

Abstract
The checkpoint kinase 1 (Chk1) preserves genome integrity when replication is performed on damaged templates. Recently, Chk1 has also been implicated in regulating different aspects of unperturbed S phase. Using mammalian and avian cells with compromised Chk1 activity, we show that an increase in active replicons compensates for inefficient DNA polymerisation. In the absence of damage, loss of Chk1 activity correlates with the frequent stalling and, possibly, collapse of active forks and activation of adjacent, previously suppressed, origins. In human cells, super-activation of replication origins is restricted to pre-existing replication factories. In avian cells, in contrast, Chk1 deletion also correlates with the super-activation of replication factories and loss of temporal continuity in the replication programme. The same phenotype is induced in wild-type avian cells when Chk1 or ATM/ATR is inhibited. These observations show that Chk1 regulates replication origin activation and contributes to S-phase progression in somatic vertebrate cells.
Keywords: checkpoint proteins, DNA foci, DNA replication, replication origins, S-phase programme
Introduction
During cell proliferation, eukaryotic genomes are replicated with high fidelity in order to ensure their genetic integrity. Because of their size, the duplication of eukaryotic genomes demands that DNA synthesis is activated at numerous replication ‘origins' (Machida et al, 2005). In mammalian cells, replication origins are activated in small groups (Jackson and Pombo, 1998; Berezney et al, 2000) that are replicated together within individual replication factories. These replicon clusters can be visualised as DNA foci and the sequential activation of potential replication origins within groups of these sub-chromosomal units is thought to play a direct role in defining the S-phase programme (Sadoni et al, 2004).
In human cells, DNA is duplicated within an S phase of about 10 h and during this time, roughly 50 000 replication origins are used to activate synthesis of replicons that are typically ∼100 kb in length (Jackson and Pombo, 1998; Berezney et al, 2000). Potential origins are activated throughout S phase so that ∼10% of replicons are engaged in DNA synthesis at any time. Single-molecule analysis of DNA fibres shows that specific origins or groups of origins function at specific times of S phase and can be developmentally regulated (Jackson and Pombo, 1998; Norio et al, 2005). In addition, the temporal activation of specific regions of the genome correlates with the appearance of distinct patterns of replication sites as cells progress from early to middle and then late S phase (reviewed in Jackson, 1995).
In somatic vertebrate cells, almost nothing is known about the mechanisms that regulate the temporal activation and suppression of potential replication origins (Gilbert, 2002; Machida et al, 2005). However, recent studies have suggested that the replication checkpoint pathways contribute to the regulation of origin activation during normal S phase (reviewed in Fisher and Mechali, 2004; Kastan and Bartek, 2004). Although checkpoint pathways are activated in response to DNA damage (reviewed in Sancar et al, 2004), a function in unperturbed S phase is implied by the fact that checkpoint kinase 1 (Chk1) is essential for normal development (Liu et al, 2000; Takai et al, 2000). In the absence of DNA damage, Chk1 has been shown to regulate the physiological turnover of Cdc25A, which in turn regulates the activation of replication origins through the action of CDK2, associated A and E type cyclins and Cdc45 (Zhao et al, 2002; Sorensen et al, 2003). This pathway, in principle, provides a mechanism for regulating origin activation throughout S phase (Fisher and Mechali, 2004; Shechter and Gautier, 2005), although how a spatially ordered programme of origin activation might be established remains unclear.
In this report, we have used cells with compromised ATM/ATR and Chk1 function to explore how the checkpoint pathways contribute to the local and long-range activation of DNA synthesis. We have recently shown that Chk1 is required to maintain normal rates of replication fork progression in human and avian cells in the absence of DNA damage (Petermann et al, 2006). In the present study, we have analysed how ATM/ATR and Chk1 influence the local activation of replication origins within replicon clusters and the spatial organisation of replication foci throughout S phase. When Chk1 function is compromised, a 2- to 3-fold increase in origin density is seen, showing that Chk1 normally functions to regulate the density of active origins by suppressing many of the potential origins that could be activated within each replicon cluster. In human cells, this super-activation of replication origins in cells with reduced Chk1 activity is restricted to replicons that are already associated with active replication sites, suggesting that the checkpoint proteins must function in the context of spatial constraints that are defined by the programmed assembly of replication factories.
Results
Replicon structure
Cells with perturbed Chk1 function display a small global increase in DNA synthesis and chromatin-associated Cdc45, consistent with Chk1 contributing to the regulation of replication origin activation (Miao et al, 2003; Syljuasen et al, 2005). However, formal proof for the super-activation of replication origins was not presented in these studies.
Using spread DNA fibres (Jackson and Pombo, 1998), we first mapped the structure of active replicons in cells that are deficient for ATR and Chk1 function (Figures 1 and 2). Active replicons were labelled with biotin-dUTP following introduction of a very low concentration of precursor into cells. As the precursor is consumed during the labelling period, immunodetection of biotin reveals comet-like labelled tracks, where the head of the comet represents the position of replication at the beginning of the labelling pulse and the tail the point when the label was consumed. Hence, the structure of labelled tracks allows unambiguous assignment of the daughter forks that emanate from a common, central replication origin (Figure 1A and B).
Figure 1.
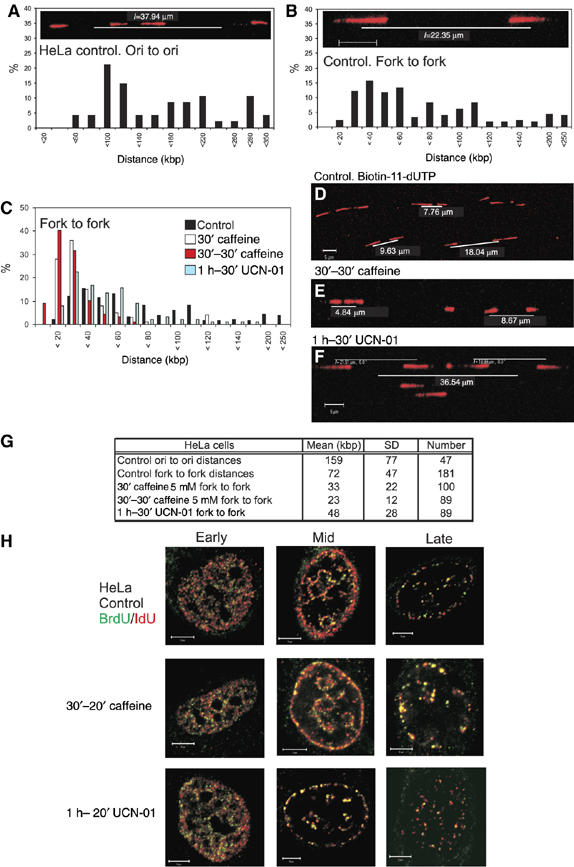
Chk1 regulates the density of active replication origins in HeLa cells. Replication structures were visualised on spread DNA fibres (A–F) using biotin incorporation (shown in red) to assign replication fork polarity. After spreading and immunolabelling, replicon structure was defined by the distance between (panel A) adjacent origins in replicon clusters and (panel B) sister replication forks. Changes in replicon structure induced by treatment with caffeine (panels C–E) and UCN-01 (panels C, F) were determined. Frequency histograms (panels A–C) show the distribution of separation in kilobase pairs, assigned using spreads like the typical examples shown (panels A, B, D–F). Parameters defining average replicon structure under different conditions are shown (G). To monitor the affect of checkpoint protein inhibitors on replication foci (H), replication sites were pulse labelled for 20 min with BrdU (green) and then for 25 min with IdU (red). Unsynchronised cultures were treated with caffeine or UCN-01 before and throughout first labelling and S-phase cells assigned to early, mid or late S phase. Images are merges of confocal sections, with areas of colocalisation shown in yellow. Scale bars (5 μm) are shown on individual images.
