

Abstract
The human interferon-induced protein kinase, PKR, is an antiviral agent that is activated by long stretches of double-stranded (ds)RNA. PKR has an N-terminal dsRNA-binding domain that contains two tandem copies of the dsRNA-binding motif and interacts with dsRNA in a nonsequence-specific fashion. Surprisingly, PKR can be regulated by certain viral and cellular RNAs containing non-Watson–Crick features. We found that RNAs containing bulges in the middle of a helix can bind to p20, a C-terminal truncated PKR containing the dsRNA-binding domain. Bulges are known to change the global geometry of RNA by bending the helical axis; therefore, we investigated the conformational changes of bulged RNA caused by PKR binding. A 66-mer DNA-RNA(+/− A3 bulge)-DNA chimera was constructed and annealed to a complementary RNA strand. This duplex forces the protein to bind in the middle. A 66-mer duplex with a top strand composed of DNA-DNA(+/−A3 bulge)-RNA was used as a control. Gel mobility-shift changes among the RNA-protein complexes are consistent with straightening of bulged RNA on protein binding. In addition, a van't Hoff analysis of p20 binding to bulged RNA reveals a favorable ΔΔH° and an unfavorable ΔΔS° relative to binding to straight dsRNA. These thermodynamic parameters are in good agreement with predictions from a nearest-neighbor analysis for RNA straightening and support a model in which the helical junction flanking the bulge stacks on protein binding. The ability of dsRNA-binding motif proteins to recognize and straighten bent RNA has implications for modulating the topology of RNAs in vivo.
The human interferon-induced double-stranded RNA (dsRNA)-dependent protein kinase PKR can regulate gene expression in several ways. PKR is an antiviral agent that can inhibit translation via phosphorylation of initiation factor eIF2α, affect cytokine signaling and transcription activation, and promote apoptosis (1, 2). In addition, PKR itself can be regulated by a number of factors including cytokines, growth factors, stress signals, and dsRNA (2). PKR senses dsRNA by a dsRNA-binding domain (dsRBD) that consists of two tandem dsRNA-binding motifs (dsRBMs).
dsRBM is a conserved stretch of 65–75 amino acids originally identified by sequence alignment of functionally diverse proteins from a wide range of organisms (3, 4). Approximately 100 dsRBMs in over 50 different proteins are currently known (4, 5). These include PKR, Escherichia coli RNase III, Drosophila staufen protein required for mRNA localization, mammalian dsRNA adenosine deaminases, and putative mammalian RNA interference factors (4, 6). Structural studies on dsRBM proteins include NMR structures of a dsRBM from staufen protein without (7) and with (8) a 12-bp dsRNA, the dsRBM from RNase III (5), the dsRBD from PKR (9), and a crystal structure of a dsRBM from Xenopus laevis RNA-binding protein A complexed with a 10-bp dsRNA (10). These structures revealed that the dsRBM has an α-β-β-β-α secondary structure, with the α-helices packed on one face of a three-stranded antiparallel β sheet. Interaction between the dsRBM and dsRNA is mediated in large part by nonsequence-specific contacts with the dsRNA (3, 11) involving 2′-OHs and phosphates (8, 10, 12). Lack of obvious RNA sequence specificity is consistent with the outcome of biochemical experiments (11). In addition, PKR does not bind to and is not activated by dsDNA or RNA–DNA hybrids (12).
Surprisingly, PKR can also interact with and be regulated by viral and cellular RNAs containing non-Watson–Crick features, including human hepatitis δ virus RNA (13), adenovirus virus-associated (VA) RNA1 (14), Epstein–Barr viral RNAs (EBERs) (15), and HIV-1 transactivation response element (TAR) RNA (16, 17). These RNAs contain various internal loops and bulges. In particular, VA RNA1 has a GAUAAAU bulge (14), EBER-2 RNA has an AA bulge (18), and TAR RNA has C, A, and UCU bulges (12, 16, 17). In addition, in vitro selection experiments revealed that RNAs containing internal loops, bulges, and noncontiguous helices can bind PKR (19). In several cases, these RNAs maintain an overall A-form geometry, but no sequence-specific motifs were evident (19). These findings indicate that dsRBM proteins have important roles involving interactions with RNAs containing non-Watson–Crick features. These interactions and any associated conformational changes remain largely unexplored. Because most viral and cellular RNAs contain non-Watson–Crick features, understanding how dsRBM proteins interact with such RNAs may have widespread biological significance.
