

Abstract
Steroid receptor coactivators (SRCs) exert profound effects on animal development and physiology. These coactivators are nuclear proteins and transcription co-regulators that function to facilitate the transcription initiation mediated by nuclear receptors, as well as by other well-known transcription factors. However, how these co-regulators are functionally regulated is poorly understood. During genome-wide screening for SRC-interacting proteins, we identified a novel ankyrin repeat containing protein, SIP (SRC-Interacting Protein), which interacts with SRC coactivators in the cytoplasm. We demonstrated that extracellular stimuli such as the addition of estrogen, induced phosphorylation of SIP in its PEST (Proline, Glutamate, Serine, and Threonine rich) domain by casein kinase II. The phosphorylation of SIP resulted in dissociation of SRC proteins from SIP in the cytoplasm and led to subsequent nuclear translocation of SRC proteins and gene coactivation. Both gain-of-function and loss-of-function experiments indicate that SIP functions to sequester SRC coactivators in the cytoplasm and buffer the availability of these coactivators, thus providing a mechanism for the regulation of the transcription regulators.
Keywords: breast cancer, coactivator, estrogen receptor, gene transcription
Introduction
Gene transcription activation by transcription factors such as nuclear receptors is accomplished through the recruitment of a series of coactivators or co-regulators. Coactivator proteins fulfill their functions through protein–protein interactions that bridge transcription factors and the basal transcription machinery and through their chromatin remodeling activities that facilitate the assembly of a transcription initiation complex (Glass and Rosenfeld, 2000). Several general coactivators, such as CBP/p300 and pCAF, are recruited by transcription factors to mediate their transcriptional activities (Glass and Rosenfeld, 2000). These proteins are known to possess intrinsic histone acetyltransferase (HAT) activity and are capable of chromatin modification by histone acetylation. The general coactivators can also be recruited by interaction with more specific coactivators, including members of the steroid receptor coactivator (SRC) family, which have been shown to enhance nuclear receptor-mediated gene transcription as well as gene transactivation by other transcription factors such as AP-1, STATs, ETS, p53, E2F, and NFκB (Shang, 2006). Previously, we reported that SRC coactivators are both necessary and sufficient for estrogen receptor (ER)-mediated gene transcription (Shang et al, 2000; Shang and Brown, 2002; Zhang et al, 2006), and we demonstrated that members of the SRC protein family are differentially involved in gene regulation (Yin et al, 2004; Zhang et al, 2004; Wu et al, 2006).
The SRC family includes SRC-1, TIF2/GRIP1/SRC-2 (hereafter referred to as GRIP1), and pCIP/ACTR/AIB1/RAC3/TRAM1/SRC-3 (hereafter referred to as AIB1) (Glass and Rosenfeld, 2000). Structurally, SRC-1, GRIP1, and AIB1 share 40% overall sequence identity, and they all contain, at the amino (N)-terminus, a basic-helix–loop–helix (bHLH) domain and a Per-Arnt-Sim (PAS) domain that are implicated in specific DNA binding and/or heterodimerization with other bHLH–PAS containing proteins (Belandia and Parker, 2000; Kim et al, 2003), and in carboxyl (C)-terminus, these proteins harbor domains that interact with other coactivators such as p300 and CARM1 (Kamei et al, 1996; Chen et al, 1999a). Three LXXLL motifs, also known as NR boxes (for ‘nuclear receptor' binding), are centrally located in all three proteins (Heery et al, 1997; Darimont et al, 1998; McInerney et al, 1998).
SRC coactivators play important roles in animal development and physiology. Genetic studies with gene ablation showed that although both male and female SRC-1-null mice are viable and fertile, they exhibit partial resistance to several hormones, including estrogen, progestin, androgen, and thyroid hormones (Xu et al, 1998; Weiss et al, 1999); elimination of AIB1 has revealed that it is required for normal growth of mouse (Wang et al, 2000; Xu et al, 2000), as well as for some female reproductive functions (Xu et al, 2000), and there is impairment of fertility in both male and female GRIP1-null mice (Gehin et al, 2002), and GRIP1-null mice also display enhanced adaptive thermogenesis (Picard et al, 2002). On the other hand, SRC proteins are also implicated in the pathogenesis of several disease states, especially malignancies. AIB1 has been found to be strongly amplified and/or overexpressed in 64% of primary breast cancers, as well as in some primary ovarian tumors (Anzick et al, 1997; Bautista et al, 1998), and translocation between the GRIP1 gene and the MOZ (monocytic zinc finger) gene has been identified in human acute myeloid leukemia (Carapeti et al, 1998).
Although the mechanistic role of SRC coactivators in gene transcriptional regulation is relatively well characterized, how the function of these proteins themselves is regulated is not clear. SRC proteins are mainly localized and function in the nucleus. Nonetheless, numerous studies indicate that these proteins are constantly shuttling between the nucleus and the cytoplasm (Stenoien et al, 2001; Qutob et al, 2002; Amazit et al, 2003; Wu et al, 2004). How the dynamic changes in subcellular distribution occur and how these changes are correlated to the activity of these proteins remains poorly understood. Here, we report the identification and functional characterization of a novel SRC-interacting protein, designated SIP for SRC-1-Interacting Protein. We have shown that SIP functions to sequester SRC coactivators in the cytoplasm and consequently regulates the transactivation activity of SRC-1 in the nucleus, thus providing a mechanism for regulating the activity of the transcriptional regulators.
Results
Cloning and characterization of SIP
As stated above, SRC coactivators share several important functional domains. The nuclear receptor-interacting domains as well as the carboxy-terminal coactivator-interacting domains are well characterized. Thus, in an effort to identify proteins that interact with different members of the SRC family to define differential biological activities of these proteins, we utilized the amino-terminal (N) fragments of the SRC proteins (SRC-N), which include the bHLH–PAS domains (Figure 1A), to screen for new potential proteins that might be associated with different members of the SRC family. The GAL4-based yeast two-hybrid system 3 from Clontech was used with yeast GAL4 BD (DNA binding domain) fusion constructs, pGBKT7-SRC-1-N, pGBKT7-GRIP1-N, and pGBKT7-AIB1-N, to screen a human mammary cDNA library (Clontech) in the yeast strain AH109. After a four-round screening of 1 × 106 clones with each of the three BD fusion constructs, positive results were obtained for clones containing an ankyrin repeat sequence with SRC-1-N (Figure 1B, left panel). To confirm the interaction of these clones with SRC-1, the cDNA was rescued from the library and retransformed into yeast cells together with the pGBKT7-SRC-1-N construct. The back hybridization experiments confirmed the interaction between this cDNA and SRC-1 (Figure 1B, right panel). In addition, back hybridization experiments in yeast cotransformed with AD (activation domain) construct containing this cDNA and pGBKT7-AIB1-N BD and pGBKT7-GRIP1-N BD plasmids indicated that the protein encoded by this cDNA also interacts with AIB1 and GRIP1.
Figure 1.