Figure 2.
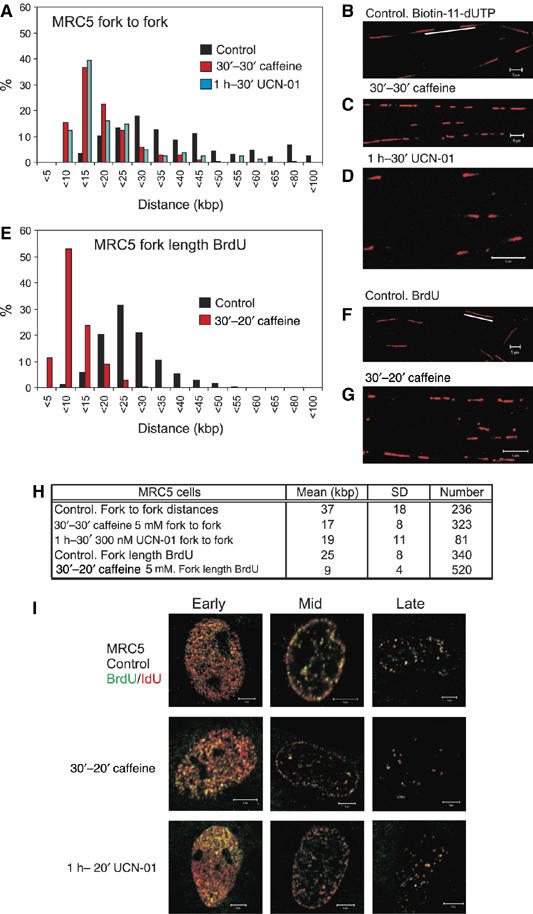
Chk1 regulates replication origin density in diploid human fibroblasts. Replication structures of MRC5 human fibroblasts were visualised using the procedures described in Figure 1. Replicon size (A–D) was measured using the separation of sister forks (panel A) after labelling with biotin-dUTP (panels B–D) and structures in untreated cells compared with those in cells treated with caffeine or UCN-01. BrdU labelling was also used to determine the rate of fork elongation (E–G) under the same conditions. Parameters defining average replicon structure under different conditions are shown (H). The organisation of active replication foci in cells treated with caffeine and UCN-01 was compared with untreated cells (I), as described in Figure 1. Scale bars, 5 μm.
As the majority of replicons in vertebrate cells are co-replicated in small replicon clusters, the origin spacing within these clusters provides a direct measure of the replicon length (Figure 1A). In unsynchronised HeLa cells, adjacent origins were, on average, 159 kb apart (Figure 1G), similar to the value of 145 kb during early S phase (Jackson and Pombo, 1998). Although inter-origin distance measurements are robust, this analysis neglects isolated replicons and the outgrowing forks of replicon clusters, which can be much longer than those that are located between the active origins (Berezney et al, 2000). To incorporate these forks, we also measured the separation of the daughter forks of individual replicons (Figure 1B). As the cells are unsynchronised, labelled replicons are visualised at steady state so that the average separation of daughter forks at the time of labelling will approximate to half the replicon length. In unsynchronised cells, the expected wide range of daughter fork separations was seen, with an average of 72 kb (Figure 1B and G), roughly half the average inter-origin spacing.
The distribution of daughter forks was also assessed in unsynchronised cells that had been treated with the kinase inhibitors caffeine-general inhibitor ATR/ATM kinase activity—and UCN-01—a specific inhibitor of Chk1 autophosphorylation (Figure 1C–G). When caffeine was added either 30 min before the time of replication labelling (designated 30′ caffeine) or 30 min before and throughout labelling (designated 30′–30′ caffeine; Figure 1C) a clear approximately 2- to 3-fold decrease in fork separation was seen (Figure 1G). Similar results were observed when the Chk1 inhibitor UCN-01 was added 60 min before and throughout labelling, with replication structures ∼2-fold closer than in untreated cells (Figure 1C and G); slight differences in the caffeine- and UCN-01-induced responses may imply that ATM/ATR also inhibit fork movement through other mechanisms that are independent of Chk1. In both cases, these increases in origin density are conservative estimates because the analysis monitors paired sister forks but does not score conjoined forks from origins that initiated synthesis during the labelling pulse.
A notable feature of this experiment is that the increased origin density results in a clear decrease in the proportion of widely spaced forks. While cells must contain the natural distribution of forks at the beginning of this experiment, the relative decrease in remote daughter fork results from two factors: (1) novel initiation events dominate the analysis because of the high frequency of caffeine-induced origin activation. These recently activated origins with close sister forks (Figure 1E and F) represent the major population seen in the randomly selected fields analysed and (2) widely separated forks are inevitably from long replicons, with replication complexes engaged throughout the labelling protocol. Reduced efficacy of replication at some forks compromises the structure and assignment of these forks as bona fide sister pairs. Even so, if spreads are scanned using biased selection criteria, the active forks in long replicons can be found.
Global analysis of replication in caffeine-treated cells
We next characterised how defects in the ATM/ATR pathways influence the global structure of replication sites, in order to evaluate if the initiation events induced by caffeine and UCN-01 were distributed throughout the nucleus. Changes in the structure of the replication programme were assessed using simple labelling and indirect immunofluorescence (Supplementary Figure S1), and double labelling (Figure 1H). This analysis demonstrates that whereas DNA synthesis was profoundly altered in the presence of caffeine at the level of individual forks (Figure 1A–G), no defects were seen at the level of changes in global sites of DNA synthesis. Hence, in the presence of inhibitor, colocalisation of the two labels and the maintenance of replication patterns with the normal number and structure of replication foci shows that inhibitor-induced super-activation of latent origins was restricted to replicons that were engaged with active replication factories (Figure 1H). Following the most effective caffeine treatment, almost all active centres contained the first and second labels (P<0.47 (n=26) using Student's t-test to compare control and caffeine-treated cells).
The super-activation of latent origins seen in HeLa cells was confirmed in MRC5 cells, which are normal human diploid fibroblasts from lung (Figure 2). These diploid fibroblasts have a more uniform origin spacing and average separation that is about half that in transformed HeLa cells. Even so, DNA spreads from control and caffeine- or UCN-01-treated MRC5 cells (Figure 2A–H) showed a clear 2- to 3-fold increase in origin activation (Figure 2A–D) and a corresponding fall in the rate of fork migration (Figure 1E–H) upon inhibition of the ATR/ATM pathways. As for HeLa cells (Figure 1), the observed super-activation of latent origins in MRC5 fibroblasts was restricted to pre-established replication factories (Figure 2I).