Bulges are among the most common non-Watson–Crick features in RNA. A bulge occurs when a duplex is interrupted by single-stranded nucleotides on only one strand. In RNA, bulges are important for secondary and tertiary structure formation (20) and can form specific interactions with proteins (21). Studies using PAGE (22–24) and transient electric birefringence (TEB) (25) have indicated that bulged nucleotides can bend the helical axis of RNA and DNA. This contrasts with internal loops, which do not significantly bend the helix (24, 26). It has been found that A bulges have greater effects on electrophoretic mobility than do U bulges (22, 23, 25). By using TEB measurements, An-bulge-induced bend angles of 0° (n = 0), 15° (n = 1), 42° (n = 2), 58° (n = 3), and 93° (n = 6) have been measured in the absence of Mg2+ (25). Experiments involving separation of two bulges by a variable number of base pairs result in strong phase dependence, suggesting that bulge-induced bends are relatively rigid and not just sites of flexibility (22, 23, 27).
It is possible that conformational changes of bulged RNA occur on binding of dsRBM proteins. To address this issue, we placed An bulges at the center of RNA duplexes and examined the effects of binding of the dsRBD from PKR. We compare the electrophoretic mobility of the bulged RNA-protein complex with that of a dsRNA-protein complex and carry out a thermodynamic study of binding to straight and bent RNAs. Results support a model in which binding of the dsRBD induces straightening of bent dsRNA.
Materials and Methods
Expression and Purification of Proteins.
The ≈20-kDa construct H6Tp20 has an N-terminal (His)6 and residues 1–184 of PKR, which contain the dsRBD (12). H6Tp20 was overexpressed and purified as described (12). The concentration of p20 was determined spectrophotometrically. It has been demonstrated that the (His)6 tag does not appreciably affect binding of p20 to dsRNA (12).
Shorthand Notation for Oligonucleotides.
Experiments used a variety of strands composed of DNA, RNA, or both (=an RNA-DNA chimera). Each oligomer is referenced with some or all of the following information: strand, length, radiolabel, composition and order of the segments, and number of bulges. In all cases, the radioactive phosphorous is at the 5′ end of the designated segment, and the bulge is in the center of the segment. The length of each segment is 22 nt plus the A bulges. For example, TS66-Dp*R(A3)D refers to a top-strand chimera that is 69 nt (=66 mer + A3 bulge) and has a 22-nt 5′ DNA segment, a 25-nt internal RNA segment that is labeled at its 5′ end and 3 As in its center, and a 22-nt 3′ DNA segment. The 3 As become bulged on annealing to the bottom strand.
Preparation of Oligonucleotides.
DNA oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA), chemically 5′ phosphorylated as necessary, and PAGE purified.
(i) Preparation of RNA segments.
RNA transcripts (Fig. 1, 2) were prepared from oligomer templates by using phage T7 RNA polymerase (Ambion, Austin, TX) (28). Transcripts were purified by 12% PAGE and eluted by crushing and soaking for 16 h at 4°C in TEN250 [10 mM Tris (pH 7.5)/1 mM EDTA/250 mM NaCl]. RNAs were concentrated by ethanol precipitation, washed with 70% ethanol, stored in TE [10 mM Tris (pH 7.5)/0.1 mM EDTA], and concentrations were determined spectrophotometrically. As necessary, transcripts were dephosphorylated with calf intestinal phosphatase, 5′-end radiolabeled via [γ-32P] ATP and polynucleotide kinase, and repurified by PAGE.
Figure 1.

Construction of chimeric duplexes. Oligonucleotides used in chimeras and splints are shown. RNA is bold, DNA is in normal lettering, and bulges and ligation junctions are indicated. “p” represents a 5′-phosphate and “OH” a 3′-hydroxyl. (A) Preparation and sequence of DRD chimeras. Shown is base pairing to splints and BS66-R-1. (B) Preparation and sequence of DDR chimeras. Shown is base pairing to splint 1 and BS66-R-2.
