2006) Regulation of epithelial wound closure and intercellular adhesion by interaction of AF6 with actin cytoskeleton. J Cell Sci 119: 3385–3398 [DOI] [PubMed] [Google Scholar]
- Lua BL, Low BC (2005) Cortactin phosphorylation as a switch for actin cytoskeletal network and cell dynamics control. FEBS Lett 579: 577–585 [DOI] [PubMed] [Google Scholar]
- Martin GS (2001) The hunting of the Src. Nat Rev Mol Cell Biol 2: 467–475 [DOI] [PubMed] [Google Scholar]
- Martinez-Quiles N, Ho HY, Kirschner MW, Ramesh N, Geha RS (2004) Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol Cell Biol 24: 5269–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourry C, Grant SG, Borg JP (2003) PDZ domain proteins: plug and play!. Sci STKE 2003: RE7. [DOI] [PubMed] [Google Scholar]
- Otte L, Wiedemann U, Schlegel B, Pires JR, Beyermann M, Schmieder P, Krause G, Volkmer-Engert R, Schneider-Mergener J, Oschkinat H (2003) WW domain sequence activity relationships identified using ligand recognition propensities of 42 WW domains. Protein Sci 12: 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziwill G, Erdmann RA, Margelisch U, Moelling K (2003) The Bcr kinase downregulates Ras signaling by phosphorylating AF-6 and binding to its PDZ domain. Mol Cell Biol 23: 4663–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ress A, Moelling K (2006) Interaction partners of the PDZ domain of Erbin. Protein Pept Lett 13: 877–881 [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr (2004) Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun 324: 1155–1164 [DOI] [PubMed] [Google Scholar]
- Schneider S, Buchert M, Georgiev O, Catimel B, Halford M, Stacker SA, Baechi T, Moelling K, Hovens CM (1999) Mutagenesis and selection of PDZ domains that bind new protein targets. Nat Biotechnol 17: 170–175 [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275: 73–77 [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A (1991) Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64: 693–702 [DOI] [PubMed] [Google Scholar]
- Sun G, Sharma AK, Budde RJ (1998) Autophosphorylation of Src and Yes blocks their inactivation by Csk phosphorylation. Oncogene 17: 1587–1595 [DOI] [PubMed] [Google Scholar]
- Takai Y, Nakanishi H (2003) Nectin and afadin: novel organizers of intercellular junctions. J Cell Sci 116: 17–27 [DOI] [PubMed] [Google Scholar]
- Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, Volkmer-Engert R, Oschkinat H, Ay B, Radziwill G, Dong L (2004) Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. An improved method for the synthesis of cellulose membrane-bound peptides with free C termini is useful for PDZ domain binding studies. J Mol Biol 343: 703–718 [DOI] [PubMed] [Google Scholar]
- Yeatman TJ, Roskoski R Jr (2004) A renaissance for SRC. Src protein-tyrosine kinase structure and regulation. Nat Rev Cancer 4: 470–480 [DOI] [PubMed] [Google Scholar]
- Ziogas A, Moelling K, Radziwill G (2005) CNK1 is a scaffold protein that regulates Src-mediated Raf-1 activation. J Biol Chem 280: 24205–24211 [DOI] [PubMed] [Google Scholar]