Caffeine-induced activation of latent replication origins
We next used a double labelling strategy to define the relationship between origins that were induced in response to replication stress and the previously active forks (Figure 3). Cells were preincubated in caffeine and pulse labelled for 20 min with bromo-deoxyuridine (BrdU) in presence of caffeine, washed and then pulse labelled with biotin-dUTP (Figure 3A). This labelling protocol defines five classes of replication structure (Figure 3B). For both HeLa and MRC5 (Figure 3C), ∼67% of forks were labelled consecutively with the first and second labels (class 1), as expected for forks that were elongating throughout the labelling period. In both cell types, 16% of labelled structures contained two forks growing from a common origin (class 2) and a similar number of growing forks fused and so terminated during labelling (class 3). In controls, very few tracks were labelled uniquely with the first or second label (class 4).
Figure 3.
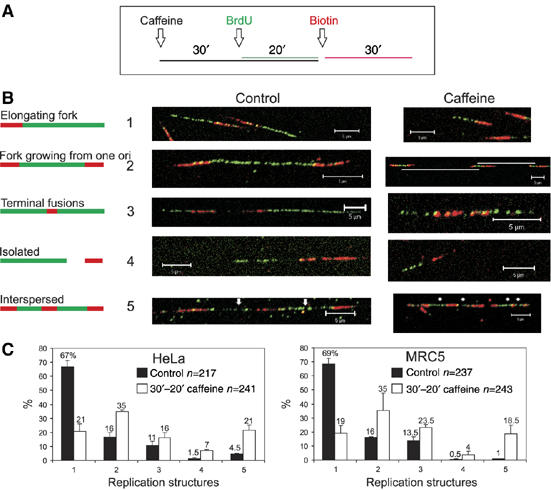
Activation of latent origins in human cells treated with caffeine. HeLa and MRC5 cells were treated with caffeine and pulse labelled with BrdU and biotin-11-dUTP, as shown (A). DNA fibres were prepared, treated with acid and immunolabelled to visualise BrdU (green) and biotin-dU (red). Five classes of replication structure were defined (B; classes 1–5) and the relative frequency of occurrence of the different classes was scored (frequency histograms (C)). Scale bars, 5 μm.
In the presence of caffeine, the rate of fork elongation was reduced by at least two-fold throughout the labelling period (compare control and caffeine class 1 forks in Figure 3B). In addition, quite different proportions of replicating structures were seen in caffeine treated samples (Figure 3C). Strikingly, the proportion of consecutively labelled forks (class 1) fell by >3-fold as a result of two factors. First, caffeine treatment resulted in a >2-fold increase in the number of new initiation events during both the first (class 2) and second (class 4) labelling periods. Second, a dramatic increase in the frequency of closely spaced active origins (class 5) was also seen. Despite a clear fall in the proportion of elongation forks, <5% of forks (class 4—green only) stalled during the first pulse and remained inactive during the second. These observations show that in the presence of caffeine, active forks frequently stall and sometimes collapse, and that these defects in replication are compensated for by the activation of novel, caffeine-dependent replication origins. A diagnostic feature of novel origin activation is the dramatic increase in interspersed labelling (class 5). Figure 3B (class 5 caffeine) shows a typical extreme case with a 75 kb region containing four active origins. Clearly, this origin density is very much higher than is normally seen in control cells (Figures 1 and 2).
In the presence of caffeine, this increased density of active forks, together with a high frequency of clustered caffeine-dependent initiation events in the vicinity of previously active forks, confirms that the vast majority of novel initiation events occur in already active replicon clusters. Under these conditions, the efficacy of replication is severely compromised, with all forks showing reduced rates of elongation, consistent with frequent stalling during synthesis. Despite this reduced rate, a majority of forks continue synthesis and only a minority collapse (Figure 3). Importantly, this experiment shows that active replicons contain latent origins ahead of the growing fork, which are suppressed under normal conditions but activated in response to replicative stress.
Organisation of DNA replication in Chk1−/− cells
Because inhibitors such as caffeine might alter or stress the normal pathways that regulate the activation of replication origins, we next wanted to analyse if DNA synthesis was perturbed in Chk1-deficient cells. For this purpose, we chose to use Chk1-deficient DT40 (Zachos et al, 2003), as Chk-1 depletion in mammalian model systems is known to be cell lethal (Liu et al, 2000). Previously, we showed that Chk1-deficient DT40 cells have a reduced rate of replication at individual forks (Petermann et al, 2006), which appears contrary to the observed increase in global DNA synthesis when UCN-01 was used to disrupt Chk1 function in mammalian cells (Syljuasen et al, 2005).
Detailed analysis of replicon structure in DT40 cells showed that an increased origin density compensates for the reduced fork rate (Figure 4). Replicons of wild-type (wt) DT40 cells were smaller than those of HeLa cells, with an average daughter fork separation of 36.7 kb and origin spacing of 67.8 kb (Figure 4). Replicons in Chk1-deficient DT40 cells were even smaller than those in wt control cells (P<2.7E-19), with an average daughter fork separation of 24.2 kb and average origin spacing of 47.3 kb. This represents a 1.5- to 2-fold increase in the number of active origins in Chk1-deficient DT40 cells, which is sufficient to maintain the normal S-phase duration when individual forks polymerise nucleotides at ∼50% of the rate in wt cells (Petermann et al, 2006).
Figure 4.
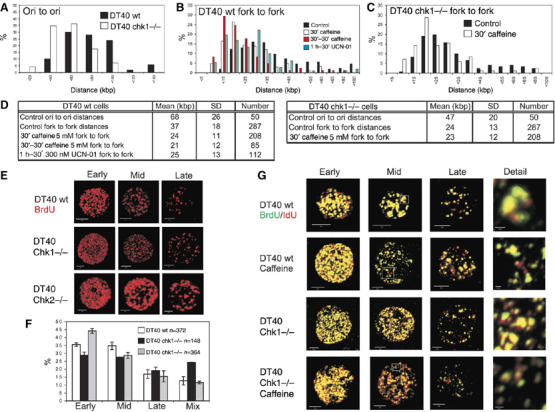
Inhibition of ATR or Chk1 mimics the replication phenotype of Chk1-deleted DT40. Active replicons in Chk1−/− and isogenically matched wt DT40 cells were analysed after pulse labelling with biotin-11-dUTP. Replication structures were visualised on spread DNA fibres as in Figure 1. Changes in replicon structure in cells with compromised Chk1 activity are shown (frequency histograms (A–C)). In wt DT40, caffeine and UCN-01 (panel B) produced a significant decrease in replicon size (P<1.197E-21 and 2.52E-12, respectively, versus controls). No difference was seen when Chk1−/− DT40 cultures (panel C) were treated with caffeine (P=0.541). Parameters defining average replicon structure under different conditions are shown (D). The S-phase programme of wt DT40 was also compared with Chk1- and Chk2-deficient cells. Cells were pulse labelled for 20 min with BrdU, immunolabelled (E) and the proportion (F) of early-, mid- and late-S-phase patterns determined. Patterns that did not conform to the recognised structural and spatial criteria were designated as mixed. Caffeine-induced changes in the organisation of replication foci (G) were assessed by double labelling, as described in Figure 1H. High-power details are from the boxed areas shown. Scale bars (5 and 0.5 μm in Detail) are shown on individual images.