Figure 2.

Mobility-shift assays of An-bulged duplexes. RNA is bold, DNA is in normal lettering, and bulges are indicated. p*dsR22-An and p*ds66-An represent 5′-labeled top strand annealed to bottom strand. (A) Electrophoresis was on a native 10% polyacrylamide gel, with 0 or 0.3 μM p20. Gel was run at 22°C, resulting primarily in the 1:1 protein-RNA complex at this [p20]. (B) Electrophoresis was on a native 15% polyacrylamide gel with 0, 0.01, 0.03, and 0.1 μM p20. Gel was run at 4°C. Positions of 1:1 and 2:1 RNA-protein (RNP) complexes are shown. Duplex structures in the 1:1 complex are drawn on the basis of the best model. Straightening may not be complete. Protein is depicted as an oval and the duplex as a rectangle with dsRNA segments filled and RNA–DNA hybrid segments open.
(ii) Construction of chimeras and chimeric duplexes.
DNA-RNA-DNA (DRD) chimeras (66 or 69 mers) were prepared on the basis of a published method (29). DRD chimeras were constructed in two steps (Fig. 1A). Step 1: TS22-p*R(An) (22 or 25 mer, n = 0 or 3), pD3′ (22 mer), and splint 2 were heated at 90°C for 2 min in TE and cooled to 22°C over 20 min. For the A3 RNA, a longer splint was used that pairs with the extra As. After hybridization, ligation buffer and T4 DNA ligase (Promega) were added and incubated for 16 h at 22°C. Ligated product, p*R(An)D3′ (44 or 47 mer) was separated by 6% PAGE, visualized by autoradiography, and purified. Step 2: Purified p*R(An)D3′ and splint 1 were heated to 90°C for 2 min in TE and cooled to 22°C over 20 min. D5′-1 (22 mer) was added and incubated at 22°C for 30 min. After hybridization, T4 DNA ligase and ligation buffer were added and incubated as above. Ligated product, TS66-Dp*R(An)D (66 or 69 mer), was separated and purified as above. DNA-DNA-RNA (DDR) chimeras (66 or 69 mers) were also constructed. The procedure was similar to DRD but involved only one ligation of D5′(An)-2 (44 or 47 mer, n = 0 or 3) to the 5′ end of TS22-p*R(A0) (22 mer) RNA by using splint 1 (Fig. 1B). D5′(An)-2 are single oligonucleotides but are referred to as “D-D” in the shorthand notation, to be consistent with each segment being a 22 mer.
Chimeric duplexes were prepared by annealing a radiolabeled top-strand chimera, ≈1 nM, with 0.2 μM bottom-strand RNA (Fig. 1). Annealing was in TEN100 [10 mM Tris (pH 7.5)/1 mM EDTA/100 mM NaCl] at 95°C for 3 min, followed by cooling to 22°C for 20 min.
Characterization of Chimeras and Chimeric Duplexes.
RNA sequencing lanes of Dp*RD and DDp*R chimeras were prepared by cleavage with nuclease RNase T1 under denaturing conditions. All-nucleotide lanes of BS66-p*R-1 and the RNA segment in the chimeras were prepared by alkali treatment. Structure mapping of the duplexes was performed under native conditions. The chimeric duplex was prepared by annealing, and digested with RNase T2 for 30 min at 22°C in 150 mM NaCl/25 mM Hepes (pH 7.5). The reaction was quenched on dry ice and loaded on a denaturing gel.
Mobility-Shift Assays.
Native-gel mobility-shift assays were performed with limiting concentrations of radiolabeled dsRNA, as described (12). Binding was in 25 mM Hepes (pH 7.5)/10 mM NaCl, 5% glycerol/5 mM DTT/0.1 mM EDTA/0.1 mg/ml herring sperm DNA (Sigma). Binding reactions were allowed to equilibrate at the temperature of the experiment for at least 10 min and were loaded on a 15% (79:1 acrylamide/bis)/0.5 × TBE native gel. Identical results were obtained for a 30-min preincubation (4°C). Electrophoresis was at 330 V for 3–5 h. The temperature of the apparatus was controlled by an external bath that flowed into a heat exchanger located behind the gel plates. External bath temperatures were 4, 10, 16, or 20°C, and the actual temperature in the gel was measured by insertion of a thermocouple probe into the gel (30). A minimum of two titrations were performed for each RNA at each temperature; each titration was loaded on two gels to give a minimum of four gels per condition. The fraction of bound RNA in all complexes, f, was the average from the gels.