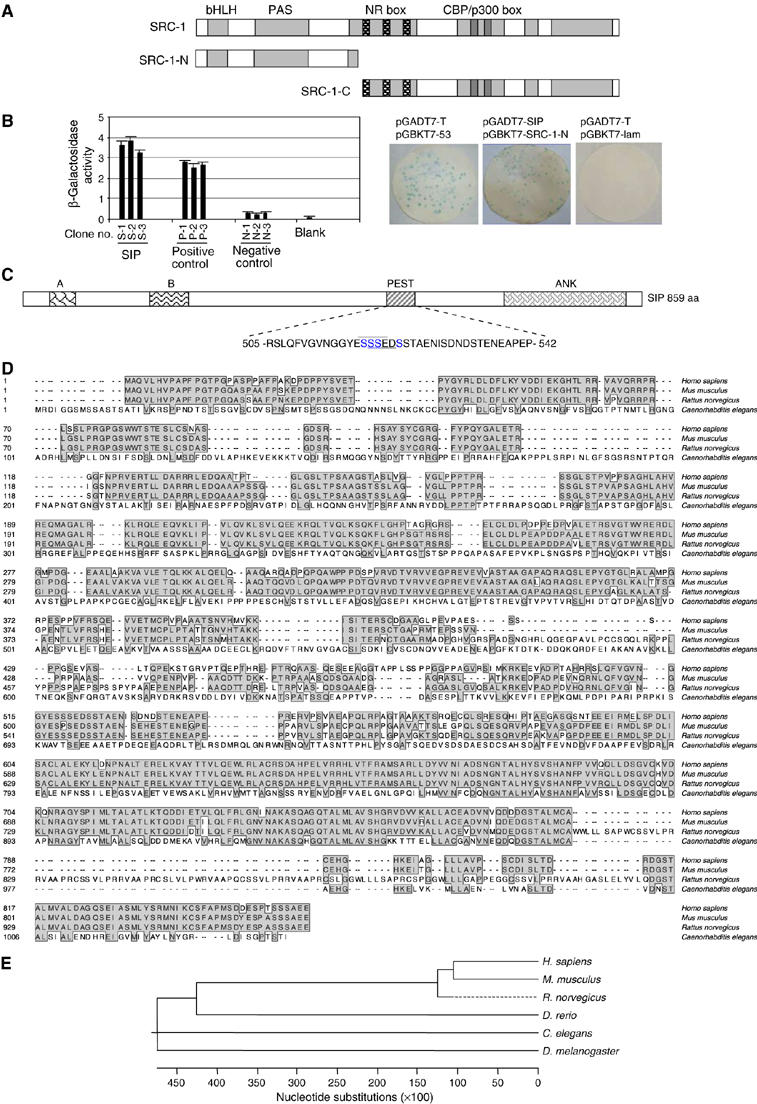
Cloning and characterization of SIP. (A) A schematic representation of the structure of SRC proteins and their deletions. (B) The detection of SIP and SRC-1 interaction by yeast two-hybrid experiments. Left panel: yeast colonies (S, SIP; P, positive control, and N, negative control) were assayed for β-galactosidase activity from liquid culture. Right panel: yeast colonies from back hybridization were assayed for β-galactosidase activity using a colony-lift filter assay; positive and negative controls are shown in left and right, respectively, and positive clones are shown in blue. (C) Schematic representation of SIP protein structure. The following conserved domains are shown: A, B, PEST, and ankyrin repeats (ANK). Overscored and underscored residues within the PEST domain indicate two overlapping putative CK II phosphorylation sequences. (D) Amino-acid sequence alignment of SIP from different species. Shaded residues represent conserved regions. (E) Phylogenetic analysis of the evolutionary relationship among SIP proteins from different species.
DNA sequencing revealed that the cDNA is 2580 bp in length (GenBankTM AY639929) and contains an open reading frame encoding for a protein of 859 amino acids. The predicted molecular mass of this protein is ∼92.2 kDa, with a theoretical isoelectric point 5.28. The corresponding gene was mapped to chromosome 19p13.2 and consists of eight exons and seven introns. We named this gene as SIP for its encoded SRC-Interacting Protein. SIP has been described previously as Ankrd25 (Harada et al, 2005). Bioinformatics analysis indicated that SIP contains an ankyrin repeat domain (668–836), a PEST (Proline, Glutamate, Serine, and Threonine rich) domain, and two additional conserved regions with unknown functions (residues 37–72 and 176–229) (Figure 1C). In addition, the PEST domain harbors two overlapping casein kinase II (CK II) phosphorylation sites (residues 518–521, 519–522). Amino-acid sequence alignment indicated that human SIP shares 81% identity with its mouse homologue, BAC33660.1, and the similarity of the amino-acid sequence of SIP with homologues in other organisms was as following: 75% in Rattus norvegicus, 46% in Danio rerio, 37% in Caenorhabditis elegans, and 36% in Drosophila melanogaster (Figure 1D). Phylogenetic analysis also indicated that SIP is an evolutionarily well-conserved gene (Figure 1E).
In order to confirm the existence of SIP transcript(s) and to examine the expression profile of SIP, we next analyzed the expression of SIP by Northern blotting with Clontech's human multiple tissue blots. The results indicated that SIP was expressed as a single message of ∼5.0 kb in length, with the highest expression in heart, relatively high expression in lung, liver, and skeletal muscle, and low expression in placenta, kidney, and pancreas; no SIP transcript was detected in brain (Figure 2A).
Figure 2.
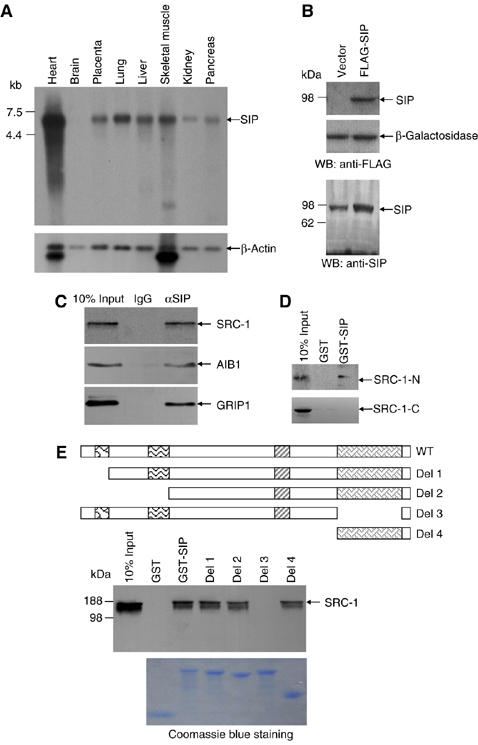
The expression profile of SIP in human tissues and the physical interaction of SIP with SRC-1. (A) Northern blot analysis of SIP expression. (B) Western blotting analysis of SIP expression. MCF-7 cells were transfected with empty vector or a FLAG-tagged SIP expression vector and cellular proteins were prepared for Western blotting with anti-FLAG (upper panel) or anti-SIP (lower panel). (C) Co-immunoprecipitation of SIP and SRC coactivators. Whole-cell lysates from MCF-7 cells were first immunoprecipitated with antibodies against SIP and then immunoblotted with antibodies against SRC-1, GRIP1, or AIB1. (D) SIP interacts with the N-terminus of SRC-1. GST pull-down assays were performed with GST-SIP and 35S-labeled SRC-1-N (1–1896) or SRC-1-C (1897–4326). (E) Mapping the domain of SIP that interacts with SRC-1. GST pull-down experiments were performed with 35S-labeled SRC-1 and GST, GST-SIP, GST-SIP-Del 1 (73–859), GST-SIP-Del 2 (230–859), GST-SIP-Del 3 (1–668+836–859), or GST-SIP-Del 4 (669–859). Schematic representations of the deletion constructs are shown. The bottom panel shows the Coomassie blue staining of the purified GST fusion proteins.
To examine the expression of SIP protein, a FLAG-tagged SIP expression construct was transfected into MCF-7 cells. Forty-eight hours after the transfection, cellular proteins were analyzed by Western blotting with a monoclonal antibody against FLAG. As shown in Figure 2B (upper panel), Western blotting indicated that SIP was expressed as a protein of ∼92 kDa in SIP-transfected MCF-7 cells. Western blotting analysis of the endogenous as well as the overexpressed SIP protein with polyclonal antibodies against SIP that we generated in the late phase of the study with recombinant SIP (1–266 aa) indicated that SIP is a protein with an Mr of ∼92 kDa (Figure 2B, lower panel), confirming its predicted molecular weight.