As for HeLa and MRC5 cells, clear reductions in daughter fork separation were seen in DT40 cells treated with UCN-01 (P<2.52E-12) or caffeine (P<1.19E-21) (Figure 4B and D); similar reductions in fork to fork distance were seen following treatment with caffeine and UCN-01 (P<0.43). A very similar replicon structure was also seen in Chk1−/− DT40 cells (P<0.548 Chk1−/− versus wt DT40 incubated with caffeine; Figure 4C and D). This shows that DT40 cells have a very similar density of active replication origins when either ATR/ATM or Chk1 function is compromised and implies that Chk1 plays a major role in regulating active origin density in these cells.
The replication programme in DT40 cells
Defects in origin activation in Chk1-deficient DT40 cells prompted us to evaluate how the absence of Chk1 influenced the S-phase programme in these cells. As for HeLa cells (Figure 1), the S-phase programme of wt DT40 cells (Zachos et al, 2003) can be described by the size, complexity and nuclear distribution of their replication foci (Figure 4E–G and Supplementary Figure S2). Using wt DT40, patterns that are characteristic for this cell type were seen using different labelling protocols, which allow multiple labelling without reagent cross-talk in fixed or living cells (Figure 4E and Supplementary Figure S2). Chk1-deficient cells had the same range of replication structures as parental DT40 controls. However, it was immediately obvious that the complexity of foci in these cells was greater than in controls. In early S phase, high-resolution 3D analysis of replication foci showed DT40 cells to have 390±176 (n=10) foci, whereas Chk1-deficient cells, with 755±425 (n=7) foci, had roughly two-fold more active sites. The replication patterns in Chk1−/− and wt cells were shown to be statistically different using the Mann–Whitney test (P<0.02; U-value 59). Notably, the foci in Chk1-deficient cells were smaller and more homogeneous in structure, independently of labelling intensity (Figure 4E and Supplementary Figure S2). Chk2-deficient DT40 cells, in contrast, displayed the normal distribution of replication patterns, with no significant changes in the structure or number of replication foci (Figure 4E and Supplementary Figure S2). These observations imply that the natural link between DNA synthesis and the spatial organisation of active replication foci is compromised in Chk1-deficient DT40 cells.
Defects in the DT40 replication programme
Quantitative analysis of early-, mid- and late-S-phase patterns showed that Chk1-deficient cells had a higher proportion of mixed S-phase structures than wt controls (Figure 4F). To extend this observation, we used double labelling protocols to describe the spatial architecture of the S-phase programme (Figure 4G). The vertebrate S phase is structured so that the sequential activation of replicon clusters occurs at spatially adjacent sites as S phase proceeds (Sadoni et al, 2004). This spatial relationship is maintained in wt DT40 (Figure 4G), where 81±7% of foci showed colocalisation when the two pulse labels were applied consecutively. Notably, when double labelling was performed in Chk1-deficient cells, colocalisation was only seen at 59±16% of foci (Figure 4G, Detail and Supplementary Figure S2D), which was very similar to the colocalisation in both wt and Chk1−/− DT40 treated with caffeine (Supplementary Figure S2D). Hence, Chk1-deficient cells show a >2-fold increase in the number of active foci that are spatially uncoupled from foci that were labelled immediately upstream in the replication programme. From these experiments, we conclude that Chk1 contributes to the mechanism that normally regulates S-phase progression in DT40 cells.
Following treatment with caffeine, the structure of replication foci in wt DT40 cells mimicked those seen in Chk1-deficient cells (Figure 4G). After 50 min in the presence of caffeine, a ∼2-fold increase in the complexity of early-S-phase foci was seen, although no increase was seen when Chk1-deficient cells were treated with caffeine (Figure 4G and Supplementary Figure S3). Similar observations were recorded in cells treated with UCN-01 (Supplementary Figure S3). As for Chk1−/− DT40, double labelling of replication foci in DT40 cells treated with Chk1 inhibitors showed a ∼2-fold increase in foci that were not colocalised during consecutive pulse labels (Figure 4G, Detail and Supplementary Figure S2). This is consistent with disorganisation of the replication programme in the presence of Chk1 inhibitors and shows that caffeine and UCN-01 phenocopy Chk1 deletion in DT40 cells.
Unprogrammed initiation of DNA synthesis in Chk1−/− DT40
To look more precisely at the specific roles of Chk1, we also analysed if the activation of DNA synthesis was perturbed in Chk1-deficient DT40 cells under normal—unstressed—growth conditions (Figure 5). Cells were labelled for 24 h with BrdU before pulse labelling with biotin-dUTP. After spreading, both analogues were visualised by indirect immunolabelling and the distribution of biotin-labelled replication forks was analysed in single, unbroken DNA fibres of ∼250 kb. In a typical experiment (Figure 5A and B), labelled regions of isolated (single), unbroken DNA strands were selected at random (using BrdU labelling) and aberrant replication structures within 25 kb of the paired forks of replicon clusters with two active replicons were recorded (Figure 5A). In wt DT40, 93.5% of labelled replicons conformed to the expected structure, with pairs of divergent sister forks. Only 6.5% had any associated unexpected labelling (n=46; average length of labelled region 93.9 kb). In contrast, 35% of randomly selected replication structures from Chk1-deficient DT40 (Figure 5A and B) had associated aberrant replication (n=40; average length of labelled region 56.5 kb).
Figure 5.