For the mobility-shift assays with DRD and DDR chimeric duplexes, electrophoresis was for 8 h at 330 V and 4°C. Mobility is defined as the distance from the well to the middle of a band.
Determination of ΔH° and ΔS° from van't Hoff Plots.
Data were quantified by a PhosphorImager (Molecular Dynamics) and fit by nonlinear least squares (kaleidagraph, Synergy Software, Reading, PA) to Eq. 1:
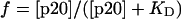 |
1 |
to yield the dissociation constant KD and an associated error, σKD. Under conditions of low temperature and/or high p20 concentration, binding to unbulged and bulged duplexes gave rise to a second gel shift (12). However, the intensity of the upper gel shift was less than that of the lower shift, suggesting binding was dominated by the 1:1 protein-RNA complex. Conditions in which saturation of RNA was achievable (e.g., low temperature) revealed f values ≈1; conditions in which saturation of RNA was unachievable were also fit well by Eq. 1. The fraction of bound RNA at saturation under all conditions was therefore assumed to be 1. On converting KD to KA, σKA was calculated from σKD by maintaining percent errors (31). Thermodynamic parameters were determined from a van't Hoff analysis by plotting ln KA vs. T−1 and by using a fit (kaleidagraph) in which the ln KA points were weighted by σKA/KA (31), shown as error bars in the plot. ΔH° was calculated as ΔH° = −slope × 0.001987 kcal K−1 mol−1, ΔS° was calculated as ΔS° = intercept × 1.987 entropy units, and errors in ΔH°, ΔS°, ΔΔH°, and ΔΔS° (Table 2) were propagated by standard methods from errors in the slope and intercept (31). ΔS° is subject to substantial error, because it is calculated by a long extrapolation to infinite temperature. Nevertheless, the signs and values of ΔS° and ΔΔS° are deemed reliable.
Table 2.
Thermodynamic parameters for RNA-protein complex formation
| RNA | ΔH°, kcal/mol | ΔS°, eu |
|---|---|---|
| dsR22-A0 | −21.1 ± 2.1 | −41.7 ± 7.2 |
| dsR22-A3 | −28.3 ± 2.0 | −70.0 ± 7.2 |
| ΔΔX(A3-A0) | −7.3 ± 2.9 | −26.3 ± 10.2 |
Experiments were at 5.5, 11.4, 17.3, and 22°C, with temperatures measured in the gel. Sequences are at the top of Fig. 2A. ΔH° and ΔS° were calculated from a van't Hoff analysis. ΔΔX(A3-A0) is ΔX of dsR22-A3 minus ΔX of dsR22-A0, where X denotes H° and S°. Error analysis is described in Materials and Methods.
Results and Discussion
Construction of Chimeras and Chimeric Duplexes.
Previous studies on DNA bending showed that the shape of the DNA molecule affects its electrophoretic mobility (32, 33). This holds true even when a protein is bound, as long as the overall DNA molecule is several times longer than the segment that interacts with the protein (32, 33). By comparing the electrophoretic mobility of various free and bound DNA species, protein-induced bending of the DNA can be inferred. In principle, comparative electrophoretic methods could be applied to elucidate the effect of p20 binding on the conformation of bulged RNA. However, dsRBM proteins interact with RNA in a nonsequence-specific fashion, necessitating construction of an alternative duplex.