Physical and functional interaction between SIP and SRC coactivators
To further confirm in vivo interaction between endogenous SIP and SRC coactivators, total proteins from MCF-7 cells were extracted and immunoprecipitated with the antibodies against SIP, and Western blottings were performed with antibodies against SRC-1, AIB1, or GRIP1. The results of these experiments indicated that SIP indeed was co-immunoprecipitated with all three SRC proteins (Figure 2C).
Next, we performed GST pull-down assays to examine the nature (direct or indirect) of the interaction between SIP and SRC coactivators, and, if the interaction is direct, to map the domain(s) in SIP that mediate(s) the interaction with SRC coactivators. In these experiments, wild-type SIP and its deletion mutants were expressed as GST fusion proteins and SRC-1 was in vitro transcribed/translated in the presence of [35S]methionine. The results of the GST pull-down experiments indicated that SIP and SRC-1 interacted directly and that the ankyrin repeats in SIP mediate SIP's interaction with SRC-1 (Figure 2E). GST pull-down experiments also indicated that the SRC-1-N but not SRC-1-C interacted with SIP (Figure 2D), supporting the results obtained from the yeast two-hybrid experiments.
To gain insight into the biological function of the SIP protein, we next analyzed the subcellular localization of this protein. Fluorescent imaging of GFP-SIP in MCF-7 cells indicated that SIP was primarily a cytosolic protein (Figure 3A), implying that SIP may mainly function in the cytoplasm.
Figure 3.
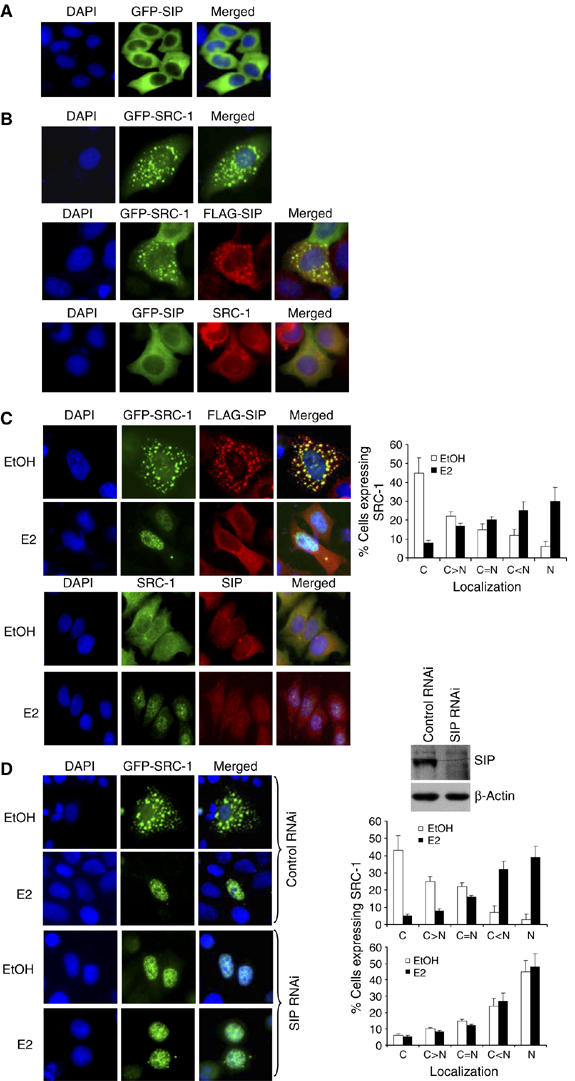
Subcellular localization of SIP and SRC-1. (A) Subcellular localization of SIP protein. MCF-7 cells were transfected with pEGFP-SIP and were visualized by fluorescence microscopy 24 h after the transfection. DAPI staining was also included to visualize the cell nucleus. (B) Subcellular colocalization of SIP and SRC-1. MCF-7 cells were transfected with pEGFP-SRC-1 alone (upper panels), pEGFP-SIP alone (bottom panels), or cotransfected with pEGFP-SRC-1 and pcDNA3.1-FLAG-SIP (middle panels). Twenty-four hours after transfections, EGFP fluorescence and rhodamine staining of FLAG (middle panels) or endogenous SRC-1 (bottom panels) were visualized by fluorescence microscopy. (C) Estrogen stimulates SRC-1 nuclear translocation. MCF-7 cells were cotransfected with pEGFP-SRC-1 and pcDNA3.1-FLAG-SIP (upper two panels) or untransfected (lower two panels), and grown in estrogen-depleted media for at least 2 days. The cells were then left untreated or treated with 100 nM of 17β-estradiol (E2) for 30 min, and the fluorescent imaging of SRC-1 and rhodamine staining of FLAG were performed (upper two panels). The endogenous SIP and SRC-1 were also immunostaining with rhodamine and FITC, respectively (lower two panels). Cellular distribution of SRC-1 was scored in at least 100 cells. The results are means±s.d. of three independent determinations (right panel). (D) Subcellular localization of SRC-1 under SIP knockdown. MCF-7 cells were transfected with pEGFP-SRC-1 and a vector designed for SIP RNAi or a nonspecific control RNAi vector. Forty-eight hours after the transfections, EGFP fluorescence was visualized by fluorescence microscopy in the presence or absence of E2 treatment. Cellular distribution of SRC-1 was scored in at least 100 cells. The results are means±s.d. of three independent determinations (right panels).
The cytoplasmic localization of SIP and the direct interaction between SIP and SRC coactivators suggest that SIP and SRC-1 could colocalize in the cytoplasm. To test this hypothesis, FLAG-SIP and pEGFP-SRC-1 constructs were cotransfected into MCF-7 cells and immunostaining of FLAG-SIP and GFP-SRC-1 fluorescence were examined (Figure 3B, middle panel). In addition, images were also obtained of transfected GFP-SIP and immunostained endogenous SRC-1 (Figure 3B, bottom panel). The results of these experiments indicated that SIP and SRC-1 were indeed colocalized in the cytoplasm.
As mentioned before, as transcriptional co-regulators, SRC proteins are mainly localized and function in the nucleus. However, numerous studies indicate that these proteins are constantly shuttling between the nucleus and the cytoplasm (Stenoien et al, 2001; Qutob et al, 2002; Amazit et al, 2003; Wu et al, 2004). Thus, the subcellular distribution of SRC coactivators could constitute a mechanism by which the activity of these co-regulators is regulated. In light of the observation that SIP interacted with SRC-1 in the cytoplasm, it is reasonable to postulate that SRC-1 is sequestered by SIP in the cytoplasm, when there is no extracellular stimulus. Upon an extracellular stimulation such as estrogen addition, signal transduction pathway(s) is activated that leads to the dissociation of SIP and SRC proteins and subsequently to SRC translocation into the nucleus. In order to test this hypothesis, FLAG-SIP and pEGFP-SRC-1 were cotransfected into MCF-7 cells and immunostaining of FLAG-SIP and imaging of EGFP fluorescence were performed in the presence or absence of estrogen stimulation. As shown in Figure 3C (upper two panels), without estrogen stimulation, SIP and SRC-1 were colocalized in the cytoplasm. Under estrogen stimulation, most of the SRC-1 was detected in the nucleus, whereas SIP remained in the cytoplasm. Immunostaining of endogenous SIP and SRC-1 yielded similar results (Figure 3C, lower two panels). In the meanwhile, SIP silencing by RNA interference (RNAi) resulted in SRC-1 nuclear localization, independent of E2 treatment (Figure 3D). These experiments support the hypothesis that SIP functions to sequester SRC coactivators in the cytoplasm.