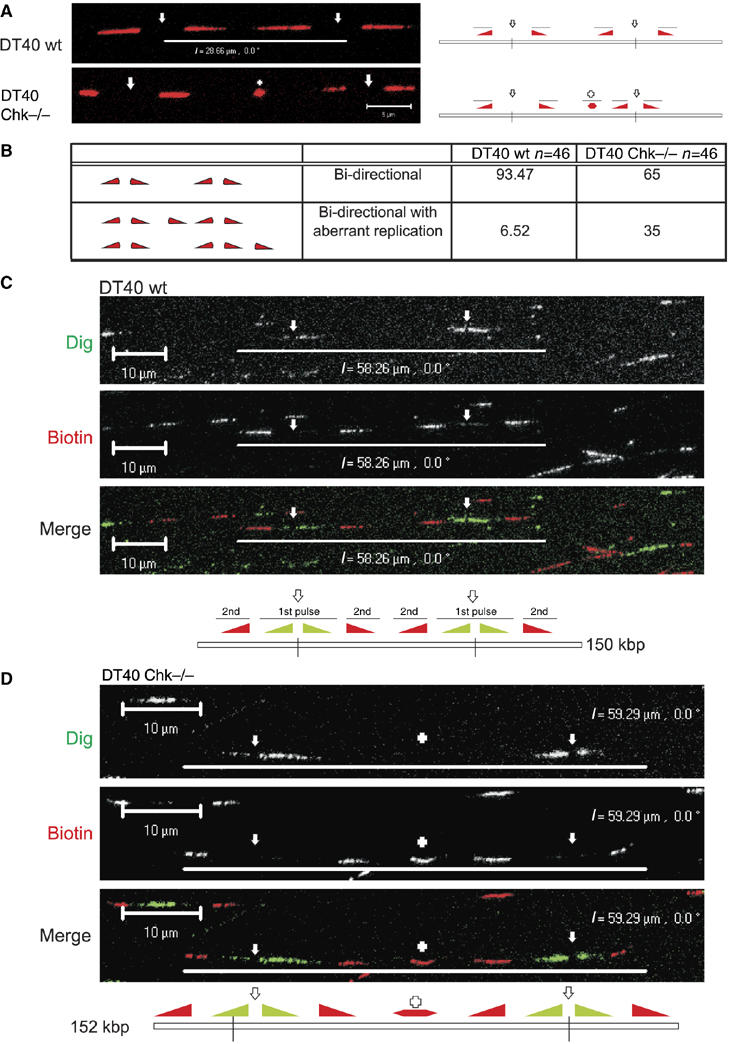
Unprogrammed activation of DNA synthesis in Chk1-deficient DT40 cells. Chk1−/− and isogenically matched wt DT40 cells were labelled with BrdU for 24 h and pulse labelled with biotin-dUTP (30 min; (A, B)) or Dig-dUTP, followed by biotin-11-dUTP (both 30 min; (C, D)). In panel (A) are shown sites of biotin-dU incorporation (red) in single, isolated DNA fibres from wt and Chk1−/− DT40 cells. DNA fibres were labelled with BrdU to ensure that only single fibres were used for the analysis; for simplicity the BrdU labelling is not shown (some examples are provided in Supplementary Figure S4 and S5). Replicon structure was defined by the organisation of the growing forks (diagram (panel A); filled triangles represent the direction of fork growth from central origins, depicted by open arrows; + represents a novel or aberrant replication structure) using fibres with two active replicons (four labelled forks). The relative frequency of normal (bidirectional) and aberrant replication structures (within 25 kb of normal forks) was scored (panel B). Unprogrammed initiation in Chk1−/− cells was confirmed using a combination of Dig-dU and biotin-dU to define replicon structure (panels C, D). DNA fibres were immunolabelled to visualise isolated fibres with two active origins (∼250 kb DNA). Such replicons contain two pairs of growing forks that were labelled first with Dig and then with biotin (panels C, D). In Chk1−/− DT40 (panel D), but not in wt controls (panel C), unprogrammed initiation contributes to synthesis of the unreplicated DNA between the active growing forks—in this example, a new initiation event (+) occurred in the centre of the fibre during the second pulse. Diagrams of the structure of the labelled replicons are shown (panels C, D), using nomenclature described in panel A. Scale bars (5 μm (panel A) and 10 μm (panels C, D)).
This ∼5-fold increase of aberrant replication structures in Chk1-deficient cells is consistent with the activation of novel origins, which normally would only occur under conditions of replicative stress (Woodward et al, 2006). To emphasise this point, we developed a triple labelling protocol, which provides a more precise description of active replicon structure (Figure 5). Cells were labelled for 24 h with BrdU and pulse labelled sequentially with digoxigenin-dUTP and biotin-dUTP, and spread DNA fibres were immunolabelled (Figure 5C and D and Supplementary Figure S4). Undamaged DNA fibres of ∼250 kb, with two adjacent active replicons, were analysed. Typical fibres from DT40 cells displayed four active forks that were each labelled throughout both the first and second pulses (Figure 5C). The two origins in this cluster are 32.84 μm (85 kb) apart and all four of the active forks show similar rates of elongation (Figure 5C). Figure 5D shows a similarly labelled DNA fibre from Chk1-deficient cells, in which an additional origin (+) was activated between the two already active origins, during the second pulse label. Such initiation within ∼10 kb of two converging forks is extremely uncommon in wt cells, but not unusual, if Chk1 function is perturbed (e.g. Figures 1F and 5A) and is consistent with a role of Chk1 in regulating the activation of potential replication origins during the normal S phase.
Aberrant initiation events in Chk1-deficient cells are linked to defects in active replication forks
We next explored if aberrant initiation events in cells with compromised Chk1 activity were related to intrinsic defects in DNA replication (Syljuasen et al, 2005; Petermann et al, 2006). Normally, BrdU in DNA is only detected by immunolabelling if DNA is first denatured (typically by HCl treatment). However, BrdU incorporated into the DNA of Chk1-deficient cells is readily detectable without denaturation and TUNEL analysis confirms that this is due to persistent DNA nicks (Syljuasen et al, 2005; Petermann et al, 2006).
Cells were pulse labelled consecutively with BrdU and biotin-dUTP and labelled tracks of spread DNA fibres visualised either without or with prior acid treatment (Figure 6 and Supplementary Figure S5). When immunolabelling was performed after acid treatment, >90% of forks had adjacent tracks labelled with BrdU and biotin-dU, in both wt and Chk1-deficient cells (Figure 6; (+)HCl). However, when acid treatment was omitted, incorporated BrdU was almost completely undetected in spreads from wt cells, with <2% of biotin tracks showing very weak (background) BrdU immunostaining (Figure 6A, (−)HCl). In contrast, 16% of biotin tracks in spreads from Chk1-deficient cells had adjacent regions of BrdU immunostaining (Figure 6B, (−)HCl and Supplementary Figure S5). These labelled regions were typically 2.5–10 kb long and the fragmented labelling was clearly different in structure to the typical ∼25 kb tracks seen after HCl treatment. Notably, the acid-independent BrdU labelling in Chk1-deficient cells was frequently adjacent to a newly activated origin. A typical example is shown (Figure 6B, (−)HCl and bottom diagram in 6C), where fragmented BrdU labelled over ∼10 kb lies adjacent to a pair of divergent, biotin-labelled forks. Clearly, these biotin-labelled forks resulted from the activation of a novel replication origin towards the end of the BrdU labelling period, at a position that is ∼12 kb from the boundary of the BrdU labelling. As the DNA adjacent to the BrdU boundary is labelled with biotin, the boundary region must represent a termination site, where the biotin-labelled and stalled or aberrant BrdU-labelled forks met (Figure 5C).
Figure 6.

Novel origin activation is spatially coupled to aberrant replication structures in Chk1−/− DT40 cells. wt and Chk1-deficient DT40 cells were pulse labelled consecutively with BrdU (20 min) and biotin-dUTP (30 min). Spread DNA fibres were immunolabelled to visualise sites of incorporation of BrdU (green) and biotin-dU (red). Labelling was performed either with or without prior treatment with HCl, as shown (A, B). Merged images show overlays of green and red channels (panels A, B). In panel B, the merged image and individual channels are shown for a high-power view of a single fibre. A diagram describing the analysis is shown (C), using the nomenclature described in Figure 5. X indicates a boundary between regions labelled in the first pulse and a nearby replicon that was labelled predominantly during the second, as seen in panel B (high-power example). Scale bars (5 or 10 μm) are shown on individual images.