To force p20 to bind only to a dsRNA segment within the context of a larger duplex, we took advantage of p20's lack of interaction with RNA–DNA hybrids (12). Chimeric DRD strands were constructed by ligating 22- or 25-mer RNAs to two 22-mer DNAs in two splint-mediated ligations to give TS66-Dp*R(A0)D and TS66-Dp*R(A3)D (Fig. 1A). This approach utilizes T4 DNA ligase, which catalyzes formation of a phosphodiester bond between a 3′-hydroxyl and a 5′-monophosphate juxtaposed by base pairing to a splint (29). The yield for the first ligation was ≈40%, with an overall yield after two ligations of ≈10–15%. Numerous attempts were made to prepare the DRD chimeras in one step but were largely unsuccessful, presumably because of misannealing. The DRD chimeras were annealed to a 66-mer RNA bottom strand (BS66-R-1) to prepare a chimeric duplex with an internal 22-bp dsRNA segment (+/−an A3 bulge) flanked by two 22-bp RNA–DNA hybrid segments (Fig. 1A). In addition, chimeric DDR strands were constructed by ligating a 22-mer RNA to the 3′-end of a 44- or 47-mer DNA in one step, to give TS66-DD(A0)p*R and TS66-DD(A3)p*R (Fig. 1B), with yields of ≈40%. The DDR chimeras were annealed to a 66-mer RNA bottom strand (BS66-R-2) to prepare a chimeric duplex with an external 22-bp dsRNA segment flanked by a 44-bp RNA–DNA hybrid segment (+/−a dA3 bulge) (Fig. 1B). All the bulges were in the center of the duplex, and adenosine bulges were chosen because these have been well studied (22, 23, 25). In addition, As were not placed opposite Us or Ts to avoid migration of the bulge.
Chimeras and chimeric duplexes were characterized in several ways. DRD and DDR products from the ligation steps were run on a denaturing polyacrylamide gel and were found to migrate at expected positions relative to a ladder; unbulged DRD and DDR strands comigrated, as did bulged DRD and DDR strands (data not shown). In addition, the DRD and DDR strands were subjected to alkaline hydrolysis and RNase T1 sequencing. Results were consistent with RNA segments of the proper sequence and length at the proper position (Fig. 5, which is published as supplemental data on the PNAS web site, www.pnas.org). Chimeric duplexes were formed by annealing and were subjected to RNase T2 cleavage under native conditions. Gels revealed cleavages only for the bulged duplex at the expected position of the bulge, consistent with proper annealing of bulged and unbulged duplexes (Fig. 5).
Effect of p20 on the Gel Mobility of Chimeric Duplexes.
A bulge-induced bend in dsRNA produces retarded electrophoretic mobility during native PAGE (22, 23). RNA duplexes (22 bp) containing internal A bulges of variable size had retarded migration in a native polyacrylamide gel relative to an unbulged duplex (Fig. 2A). Mobility decreased with increasing bulge size for the series A0,1,2,3 (Fig. 2A), consistent with previous reports (22, 23, 25). On binding to p20, the electrophoretic mobility of all RNA duplexes became identical regardless of the bulge size (Fig. 2A), suggesting that p20 changes the RNAs to a similar final conformation.
Previous studies on protein-induced conformational changes of DNA indicated that comparative electrophoresis techniques are most sensitive when the overall length of the DNA is several-fold greater than the protein-binding site (32, 33). We therefore carried out comparative electrophoresis experiments on model DRD chimeric duplexes containing an internal 22-bp dsRNA-binding site (+/−an A3 bulge), flanked by two 22-bp RNA–DNA hybrid segments (Fig. 2B). Previous experiments using p20 showed that the RNA-binding site must be at least 16 bp to give a mobility shift with a 1:1 protein/RNA stoichiometry (12) and 22 bp to give an additional 2:1 complex (11, 12). We chose a 22-bp internal dsRNA segment to allow both 1:1 and 2:1 complexes to be examined. Currently, it is unclear whether both dsRBMs in p20 contact the RNA simultaneously, although mutational studies suggest they do (unpublished results).
Comparison of electrophoretic mobilities reveals that the DR(A3)D duplex migrated considerably more slowly than the DR(A0)D duplex, with a relative gel mobility of 0.79 (=mobility of A3 duplex/A0 duplex) (Fig. 2B). This is consistent with the ability of A bulges to significantly bend dsRNA (22, 23, 25). Addition of p20 resulted in the formation of two complexes, as expected (12). The electrophoretic mobility of the two duplexes became nearly identical on binding to p20, with relative mobilities of 0.94 and 0.97 for complexes 1 and 2, respectively. This is consistent with the chimeric duplexes having similar final conformations.