Functional and mechanistic roles of SIP in ER-regulated gene transcription
As stated above, SRC coactivators are nuclear proteins that participate in gene transcriptional regulation by nuclear receptors as well as other transcription factors. In order to investigate the functional consequence of the cytoplasmic interaction between SIP and SRC-1 on the SRC transactivation function, the effect of gain-of-function and loss-of-function of SIP on the transactivation activity of SRC-1 was examined in ER-regulated gene transcription. In these experiments, an estrogen responsive element (ERE)-driven luciferase reporter (ERE-luc) or luciferase constructs driven by the promoters of ER target genes c-Myc (He et al, 1998) or cyclin D1 (Tetsu and McCormick, 1999) were transfected into MCF-7 cells, together with either an SRC-1 expression construct, a SIP expression construct, or a construct containing a specific sequence for silencing the expression of SIP. Reporter activity under these experimental conditions was measured. Consistent with previous experiments, expression of SRC-1 enhanced ER-mediated transactivation (Figure 4A). Interestingly, SIP overexpression resulted in inhibition of the reporter activity, whereas silencing the expression of SIP led to an increased reporter activity (Figure 4A). The expression of the endogenous proteins (Figure 4B) and the mRNAs (Figure 4C) of c-Myc and cyclin D1 was inhibited by overexpression of SIP and was increased under SIP RNAi. Collectively, these experiments indicated that SIP functions to negatively regulate ER-mediated gene transcription, possibly through cytoplasmic sequestration of SRC coactivators.
Figure 4.
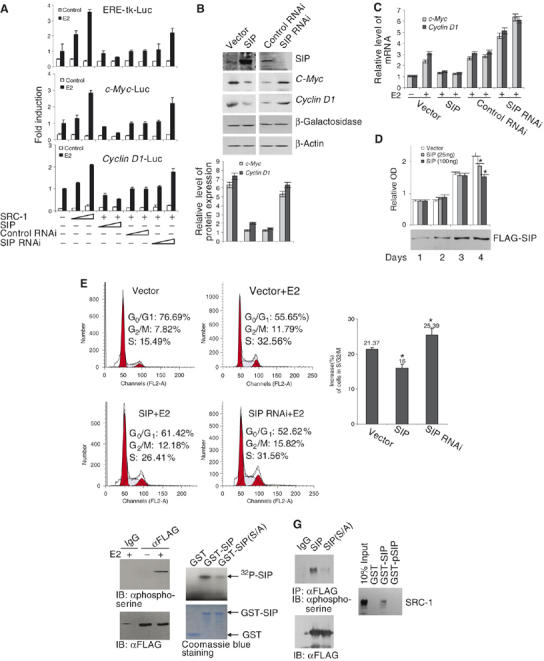
The biological function of SIP. (A) The effect of SIP on ER-regulated gene transcription. MCF-7 cells were transfected with different amounts (25 or 100 ng) of SRC-1 or SIP expression vectors, or control siRNA, or SIP siRNA constructs. The cells were then switched to estrogen-depleted media for 2 days before addition of 100 nM of E2 for 12 h and harvesting for luciferase activity assays. Each bar represents the mean±s.d. for triplicate experiments. (B) The effect of SIP on endogenous ER target gene expression. MCF-7 cells were transfected with indicated plasmids and switched to estrogen-depleted media for 2 days before treatment with 100 nM of E2 for 24 h. Cellular proteins were prepared for Western blot analysis (upper panels). The protein blots were quantitated by densitometry (lower panel). The transfection efficiency was monitored by cotransfection with an Escherichia coli lacZ construct and the expression of β-gal was detected by Western blotting with a β-galactosidase antibody. (C) Total RNAs were prepared from above described cells for real-time RT PCR analysis of the expression of c-Myc and cyclinD1. (D) The effect of SIP on estrogen-stimulated cell proliferation. MCF-7 cells were transfected with a vector or with a FLAG-SIP construct. MTT assays were performed each day after 1, 2, 3, and 4 days of the transfection. The data represent experiments performed in sextuplet; the asterisk indicates significant differences (P<0.05). The expression of transfected SIP was analyzed by Western blotting. (E) The effect of SIP on estrogen-stimulated cell cycle progression. MCF-7 cells were transfected with an SIP expression vector or a vector designed for SIP RNAi and switched to estrogen-depleted media for 2 days before addition of 100 nM of E2 for 12 h. The cells were then collected and analyzed for cell cycle profile by FACS. A representative flow cytometry data (left panels) and the average changes in the percentage of cells (treated versus untreated) in G0/G1, S, and G2/M phases from three independent experiments are shown (right panel). The asterisk indicates significant differences (P<0.01). (F) SIP is a phospho-protein. Left panel: MCF-7 cells were transfected with pcDNA3.1-FLAG-SIP and treated with E2 for 30 min. Whole-cell lysates were immunoprecipitated with anti-FLAG antibody and the immunoprecipitated materials were immunoblotted with an anti-phospho-serine monoclonal antibody. Right panel: in vitro phosphorylation of SIP by CK II. Phosphorylation mixtures containing recombinant GST, GST-SIP, or GST-SIP(S/A) were incubated at 30°C for 1 h in the presence of CK II and [γ-32P]ATP and resolved by SDS–PAGE. The lower panel shows equal amounts of recombinant GST fusion proteins used in the assay. (G) Phosphorylation of SIP. Left panel: in vivo phosphorylation of SIP. MCF-7 cells were transfected with pcDNA3.1-FLAG-SIP or pcDNA3.1-FLAG-SIP(S/A). Forty-eight hours after the transfection, whole-cell lysates were immunoprecipitated with anti-FLAG and the immunoprecipitated materials were then immunoblotted with anti-phospho-serine or with anti-FLAG. Right panel: SRC-1 interacts only with unphosphorylated SIP. GST pull-down assays were performed with in vitro translated SRC-1 and phosphorylated GST-SIP (GST-pSIP). Bound SRC-1 was detected using anti-SRC-1 antibodies.
In order to determine whether the negative effect of SIP on ER-mediated gene transcription could extend to a physiologically relevant response in cancer cells, we examined the effect of SIP expression on estrogen-stimulated cell cycle progression. Estrogen is normally required for the G1/S transition of MCF-7 cells and estrogen deprivation leads to G0/G1 arrest (Doisneau-Sixou et al, 2003). We cotransfected MCF-7 cells with the SIP expression vector or the SIP siRNA vector and cell cycle profiles were then analyzed by flow cytometry with estrogen treatment. As shown in Figure 4E, in cells transfected with SIP, the population of cells in G0/G1 phases accumulated, whereas silencing the expression of SIP by RNAi led to an increase of cells in S and G2/M phases. These experiments indicated that SIP negatively regulated estrogen-stimulated G1/S transition of MCF-7 cells, possibly through inhibition of the transactivation function of SRC-1 in ER-regulated gene transcription. Cell proliferation studies by MTT assays also supported SIP's role in negative regulation of cell proliferation (Figure 4D).