This experiment makes two key points: (a) that replication forks in Chk1-deficient cells are intrinsically unstable and prone to accumulate ssDNA breaks over many kilobase pairs and (b) that at some forks this damage correlates with the activation of latent origins that typically are located 10–20 kb away.
Structure of replicons and DNA foci in chick embryonic fibroblasts
As checkpoint pathways may be perturbed in cells, such as DT40, that are adapted to long-term culture, we next confirmed the organisation of replicons and DNA foci in primary avian cells. Early-passage chick embryonic fibroblasts (CEF) cells were treated with inhibitors of the checkpoint pathways as describe before. As for human and DT40 cells, CEFs grown in caffeine or UCN-01 showed a ∼2-fold increase in replication fork density, consistent with increased origin activation when Chk1 function was inhibited (Figure 7A and C). This increased origin density occurred in cells with ∼50% of the natural rate of fork elongation (Figure 7B and C). As with DT40 cells (Figure 4), in the presence of caffeine and UCN-01, CEFs showed a significant (∼2-fold) increase in the number of their active replication foci (Figure 7D and E and Supplementary Figure S2), which were spatially disorganised relative to untreated cells (Figure 7F).
Figure 7.
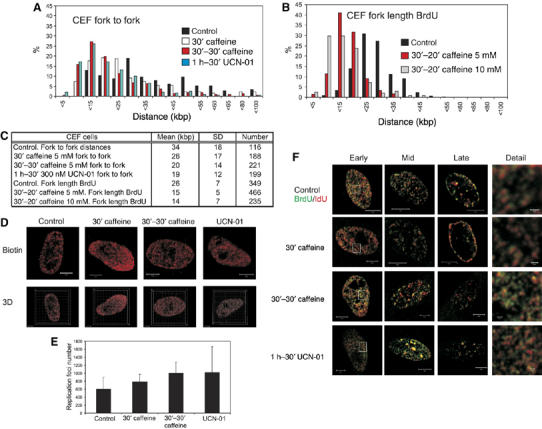
Chk1 regulates replication origin density in primary avian fibroblasts. Replication structures of chick embryo fibroblasts were visualised using the procedures described in Figure 2. Replicon size (A–C) was measured using the separation of sister forks (panel A) after labelling with biotin-dU and structures in untreated cells compared with those in cells treated with caffeine or UCN-01. BrdU labelling was also used to determine the rate of fork elongation (panel B) under the same conditions. Parameters defining average replicon structure under different conditions are shown (panel C). The number of active replication factories (D, E) and organisation of active replication foci (F) in cells treated with caffeine and UCN-01 were compared with untreated cells. Changes in the number of active DNA foci in treated cells were shown to be statistically significant using Mann–Whitney test: 30′ caffeine versus control (P<0.05; U-value 78); 30′–30′ caffeine versus control (P<0.005; U-value 88); UCN-01 versus control (P<0.02; U-value 82). Scale bars (5 and 0.5 μm in Detail) are shown on individual images.
Discussion
Under conditions of replicative stress, eukaryotic cells express sophisticated checkpoint pathways that serve to ensure the preservation of genomic integrity. If DNA is damaged during S phase, activation of the intra-S-phase checkpoint maintains replication fork stability and blocks initiation of latent replication origins (Kastan and Bartek, 2004; Sancar et al, 2004). During the damage response, activation of Chk1 leads to phosphorylation of Cdc25, which prevents downstream activation of the CDK/cyclin complexes that are required to activate latent origins. By maintaining the physiological balance of this pathway, Chk1 is also thought to contribute to the regulation of DNA synthesis in the absence of replicative stress (Zhao et al, 2002; Sorensen et al, 2003). The importance of balance within this regulatory pathway is emphasised by the phenotype of Chk1+/− cells from transgenic mice (Lam et al, 2004). Such cells maintain three clear haploinsufficiency phenotypes: inappropriate S-phase entry, accumulation of DNA damage during replication and failure to restrain mitotic entry. Inappropriate cell cycle transitions in these cells correlate with maintenance of high levels of Cdc25A.
In somatic mammalian cells, loss of Chk1 function correlates with a slight (20–30%) increase in global replication (Miao et al, 2003; Syljuasen et al, 2005), consistent with Chk1 contributing to the regulation of DNA synthesis. In support of this idea, recent studies from embryonic systems have suggested that the ATR/Chk1-dependent regulation of origin function might define the sequential activation of replication origins during S phase (discussed in Fisher and Mechali, 2004; Shechter and Gautier, 2005). However, a global increase in DNA synthesis in cells with reduced Chk1 activity conflicts with our recent observation that loss of Chk1 function reduces the rate of replication fork progression to ∼50% of that in wt cells (Petermann et al, 2006). In order to resolve this conflict, we have used quantitative analysis of nascent replicons in spread DNA fibres. Using both transformed and primary cells of human and avian origin, this approach provides the first formal proof that the density of active replication origins is increased by at least 2- to 3-fold when Chk1 activity is compromised (Figures 1, 2, 4 and 7).
Consistent with a role for Chk1 in modulating the density of active replication origins during normal S phase, our analysis of DNA fibres showed that inhibition of Chk1 compromises the efficacy of DNA replication and leads to reduced rates of elongation (Figures 2 and 7). Compromised replication at stalling forks correlates with an accumulation of aberrant DNA structures and activation of secondary, previously latent, origins (Figures 3 and 6) within the vicinity of the damaged forks. This activation of normally suppressed origins in cells with compromised Chk1 activity accounts for the observed increase in density of active origins, and implies that somatic vertebrate cells have an innate mechanism that balances the rate of elongation at replication forks with the density of active replication origins.
Alterations in the balance of the normal regulatory machinery appear to account—at least in part—for our observations. It is known that reduced expression or inhibition of Chk1 leads to accumulation of Cdc25A, which in principle would support super-activation of replication origins (Sorensen et al, 2003; Lam et al, 2004). We confirm that Cdc25A concentration increases by ∼5-fold in wt DT40 cell treated with caffeine or UCN-01 (Supplementary Figure S3), and that the origin associated protein Cdc45 is increased by about two-fold in human fibroblasts (Supplementary Figure S1) and DT40 (Supplementary Figure S3) when Chk1 activity is inhibited.
Does Chk1 contribute to S-phase progression in somatic vertebrate cells?