To test whether the electrophoretic mobility was sensitive to the shape of the duplex when bound to protein, we constructed two DDR duplexes, which contain an external 22-bp dsRNA- (no bulges) binding site flanked by a 44-bp RNA–DNA hybrid segment (+/−a dA3 bulge) (Fig. 1B). Because p20 does not bind to RNA–DNA hybrids (12), binding of the protein should not affect the bend of the duplex. Comparison of electrophoretic mobilities reveals that the DD(A3)R duplex migrated slower than the DD(A0)R duplex, with a relative gel mobility of 0.86 (=mobility of A3 duplex/A0 duplex) (Fig. 2B). This is consistent with the ability of A bulges to bend RNA–DNA hybrids (22). Faster mobility of the DD(A3)R duplex than of the DR(A3)D duplex is consistent with reports on bulged RNA–DNA hybrids and dsRNA (22), although the bulge was on the RNA strand of the hybrid of those studies. Small differences in the mobility of the DD(A0)R and DR(A0)D duplexes may reflect the order of attachment of segments with different persistence lengths.
On binding to p20, the electrophoretic mobility of the DDR duplexes remained different, with a relative gel mobility of 0.85 for complex 1, or became more different, with a relative gel mobility of 0.75 for complex 2 (Fig. 2B). The relative mobilities of the p20-bound DDR duplexes indicate that electrophoretic mobility is sensitive to the shape of the duplex even when bound to p20, similar to observations for DNA-protein complexes (32, 33). This observation indicates that the DD(A3)R duplex remains bent on binding p20, as expected. If the DR(A3)D duplex remained bent on binding p20, then the relative gel mobility of the free and bound DRD duplexes should also have remained different. However, the relative mobilities of the DRD duplexes became very similar on forming complexes 1 and 2, changing from 0.79 to 0.94 and 0.97, respectively. This observation provides strong support for straightening of the DR(A3)D duplex on binding p20.
Comparative electrophoretic studies on the lac promoter binding to the catabolite gene activator protein (CAP) or the lac repressor have been performed (32). The electrophoretic mobility of the lac promoter bound to CAP depends strongly on the position of the promoter within a longer DNA, consistent with movement in the position of a significant protein-induced DNA bend (34). In contrast, the electrophoretic mobility of lac promoter bound to lac repressor does not depend on the position of the promoter within the longer DNA, consistent with the absence of significant protein-induced DNA bending in the experiment (32). Comparison of complexes 1 and 2 for the two unbulged duplexes containing DR(A0)D and DD(A0)R revealed similar electrophoretic mobilities, with relative gel mobilities of 0.95 and 0.94 for complexes 1 and 2, respectively [=mobility of DR(A0)D duplex/DD(A0)R duplex] (Fig. 2B). This indicates that p20 does not significantly bend straight dsRNA. Apparently, p20 prefers a relatively straight substrate. Moreover, similar mobilities of the DR(A0)D and DD(A0)R duplexes in the complexes suggest that the difference in the effect of p20 on the DR(A3)D and DD(A3)R duplexes is not simply because of the location of the p20-binding site within the duplex. Instead, effects of p20 can be attributed to changes in the conformation of bulged dsRNA within the complex. Similar mobilities of p20-bound DR(A3)D-, DR(A0)D-, and DD(A0)R complexes support a model in which p20 straightens the bend of the bulged dsRNA segment.
Thermodynamic Parameters for p20 Binding.
Binding of p20 to A3-bulged and -unbulged 22-bp RNA duplexes was studied by mobility-shift assays. Increasing amounts of p20 were added to a limiting amount of preformed radiolabeled duplex, and the bound species were separated by native gel electrophoresis (Fig. 3A). The fraction of RNA bound vs. p20 concentration was fit to Eq. 1 to extract the KD. Values for KD at 22°C for the unbulged duplex were similar to those previously reported (12).
Figure 3.