Protein–protein interaction is one of the physical bases for signal transduction inside the cell. The assembly and disassembly of protein complexes is often mediated by molecular events such as phosphorylation and dephosphorylation. In light of the fact that SIP contains a PEST domain, which harbors putative CK II phosphorylation sites, it is tempting to speculate that SIP is also functionally regulated by phosphorylation. In order to further understand the molecular events that lead to the dissociation of SIP and SRC-1, we next sought to test whether or not SIP is also a phospho-protein, and, if it is, what protein kinase(s) is responsible for SIP phosphorylation. First, in order to determine whether or not SIP is a phospho-protein, the FLAG-SIP construct was transfected into MCF-7 cells with or without estrogen stimulation, then cellular proteins were immunoprecipitated with the antibody against FLAG, and the immunoprecipitates were blotted with an antibody against phospho-serine. As shown in Figure 4F (left panels), the results of these experiments indicated clearly that SIP is a phospho-protein and its phosphorylation occurred only under estrogen stimulation. In vitro phosphorylation assays were also performed using bacteria-expressed SIP and a commercial CK II kit in the presence of [γ-32P]ATP. These assays indicated that SIP could be phosphorylated in vitro by CK II (Figure 4F, right panels). SIP phosphorylation by CK II occurred at the PEST domain, as serine/alanine (S/A) mutations at S-518, S-519, S-520, and S-523 in this domain of SIP almost abolished the SIP phosphorylation by CK II (Figure 4F, right panels). In vivo phosphorylation was also detected for SIP but not for the SIP(S/A) mutant (Figure 4G, left panels), supporting the in vitro phosphorylation results. Moreover, GST pull-down experiments were performed to test the interaction between SRC-1 and in vitro phosphorylated-SIP (pSIP), and the results revealed that pSIP was no longer interacted with SRC-1 (Figure 4G, right panel), suggesting that SIP phosphorylation might trigger the dissociation of SRC-1 and SIP complex.
In order to further confirm that SIP is phosphorylated by CK II in vivo, we next examined the expression and the activity of CK II in MCF-7 cells upon estrogen stimulation. As shown in Figure 5, estrogen treatment was associated with an accumulation of CK II proteins (Figure 5A), as well as with an increased CK II activity as analyzed in vitro with a synthetic CK II substrate (Figure 5B). In addition, CK II activation resulted in an increased expression of the ER target gene cyclin D1 and silencing the expression of CK II led to a decreased cyclin D1 expression (Figure 5C). Moreover, if the activity of CK II was inhibited by its specific inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) (Sarno et al, 2001; Ruzzene et al, 2002), in vivo SIP phosphorylation was not detected, even under estrogen stimulation (Figure 5D), supporting the argument that CK II is responsible for SIP phosphorylation. Under TBB treatment, SIP and SRC-1 remained in the cytoplasm even under estrogen stimulation (Figure 5E, bottom panel). In addition, silencing the expression of CK II was also associated with cytoplasmic retention of endogenous SRC-1 (Figure 5F, bottom panels). Collectively, these experiments strongly support that estrogen stimulates CK II activation, which, in turn phosphorylates SIP, resulting in the release of SRC-1 from SIP sequestration and subsequent SRC-1 translocation into the nucleus.
Figure 5.
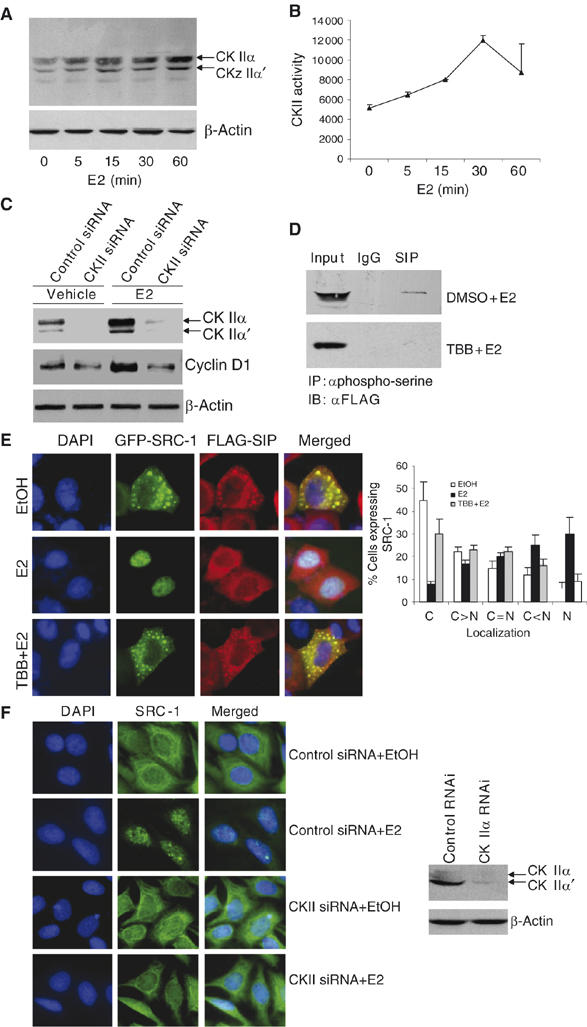
Estrogen-induced phosphorylation of SIP by CK II. MCF-7 cells were grown in the absence of estrogen for at least 2 days and treated with 100 nM of E2 for different times, and cellular lysates were then immunoblotted with specific antibodies against total CK II (A), or were analyzed for CK II activity with a CK II substrate peptide (B). (C) The expression of cyclin D1 under CK II knockdown. MCF-7 cells were transfected with a control siRNA or a siRNA against CK IIα. Forty-eight hours after the transfection, the cells were left untreated or treated with E2 for additional 24 h and the expression of cyclin D1 was analyzed by Western blotting. (D) Inhibition of SIP phosphorylation by CK II-specific inhibitor TBB. MCF-7 cells were transfected with pcDNA3.1-FLAG-SIP and treated with vehicle (DMSO) or TBB for 30 min before addition of E2 for another 30 min. Whole-cell lysates were first immunoprecipitated with anti-phospho-serine and then immunoblotted with anti-FLAG. (E) The effect of CK II-specific inhibitor on the nuclear translocation of SRC-1. pEGFP-SRC-1 and pcDNA3.1-FLAG-SIP were cotransfected into MCF-7 cells and treated with TBB before addition of E2 for 30 min. EGFP fluorescence and rhodamine staining of FLAG were visualized by fluorescence microscopy. Subcellular distribution of SRC-1 was scored in at least 100 cells. The results are means±s.d. of three independent determinations (right panel). (F) The effect of CK II knockdown on nuclear translocation of endogenous SRC-1. MCF-7 cells was transfected with a control siRNA or a CK II siRNA. Forty-eight hours after the transfections, cells were left untreated or treated with 100 nM of E2 for another 30 min. Endogenous SRC-1 was stained with FITC and visualized by fluorescence microscopy.
As shown in Figure 2E, the ankyrin repeats in SIP were responsible for the interaction of SIP with SRC-1. In order to further delineate the functional connection between SRC-1 and SIP, we transfected FLAG-tagged SIP, SIP(S/A), or SIP(ΔANK) (SIP without ankyrin repeats) into MCF-7 cells and the effect of SIP and its mutants on ER-regulated gene transcription and on estrogen-stimulated cell cycle progression were analyzed. As shown in Figure 6, SIP(S/A), which had no CK II phosphorylation residues, maintained SRC-1 sequestration function under estrogen treatment (Figure 6A, the fourth panels) and inhibited ER-mediated gene transactivation (Figure 6B). In contrast, SIP(ΔANK), which had no SRC-1 binding sequence thus no SRC-1 sequestration function (Figure 6A, the fifth panels), had no negative effect on ER-mediated gene transcription (Figure 6B). The stoichiometry of SIP-SRC-1 interaction appears to be associated with the surface charges of the PEST domain of SIP, as a phospho-serine mimetic, serine to aspartate SIP mutant SIP(S/D), in this domain was no longer able to sequester SRC-1 in the cytoplasm (Figure 6A, the sixth panels) and had only marginal effect on SRC-1-enhanced reporter activity (Figure 6B). In agreement with these observations, SIP(S/A) inhibited estrogen-stimulated cell cycle progression, whereas SIP(ΔANK) did not (Figure 6C). Collectively, these experiments strongly indicate that SIP acts to sequester SRC-1 in the cytoplasm and functions to regulate the biological activity of SRC-1 through restricting the subcellular distribution of SRC-1.