In nuclei assembled using Xenopus egg extract, ATR/Chk1 regulate the sequential activation of early and late replication origins (Marheineke and Hyrien, 2004; Shechter et al, 2004). It is important to recognise, however, that this embryonic system is very different from somatic cells in key respects. First, assembled nuclei have simple chromatin architecture and no transcription and second, the cell cycle is very rapid, with an S phase of ∼30 min and correspondingly short replicons of ∼15 kb (Blow et al, 2001). Although replicon clusters are activated throughout this short S phase (Blow et al, 2001), it is unknown if this is of physiological significance or reflects the stochastic nature of replication in this system. In clear contrast, somatic mammalian cells have been shown to have well-established S-phase programmes, with specific regions of the genome replicated at precise times (Jackson and Pombo, 1998; Sadoni et al, 2004). Progress through the programme can be correlated with the spatial architecture of the active replication sites (Jackson, 1995) and concomitantly correlated with the assembly of different classes of chromatin at different times during S phase (Zhang et al, 2002).
Despite these clear differences, it is interesting to compare our studies with in vitro observations in Xenopus extracts (Marheineke and Hyrien, 2004; Shechter et al, 2004), which suggested that ATR and Chk1 regulate S-phase progression by suppressing the activation of late origins during early S phase (Shechter et al, 2004). In human cells, we show that a mechanism that involves Chk1 suppresses the majority of potential origins that exist in active replicons; potential origins are known to outnumber those that are used by at least 10-fold (discussed in Woodward et al, 2006). When Chk1 activity was inhibited, super-activation of latent origins was restricted to regions of the genome that were already engaged with active replication factories (Figures 1H and 2I). Hence, while caffeine treatment might be expected to override Chk1-dependent suppression of late replicons in an early-S-phase cell (Shechter et al, 2004), the late origins of somatic human cells are not activated because they are unable to access active replication machinery. This implies that the temporal programme of replication factory assembly plays a dominant role in defining which potential origins can be activated at different times of S phase.
In avian cells in contrast, loss of Chk1 function correlated with a significant increase in the number of active replication foci and some disorganisation of the temporal programme of S-phase synthesis (Figures 4 and 7). This difference between human and avian cells raises interesting questions concerning the evolution of S-phase regulation. ATR and Chk1 play a key role in local (within individual replication factories) regulation of origin activation in early stages of amphibian (Xenopus) development and in avian and mammalian somatic cells. However, in the in vitro Xenopus system, the temporal activation of replicon clusters is lost if ATR is inhibited (Marheineke and Hyrien, 2004). In human cells, in contrast, S-phase timing is preserved when ATR function is compromised (Figures 1 and 2). Intriguingly, somatic avian cells seem to have evolved a hybrid organisation in which the timing programme is disordered (Figures 4 and 7) but the duration of S phase is maintained (Zachos et al, 2005). These differences presumably imply that the regulatory pathways that control S-phase progression are able to interact (i.e. complement) to different extents in mammalian and avian cells. This conclusion is not entirely unfounded, as deletion of Chk1 in the mouse is known to be cell lethal (Liu et al, 2000) whereas avian DT40 Chk1−/− cells are viable.
A model for Chk1 function during normal S phase
Experiments present here support the idea that Chk1 contributes to the efficacy of replication at active replication forks and couples defects in synthesis at active forks to the local activation of nearby latent origins. This implies that even during a normal S phase, a Chk1-dependent system continually senses the status of active replicons within replication factories and monitors the number of active forks to ensure progress of the synthetic process.
An important feature of this model is the provision of latent origins, which are suppressed during normal synthesis but serve to rescue replication if for any reason the active forks might fail. In fact, it has been known for many years that immediately before S phase, replicons have ∼10-fold excess of potential replication origins—defined by MCM2–7 complexes—which are clustered into licensing groups (discussed in Woodward et al, 2006). The redundant MCM complexes are displaced from chromatin during replication, but also appear to provide a rescue function if active forks are aborted because of DNA damage (Woodward et al, 2006). This means that under normal conditions, only a minority of potential origins is actually used, and that a mechanism must operate to limit origin activation so that once the required number of origins has been activated the use of proximal potential origins is suppressed. The data presented here support the idea (Shechter and Gautier, 2005) that the ATR/Chk1 pathway controls the activation and suppression of replication origins to regulate the density of active origins throughout S phase of somatic vertebrate cells.
Materials and methods
Cell culture
HeLa cells were grown in DMEM (Sigma) with 5% FBS and antibiotics. DT40 cells were grown in RPMI-1640 (Sigma) with 10% FBS, 1% chicken serum, 10−5 M β-mercaptoethanol and antibiotics. Early-passage MRC5 cells (ATCC cell line CCL-171—normal human diploid fibroblasts from lung) were grown in MEM, L-glutamine, sodium pyruvate, non-essential amino acids with 10% FBS and antibiotics. CEF (Kilbey et al, 1996) were grown in DMEM, L-glutamine, non-essential amino acids, sodium pyruvate, 1% of chicken serum, 10% FBS and antibiotics.
DNA fibre experiments
Replication tracks were labelled in culture medium containing 25 μM BrdU. For dual labelling, cultures were pulse labelled with 25 μM BrdU, washed and then labelled with 250 μM iodo-deoxyuridine (IdU). These conditions are known to support natural rates of fork elongation (Petermann et al, 2006). Cells were labelled with modified dUTPs as detailed below. UCN-01 was used at 300 nm and caffeine at 5 mM.
DNA fibre spreads were prepared as previously described (Jackson and Pombo, 1998). BrdU-labelled tracks were detected with BrdU anti-sheep antibody (Biodesign; M20105S; 1:1000 dilution; 1 h at 20°C) using either Cy3- or AlexaFluor-488-conjugated donkey anti-sheep secondary antibody. Biotin-11-dUTP detection was carried out using a mouse monoclonal antibody (Clone BN-34; Sigma; 1:1000 dilution; 1 h at 20°C) and Dig-11-dUTP (Roche) detection using a sheep polyclonal antibody (Roche; 1:1000 dilution; 0.5 h at 20°C) and appropriate Cy3- or AlexaFluor-488-conjugated second antibody.
Fibres were examined using a Zeiss LSM 510 confocal microscope using a × 100 (1.4 NA) lens, labelled tracks measured using the LSM software (white bars on individual images show examples of measurements recorded) and converted to kilobase pairs using a conversion factor of 1 μm=2.59 kb (Jackson and Pombo, 1998). Measurements were recorded in randomly selected fields (selected at low power) from dispersed, untangled areas of the DNA spread. As the analysis of single, unbroken fibres is a key feature of this study, routine quality control for spreading of different cell types under different experimental conditions was performed using direct DNA labelling with YOYO (Merrick et al, 2004) or BrdU immunolabelling of fibres from cells labelled with BrdU for ∼24 h.
Replication foci in situ
Modified nucleoside triphosphates were introduced into cells using FuGene (Roche). Briefly, DT40 cells were pelleted (200 g, 4 min), washed 3 × in cold PBS and 1 × 106 cells in 10 μl PBS transfected by incubating with transfection mix (12 μl PBS, 3 μl of FuGene and 1 μl (1 nmol) of the required dUTP analogue) for 10 min at 0°C. After transfection, the cells were washed in fresh medium (0.5 ml of medium, 200 g, 4 min) and incubated in medium (0.5 ml) for 30 min at 37°C. For double labelling, the same protocol was used for the second dUTP analogue. For experiments using BrdU, cultures were pulse labelled with 25 μM BrdU either before or after transfection. HeLa cells were grown on microscope coverslips and transfected with biotin-11-dUTP. Coverslips were rinsed in PBS and placed over a 7.5 μl droplet of transfection mix on parafilm and incubated for 10 min at 0°C. Coverslips were then rinsed in PBS and incubated in fresh medium for 30 min. MRC5 and CEF cells were transfected directly with 30 μl of transfection mix in 35 mm Petri dishes and incubated on ice for 10 min, rinsed with cold PBS and incubated in fresh medium for 30 min.