Temperature dependence of p20 binding to straight and bent RNAs. dsRNAs contained either no bulge (p*dsR22-A0) or an A3 bulge (p*dsR22-A3). Sequences of RNA duplexes are given in Fig. 2A. (A) Mobility-shift assay for p20 binding to dsRNA. Protein concentrations were 0, 0.002, 0.004, 0.008, 0.01, 0.02, 0.04, 0.08, 0.1, 0.2, 0.4, 0.8, 1.0, and 2.0 μM. Gel was run at 22°C. (B) Representative plots of fraction RNA bound vs. [p20]. Shown are plots at 5.5°C, p*dsR22-A0 (●) and p*dsR22-A3 (○), and 22°C, p*dsR22-A0 (■) and p*dsR22-A3 (□). Fits are to Eq. 1, and KD values are in Table 1. The fits at 5.5°C superimpose. Temperatures were determined in the gel as 5.5, 11.4, 17.3, and 22°C. (C) van't Hoff plots for p20 binding to p*dsR22-A0 (●) and p*dsR22-A3 (○). Fits are weighted, and ΔH° and ΔS° values are in Table 2.
To determine values for ΔH° and ΔS°, binding titrations were carried out at a series of temperatures (Table 1). As shown in Fig. 3B, the affinity of p20 for the unbulged duplex is similar to that for the bulged duplex at 5.5°C. As temperature increases, binding affinity to both duplexes decreases; however, it decreases faster for the bulged duplex (Fig. 3B; Table 1). This indicates that binding of p20 to the two duplexes is governed by different thermodynamic parameters. Values for ΔH° and ΔS° were determined by a van't Hoff analysis of the data (Fig. 3C; Table 2). Binding of p20 to the duplexes resulted in favorable ΔH° values, −21 and −28 kcal/mol, and unfavorable ΔS° values, −42 and −70 entropy units, for the unbulged and bulged duplexes, respectively. Comparison of the structures of free dsRBMs from several proteins (5, 7, 9) to the structures of the RNA-bound dsRBMs (8, 10) reveals little change in the structure of the dsRBM on binding to RNA, consistent with lack of a significant hydrophobic effect on RNA binding and a negative entropy.
Table 1.
Dissociation constants, KD (μM), for binding of RNAs to p20
| RNA | 5.5°C | 11.4°C | 17.3°C | 22°C |
|---|---|---|---|---|
| dsR22-A0 | 0.04 ± 0.008 | 0.068 ± 0.011 | 0.21 ± 0.032 | 0.32 ± 0.034 |
| dsR22-A3 | 0.044 ± 0.004 | 0.12 ± 0.015 | 0.45 ± 0.032 | 0.72 ± 0.079 |
The ΔH° and ΔS° values for p20 binding to bulged and unbulged duplexes can be compared (Fig. 4). The initial state, [1], of the free A3-bulged duplex is shown as bent, consistent with transient electric birefringence measurements of a bend angle of ≈58° (25), and the final state, [3], of the p20-bound complex is shown as straightened, consistent with the comparative electrophoretic experiments. In addition, state [1] is modeled without nearest-neighbor interactions of the adjacent base pairs, consistent with thermodynamic, theoretical, and structural considerations (35–37). The initial and final states are connected by a path involving state [2], with a free straightened A3 duplex in which the neighboring base pairs are stacked (Fig. 4A). This pathway is acceptable because ΔH° and ΔS° are state functions. The values used for ΔX°3–1 are from p20 binding to dsR22-A3, where ΔX°j-1 represents the change in a enthalpy or entropy on going from state [i] to [j]. The values used for ΔX°3–2 are from p20 binding to dsR22-A0. This assumes that the A3 bulge can be tolerated within a p20-bound complex. This should be a reasonable assumption, because binding to dsR22-A3 is tight and binding to dsRNA is nonsequence specific, allowing p20 to slip its binding register to minimize any steric interference. ΔX°3–2 can be subtracted from ΔX°3–1 to calculate experimental values for stacking of the helical junction, ΔX°2–1, resulting in a favorable ΔΔH° term of −7.3 kcal/mol and an unfavorable ΔΔS° term of −26.3 entropy units (Fig. 4; Table 2).
Figure 4.

Thermodynamic considerations for protein binding to RNA. (A) Three different states are shown: state [1] for bent dsRNA, state [2] for straightened RNA, and state [3] for dsRNA-protein complex. Levels are based on ΔG°22. (B) Comparison of experimental and predicted thermodynamic values. ΔX°2–1 represents the change in enthalpy or entropy on going from states [1] to [2]. Prediction 1 values come from the sum of the nearest-neighbor values for stacking of a 3′-U and 5′-A on a GC base pair, as indicated (35). Prediction 2 values are for formation of a UA base pair next to a GC base pair (35).