Figure 6.
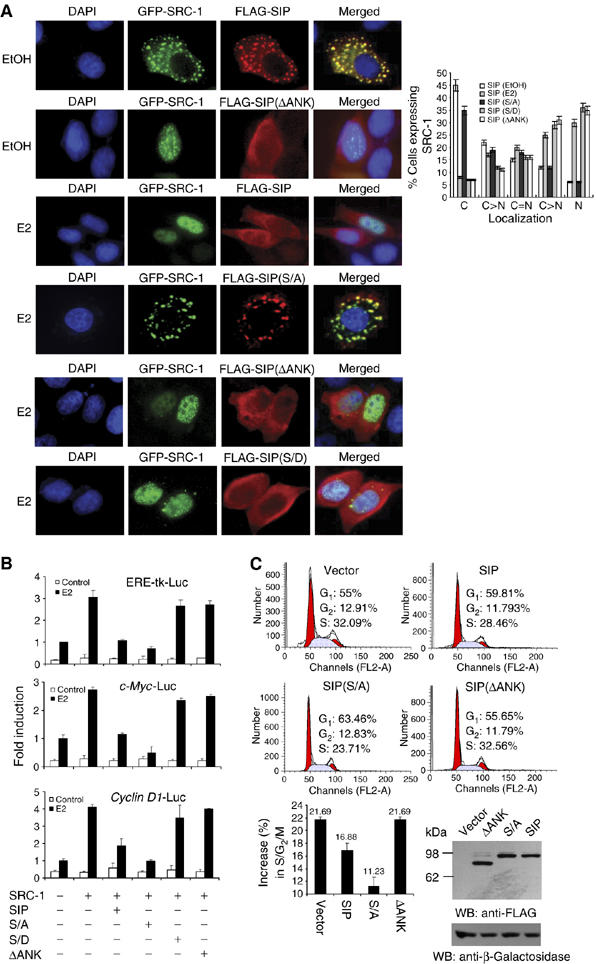
The molecular mechanism involved in sequestration of SRC-1 by SIP. (A) The effect of SIP, SIP(S/A), SIP(ΔANK), and SIP(S/D) on the subcellular localization of SRC-1. pEGFP-SRC-1 and FLAG-tagged SIP, or SIP mutants were cotransfected into MCF-7 cells. Twenty-four hours after the transfection, the cells were left untreated or treated with E2 for 30 min and EGFP fluorescence and rhodamine staining of FLAG were visualized by fluorescence microscopy. Subcellular distribution of SRC-1 was scored in at least 100 cells. The results are means±s.d. of three independent determinations (right panel). (B) The effect of SIP, SIP(S/A), SIP(ΔANK), and SIP(S/D) on ER-regulated gene transcription. MCF-7 cells were cotransfected with the indicated reporter constructs and expression plasmids. After treatment with E2 for 12 h, the cells were collected for the measurement of the reporter activity. Each bar represents the mean±s.d. for triplicate experiments. (C) The effect of SIP, SIP(S/A), and SIP(ΔANK) on E2-stimulated cell cycle progression. MCF-7 cells were transfected with wild-type SIP or SIP mutants and grown in estrogen-depleted media for 2 days. The cells were then treated with E2 for 12 h and the cell cycle profile was analyzed by FCAS. A representative flow cytometry data (upper panels) and the average changes compared with no treatment in the percentage of cells in G0/G1, S, and G2/M phases from three independent experiments (lower panel) are shown. The expression of SIP and its mutants was measured by Western blotting with cotransfection of an E. coli lacZ construct to monitor the transfection efficiency.
Finally, since SIP was able to interact with all three members of the SRC coactivators (Figure 2C), we examined the effect of SIP on the subcellular distribution of endogenous AIB1 and GRIP1 in MCF-7 cells in the presence or absence of estrogen treatment. GFP-SIP was imaged and endogenous AIB1 or GRIP1 was immunostained, and the results indicated that in the absence of estrogen, SIP was colocalized with AIB1 and GRIP1 in the cytoplasm. Under estrogen treatment, AIB1 and GRIP1 were translocated into nucleus, whereas SIP remained in the cytoplasm (Figure 7A), indicating that SIP functionally affects all three members of SRC coactivators. Furthermore, transfection of SIP(ΔANK) was associated with an increased nuclear translocation of AIB1 (Figure 7B), further supporting the argument that SIP interacts with SRC coactivators through its ankyrin repeats and functions to sequester SRC coactivators.
Figure 7.
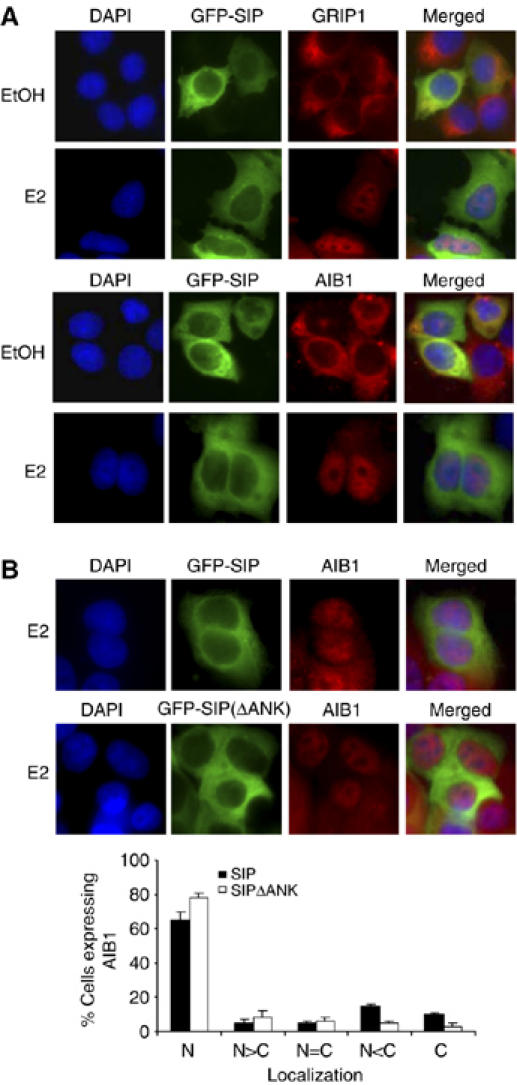
The effect of SIP on subcellular localization of GRIP1 and AIB1. (A) MCF-7 cells were transfected with pEGFP-SIP. Twenty-four hours after the transfection, the cells were left untreated or treated with E2 for 30 min, and EGFP fluorescence and rhodamine staining of endogenous GRIP1 and AIB1 were visualized by fluorescence microscopy. (B) The effect of SIP(ΔANK) mutant on subcellular localization of AIB1. MCF-7 cells were transfected with pEGFP-SIP or pEGFP-SIP(ΔANK). Twenty-four hours after the transfection, the cells were treated with E2 for 30 min, and EGFP fluorescence and rhodamine staining of endogenous AIB1 were visualized by fluorescence microscopy. Subcellular distribution of AIB1 was scored in at least 100 cells. The results are means±s.d. of three independent determinations.
Discussion
Current data indicate that SRCs, through their intrinsic HAT activity (Voegel et al, 1996; Onate et al, 1998) and/or through the recruitment of other enzymatic activities (Glass and Rosenfeld, 2000), accomplish their transactivation activity by modifying the configuration of chromatin structure. Despite the substantial progresses in our understanding of the biological functions of SRCs, several intriguing and physiologically relevant questions remain. One such question is how the functional specificity of these coactivator proteins is regulated. As mentioned before, gene ablation studies clearly indicate that the members of the SRC protein family are not functionally redundant. It is proposed based on current data that one of the mechanisms that determines the functional specificity of SRC coactivators comes from differential post-translational modifications of these coactivators. Indeed, phosphorylation, acetylation, and sumoylation have been reported for SRC proteins (Chen et al, 1999b; Kotaja et al, 2002; Chauchereau et al, 2003; Smith and O'Malley, 2004; Wu et al, 2005b, 2006), and differential chemical modifications by different enzymes would no doubt affect the functional dynamics of SRC proteins.