These loading conditions use very low precursor concentrations that were optimised to transiently support normal rates of fork elongation. The treatment has no short- or long-term effects on cell cycle duration and cell viability. For example, replication forks in DT40 cells analysed 10 min after loading travelled 12.19±2.61 kb (n=52), which corresponds with the average elongation rate of 1.2 kb/min in these cells (Petermann et al, 2006). In addition, the Chk1-dependent DNA damage checkpoint was not activated by these loading and replication conditions (Supplementary Figure S4).
Immunofluorescence and direct labelling of DNA foci
wt, Chk1−/− and Chk2−/− DT40 cells at 5 × 105 cells/ml were labelled with 25 μM BrdU, 250 μM IdU or transfected with the specific dUTP analogue. Cells were washed 3 × in cold PBS and 1 × 106 cells swollen in 0.075 M KCl for 15 min at 37°C (Jackson and Pombo, 1998). Nuclei were fixed with methanol/acetic acid (3:1), added dropwise onto washed microscope slides, and dried. HCl denaturation and immunolabelling were performed as detailed before (Jackson and Pombo, 1998).
For double labelling experiments, DT40 nuclei fixed with methanol/acetic acid or HeLa, MRC5 and CEF cells fixed in 4% PF were rinsed three times in PBS, two times in ddH2O and then incubated with HCl. The slides were rinsed in PBS and PBS+, blocked and incubated with rat anti-BrdU (Immunologicals Direct Clone BU 1/75; 1:1000 dilution; 1 h at 20°C), washed 3 × in PBS and 3 × in PBS+ and AlexaFluor-488-conjugated donkey anti-rat antibody (1:1000 dilution; 1 h; 4°C). The slides were washed 3 × in PBS and 3 × in PBS+, and IdU detected using BrdU anti-sheep antibody (1:1000 dilution; 15 h at 4°C). Slides were then washed 3 × in PBS, 3 × in PBS+ and incubated with Cy3-conjugated donkey anti-sheep antibody (1:1000 dilution; 1 h at 4°C). Finally, the slides were washed 3 × in PBS+, 3 × in PBS, incubated with 5 μg/ml Hoechst 33258 (Sigma) for 10 min, rinsed 3 × in PBS and mounted with 50:50 PBS–glycerol.
For nuclei labelled with Cy3-dUTP and/or AlexaFluor-488-conjugated dUTP, slides were washed 3 × in PBS, incubated with 5 μg/ml Hoechst 33258 (Sigma) for 10 min, and then rinsed 3 × in PBS and mounted with PBS–glycerol.
Slides were examined using a Zeiss LSM 510META confocal microscope using a × 100(1.4 NA) lens. 3D images were generated using Z stacks and processed in Imaris™ software.
Supplementary Material
Supplementary Figures
Supplementary Figure Legends
Acknowledgments
This work was funded by the BBSRC (linked project grant BBS/B/06091 to DAJ and KWC). We thank Chi Tang and other colleagues for technical advice and support.
References
- Berezney R, Dubey DD, Huberman JA (2000) Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108: 471–484 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Gillespie PJ, Francis D, Jackson DA (2001) Replication origins in Xenopus egg extracts are 5–15 kb apart and are activated in clusters that fire at different times. J Cell Biol 152: 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher D, Mechali M (2004) Sleeping policemen for DNA replication? Nat Cell Biol 6: 576–577 [DOI] [PubMed] [Google Scholar]
- Gilbert DM (2002) Replication timing and transcriptional control: beyond cause and effect. Curr Opin Cell Biol 14: 377–383 [DOI] [PubMed] [Google Scholar]
- Jackson DA (1995) Nuclear organization—uniting replication foci, chromatin domains and chromosome structure. Bioessays 17: 587–591 [DOI] [PubMed] [Google Scholar]
- Jackson DA, Pombo A (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J (2004) Cell-cycle checkpoints and cancer. Nature 432: 316–323 [DOI] [PubMed] [Google Scholar]
- Kilbey A, Black EJ, Unlu M, Gillespie DAF (1996) The v-Jun oncoprotein replaces p39 c-Jun as the predominant AP-1 constituent in ASV17-transformed fibroblasts: implications for SAPK/JNK-mediated signal transduction. Oncogene 12: 2409–2418 [PubMed] [Google Scholar]
- Lam H, Liu Q, Elledge SJ, Rosen JM (2004) Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6: 45–59 [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Machida YJ, Hamlin JL, Dutta A (2005) Right place, right time, and only once: replication initiation in metazoans. Cell 123: 13–24 [DOI] [PubMed] [Google Scholar]
- Miao H, Seiler JA, Burhans WC (2003) Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J Biol Chem 278: 4295–4304 [DOI] [PubMed] [Google Scholar]
- Marheineke K, Hyrien O (2004) Control of replication origin density and firing time in Xenopus egg extracts—role of a caffeine-sensitive, ATR-dependent checkpoint. J Biol Chem 279: 28071–28081 [DOI] [PubMed] [Google Scholar]
- Merrick CJ, Jackson D, Diffley JFX (2004) Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem 279: 20067–20075 [DOI] [PubMed] [Google Scholar]
- Norio P, Kosiyatrakul S, Yang QX, Guan ZQ, Brown NM, Thomas S, Riblet R, Schildkraut CL (2005) Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol Cell 20: 575–587 [DOI] [PubMed] [Google Scholar]
- Petermann E, Maya-Mendoza A, Zachos G, Gillespie DAF, Jackson DA, Caldecott KW (2006) Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 26: 3319–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoni N, Cardoso MC, Stelzer EHK, Leonhardt H, Zink D (2004) Stable chromosomal units determine the spatial and temporal organization of DNA replication. J Cell Sci 117: 5353–5365 [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39–85 [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6: 648–655 [DOI] [PubMed] [Google Scholar]
- Shechter D, Gautier J (2005) ATM and ATR check in on origins—a dynamic model for origin selection and activation. Cell Cycle 4: 235–238 [PubMed] [Google Scholar]
- Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Bartek J, Lukas J (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3: 247–258 [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25: 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M, Nakayama K (2000) Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev 14: 1439–1447 [PMC free article] [PubMed] [Google Scholar]
- Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ (2006) Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 173: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DAF (2003) Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J 22: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DAF (2005) Chk1-dependent S–M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol Cell Biol 25: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, Piwnica-Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 99: 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Xu F, Hashimshony T, Keshet N, Cedar H (2002) Establishment of transcriptional competence in early and late S phase. Nature 420: 198–202 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figure Legends