Another prominent question is how the availability of SRC coactivators is regulated. As we reported before, the relative cellular abundance and the availability of SRC coactivators could have a profound impact on the final output of cellular signaling (Shang and Brown, 2002; Yin et al, 2004; Zhang et al, 2004; Wu et al, 2005a; Shang, 2006). In addition, it has long been proposed that a balanced population between co-regulator species (coactivators and corepressors) could be an essential factor in determining the cellular response to a specific extracellular stimulus (Glass and Rosenfeld, 2000; Smith and O'Malley, 2004). In the current report, we identified a novel ankyrin repeat containing protein, SIP, and our data support a role for SIP in sequestering SRC coactivators in the cytoplasm, suggesting that SIP could act to buffer the availability of SRC coactivators. In so doing, the differential expression of SIP in different tissues could constitute yet another layer in the regulatory cascade of cellular signaling pathways in which the SRC proteins participate.
SIP was initially described as Ankrd25 and was shown to act as a growth promoter in U2OS osteosarcoma cells (Harada et al, 2005). In the current study, we showed that SIP, through sequestration of SRC coactivators, inhibited ER-regulated transcription and inhibited estrogen-stimulated proliferation of mammary carcinoma cells. The cell growth of different tissues is influenced by different environmental cues. In addition, it has been documented that ER exhibits tissue-specific activities (Smith and O'Malley, 2004), and we have showed in previous studies that the availability of different SRC molecules could contribute to the ER tissue specificity (Shang and Brown, 2002; Zhang et al, 2004; Shang, 2006). In light of our current observation and the observation made in U2OS cells, it is intriguing to speculate that SIP also contributes to the ER tissue specificity in different tissues under different extracellular stimuli. In this context, it will be interesting to investigate in the future studies whether or not SIP has additional biological activities.
Although SRC proteins, as transcription co-regulators, mainly function in the nucleus, studies have indicated that the subcellular localization of these proteins is regulatable. For instance, it has been reported that SRC-1 shuttles between the cytoplasm and the nucleus, depending on transcription status (Amazit et al, 2003), that a shift of compartmental equilibrium for AIB1 from the cytoplasm to the nucleus occurs upon AIB1 phosphorylation by IκB kinase (Wu et al, 2002), and that treatment of serum-starved cells with growth factors or phorbol esters promotes the translocation of AIB1 from the cytoplasm to nuclear compartments (Qutob et al, 2002). Based on our current observations, it is conceivable that in the cytoplasm, SRC proteins are associated with SIP. Upon extracellular stimulation, various signaling pathways are initiated, different protein kinases are activated, and both SRC coactivators and SIP are phosphorylated, resulting in conformational changes in and dissociation of SIP and SRC proteins. Future investigations are warranted to delineate detailed stoichiometry of SIP and SRC protein interaction.
Various signaling pathways are potentially activated by estrogen treatment. These include Ca2+ flux, cAMP and MAPK, PKA, PKC, PI3K, and Src kinase (Zhang and Trudeau, 2006). It has been abundantly documented that these signaling pathways target both ERs and their coactivators (Smith and O'Malley, 2004; Wu et al, 2005b). In addition, other kinase cascades such as CK II, have also been implicated in estrogen signaling (Arnold et al, 1994). CK II is a highly conserved and ubiquitously expressed protein serine/threonine kinase present in all eukaryotes (Seldin et al, 2005). CK II is upregulated in rapidly dividing cells, including most human tumors, and it has been implicated in critical cellular processes such as proliferation, apoptosis, differentiation, and transformation. Transgenic overexpression of CK II in lymphoid or mammary lineages predisposes to transformation (Seldin et al, 2005). The oncogenic role of CK II in mammary tissue is consistent with our observation that CK II promotes, through phosphorylating SIP, the nuclear translocation of SRC coactivators, gene activation, and cell proliferation.
In summary, we report here the identification of a novel ankyrin repeat containing protein that functions to sequester SRC coactivators in the cytoplasm, thus providing a mechanism for the regulation of transcriptional regulators.
Materials and methods
Bioinformatics
The open reading frame, conserved domains, and chromosome location of SIP were predicted with the databases in NCBI (www.ncbi.nlm.nih.gov). The theoretical molecular weight and isoelectric point of SIP were calculated with the database at www.expasy.ch/tools. The homologous alignment was analyzed by the ClustalW program (version 1.60) (Thompson et al, 1994) and phylogenetic analysis was performed using the Jotun Hein method (Hein, 1990).
Fluorescence confocal microscopy
MCF-7 cells were transfected with appropriate plasmids using Lipofectamine 2000 reagent (Invitrogen). Twenty-four hours after transfection, cells were washed with PBS, fixed in 4% paraformaldehyde, and permeablized with 1% Triton X-100. Cells were washed for four times and a final concentration of 0.1 μg/ml 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma) was included in the final wash to stain nuclei. Images were visualized with an Olympus inverted microscope equipped with a charge-coupled camera. The resulting images were deconvolved with Deltavision software. For immunostaining, 24 h after transfections, cells were washed with PBS, fixed with 4% paraformaldehyde, and stained with appropriate primary antibodies followed by rhodamine- or FITC-conjugated secondary antibodies. The subcellular localization of proteins was scored in at least 100 cells under each experimental condition. Staining was considered nuclear when it was exclusively nuclear (N) or stronger in the nucleus than in the cytoplasm (N>C). In all other cases, it was considered cytoplasmic (if C=N, if C>N, and if C<N) (Amazit et al, 2003).
RNAi
Plasmids were constructed by inserting a synthesized 64-mer oligonucleotide containing a specific sequence for the 1259–1277 bp region of SIP open reading frame into pSUPER vector (Brummelkamp et al, 2002). The sequences synthesized were: oligonucleotide 1, 5′-GATCCCCGTTCCTGCCGAATCGTCTTTTCAAG AGAAAGACGATTCGGCAGGAACTTTTTGGAAA-3′ and oligonucleotide 2, 5′-AGCTTTTCCAAAAAGTTCCTGCCGAATCGTCT TTCTCTTGAAAAGACGATTCGGCAGGAACGGG-3′. The three CK II siRNAs used were 5′-GAUGACUACCAGCUGGUUCdTdT (α siRNA), 5′-UCAAGAUGACUACCAGCUGdTdT, and 5′-CAGUCUGAGGAGCCGCGAGdTdT (α′ siRNA). The negative control siRNA was 5′-GCUCAGAUCAAUACGGAGAdTdT. The oligonucleotides were resuspended in annealing buffer (100 mM potassium acetate, 30 mM HEPES–KOH, pH 7.4, 2 mM acetate) and heated to 95°C for 4 min, 70°C for 10 min, and then cooled to room temperature to generate double-stranded DNA. The double-stranded DNA was then phosphorylated and cloned into the BglII/HindIII site of the pSUPER vector. The vector was then transfected into cells with the Lipofectamine 2000 Reagent (Invitrogen).
In vitro phosphorylation and CK II activity assay
Equal amounts of immobilized GST fusion proteins were incubated with 50 ng of CK II (Upstate Biotechnology) and 50 μM [γ-32P]ATP (5 Ci/mmol) in 20 μl of CK II buffer (20 mM MOPS, pH 7.2, 10 mM MgCl2, 5 mM EGTA, 1 mM sodium orthovanadate, and 1 mM dithiothreitol) for 1 h at 30°C. The reaction was terminated by addition of SDS sample buffer and phosphorylated proteins were separated by SDS/PAGE and detected by autoradiography (Supplementary data).
For CK II activity assay, whole cell lysates were collected after repeated freezing–thawing and used for CK II activity assay using a CK II assay kit (Upstate Biotechnology).
Supplementary Material
Supplementary data
Acknowledgments
This work was supported by grants (30621002, 30393110, and 30470912 to YS) from National Natural Science Foundation of China and grants (973 Program: 2005CB522404 and 863 Program: 2006AA02Z466 to YS) from the Ministry of Science and Technology of China.
References
- Amazit L, Alj Y, Tyagi RK, Chauchereau A, Loosfelt H, Pichon C, Pantel J, Foulon-Guinchard E, Leclerc P, Milgrom E, Guiochon-Mantel A (2003) Subcellular localization and mechanisms of nucleocytoplasmic trafficking of steroid receptor coactivator-1. J Biol Chem 278: 32195–32203 [DOI] [PubMed] [Google Scholar]
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965–968 [DOI] [PubMed] [Google Scholar]
- Arnold SF, Obourn JD, Jaffe H, Notides AC (1994) Serine 167 is the major estradiol-induced phosphorylation site on the human estrogen receptor. Mol Endocrinol 8: 1208–1214 [DOI] [PubMed] [Google Scholar]
- Bautista S, Valles H, Walker RL, Anzick S, Zeillinger R, Meltzer P, Theillet C (1998) In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin Cancer Res 4: 2925–2929 [PubMed] [Google Scholar]
- Belandia B, Parker MG (2000) Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J Biol Chem 275: 30801–30805 [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Carapeti M, Aguiar RC, Goldman JM, Cross NC (1998) A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 91: 3127–3133 [PubMed] [Google Scholar]
- Chauchereau A, Amazit L, Quesne M, Guiochon-Mantel A, Milgrom E (2003) Sumoylation of the progesterone receptor and of the steroid receptor coactivator SRC-1. J Biol Chem 278: 12335–12343 [DOI] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR (1999a) Regulation of transcription by a protein methyltransferase. Science 284: 2174–2177 [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM (1999b) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 98: 675–686 [DOI] [PubMed] [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR (1998) Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev 12: 3343–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL (2003) Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer 10: 179–186 [DOI] [PubMed] [Google Scholar]
- Gehin M, Mark M, Dennefeld C, Dierich A, Gronemeyer H, Chambon P (2002) The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol Cell Biol 22: 5923–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14: 121–141 [PubMed] [Google Scholar]
- Harada JN, Bower KE, Orth AP, Callaway S, Nelson CG, Laris C, Hogenesch JB, Vogt PK, Chanda SK (2005) Identification of novel mammalian growth regulatory factors by genome-scale quantitative image analysis. Genome Res 15: 1136–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387: 733–736 [DOI] [PubMed] [Google Scholar]
- Hein J (1990) Unified approach to alignment and phylogenies. Methods Enzymol 183: 626–645 [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85: 403–414 [DOI] [PubMed] [Google Scholar]
- Kim JH, Li H, Stallcup MR (2003) CoCoA, a nuclear receptor coactivator which acts through an N-terminal activation domain of p160 coactivators. Mol Cell 12: 1537–1549 [DOI] [PubMed] [Google Scholar]
- Kotaja N, Karvonen U, Janne OA, Palvimo JJ (2002) The nuclear receptor interaction domain of GRIP1 is modulated by covalent attachment of SUMO-1. J Biol Chem 277: 30283–30288 [DOI] [PubMed] [Google Scholar]
- McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK, Rosenfeld MG (1998) Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev 12: 3357–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onate SA, Boonyaratanakornkit V, Spencer TE, Tsai SY, Tsai MJ, Edwards DP, O'Malley BW (1998) The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors. J Biol Chem 273: 12101–12108 [DOI] [PubMed] [Google Scholar]
- Picard F, Gehin M, Annicotte JS, Rocchi S, Champy MF, O'Malley BW, Chambon P, Auwerx J (2002) SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 111: 931–941 [DOI] [PubMed] [Google Scholar]
- Qutob MS, Bhattacharjee RN, Pollari E, Yee SP, Torchia J (2002) Microtubule-dependent subcellular redistribution of the transcriptional coactivator p/CIP. Mol Cell Biol 22: 6611–6626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzene M, Penzo D, Pinna LA (2002) Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem J 364: 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA (2001) Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2'). FEBS Lett 496: 44–48 [DOI] [PubMed] [Google Scholar]
- Seldin DC, Landesman-Bollag E, Farago M, Currier N, Lou D, Dominguez I (2005) CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem 274: 63–67 [DOI] [PubMed] [Google Scholar]
- Shang Y (2006) Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nat Rev Cancer 6: 360–368 [DOI] [PubMed] [Google Scholar]
- Shang Y, Brown M (2002) Molecular determinants for the tissue specificity of SERMs. Science 295: 2465–2468 [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103: 843–852 [DOI] [PubMed] [Google Scholar]
- Smith CL, O'Malley BW (2004) Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 25: 45–71 [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA (2001) FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat Cell Biol 3: 15–23 [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H (1996) TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J 15: 3667–3675 [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rose DW, Hermanson O, Liu F, Herman T, Wu W, Szeto D, Gleiberman A, Krones A, Pratt K, Rosenfeld R, Glass CK, Rosenfeld MG (2000) Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci USA 97: 13549–13554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RE, Xu J, Ning G, Pohlenz J, O'Malley BW, Refetoff S (1999) Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J 18: 1900–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang Y, Wang D, Li R, Yi X, Zhang H, Sun L, Shang Y (2005a) Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature 438: 981–987 [DOI] [PubMed] [Google Scholar]
- Wu H, Sun L, Zhang Y, Chen Y, Shi B, Li R, Wang Y, Liang J, Fan D, Wu G, Wang D, Li S, Shang Y (2006) Coordinated regulation of AIB1 transcriptional activity by sumoylation and phosphorylation. J Biol Chem 281: 21848–21856 [DOI] [PubMed] [Google Scholar]
- Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O'Malley BW (2002) Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) coactivator activity by I kappa B kinase. Mol Cell Biol 22: 3549–3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O'Malley BW (2004) Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic responses to multiple cellular signaling pathways. Mol Cell 15: 937–949 [DOI] [PubMed] [Google Scholar]
- Wu RC, Smith CL, O'Malley BW (2005b) Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocr Rev 26: 393–399 [DOI] [PubMed] [Google Scholar]
- Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O'Malley BW (2000) The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci USA 97: 6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Qiu Y, DeMayo FJ, Tsai SY, Tsai MJ, O'Malley BW (1998) Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 279: 1922–1925 [DOI] [PubMed] [Google Scholar]
- Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B, Wu H, Wu G, Wang X, Shang Y (2004) Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res 64: 5870–5875 [DOI] [PubMed] [Google Scholar]
- Zhang D, Trudeau VL (2006) Integration of membrane and nuclear estrogen receptor signaling. Comp Biochem Physiol A Mol Integr Physiol 144: 306–315 [DOI] [PubMed] [Google Scholar]
- Zhang H, Sun L, Liang J, Yu W, Zhang Y, Wang Y, Chen Y, Li R, Sun X, Shang Y (2006) The catalytic subunit of the proteasome is engaged in the entire process of estrogen receptor-regulated transcription. EMBO J 25: 4223–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yi X, Sun X, Yin N, Shi B, Wu H, Wang D, Wu G, Shang Y (2004) Differential gene regulation by the SRC family of coactivators. Genes Dev 18: 1753–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data