Two different approaches can be used to predict values for stacking of the helical junction, both using nearest-neighbor terms. In the first prediction, stacking of hydrogen-bonded GC and UA base pairs is estimated by the sum of the terms for stacking of a 3′-U and a 5′-A on a GC base pair (Fig. 4B) (35). In the second prediction, the nearest-neighbor value for formation of a UA base pair next to a GC base pair is considered. It should be noted that neither prediction by itself can be entirely correct, because individually each would predict a favorable ΔG°37.2–1, or that the free A3-bulged helix is straight. Nevertheless, each prediction contains features that are similar to the transition from state [1] to [2]. Prediction 1 should capture the major contributions to ΔH°2–1, because it considers only stacking of the bulge-interrupted base pairs, similar to the model (Fig. 4A). Prediction 2 may overestimate the ΔH°2–1 bonus, because it also counts hydrogen bonding. Indeed, the ΔH°2–1 for prediction 1 is similar to the ΔΔH° from the experiments (Fig. 4B). Conversely, prediction 2 appears to capture the major contributions to ΔS°2–1, because it considers the loss in conformational entropy on constraining two phosphodiester bonds, similar to the two to three in the model (Fig. 4A). Prediction 1 may underestimate the ΔS°2–1 penalty, because 5′-dangling end stacking is not favorable and this phosphodiester bond may not be constrained. Indeed, the ΔS°2–1 for prediction 2 is similar to the ΔΔS° from the experiments (Fig. 4B). In summary, the thermodynamic parameters for p20 binding to unbulged and bulged duplexes support a model in which the helical junction stacks on binding of p20. The favorable ΔΔH° value is consistent with estimates of base stacking, and the large unfavorable ΔΔS° value is consistent with expectations for conformational entropy loss on straightening of the bent duplex.
Conclusions
Interactions between nucleic acids and proteins can result in conformational changes of the nucleic acid, the protein, or both. Such conformational changes can be important for regulating gene expression and assembling nucleic acid-protein complexes. For example, catabolite gene activator protein is known to bend the lac operon DNA by ≈90° (34). Arginine and transactivator protein- (Tat) derived peptides straighten the bend created by the 3-nt bulge in the transactivation response element RNA from HIV (38, 39), and S15 protein mediates a conformational rearrangement of the central domain RNA necessary for ribosome assembly (40). Considerably less is known about conformational changes for nonsequence-specific RNA-protein complexes.
The dsRBM is a common motif that occurs in proteins that bind nonsequence specifically to long stretches of perfect dsRNA (3–5). Interestingly, many dsRBM proteins are also known to interact with RNAs containing non-Watson–Crick features. For example, the dsRBD from PKR binds to RNAs containing bulges, internal loops, and multibranched loops (13–17, 19). A-rich bulges cause sharp bends in the helix axis of dsRNA and are present in several known PKR-binding RNAs (22, 23, 25). Electrophoretic and thermodynamic studies presented here indicate that the dsRBD does not bend straight dsRNA, but that it does straighten bent RNA. This result is in accord with the structures of two dsRBM-RNA complexes in which the dsRNA is not appreciably bent (8, 10). These findings suggest a general model in which bulged RNA can bind to the dsRBD and undergo straightening to a dsRNA-like state, as long as the bulge can be accommodated in the final structure without steric hindrance and the energetic penalty for straightening the RNA is not severe. Examination of the dsRBM-dsRNA structures reveals numerous sites in which a bulge might be tolerated (8, 10). It is expected that other non-Watson–Crick motifs will be tolerated in a dsRBM-binding site, as long as they obey these rules. Interactions with dsRBM proteins may induce such RNAs to undergo conformational changes to a more linear arrangement. Straightening may allow bulge-containing RNAs to mimic the conformation of perfect dsRNA and to serve as regulators of PKR. Because many viral RNAs contain bulges (12–18), straightening would serve to greatly increase the number of RNAs that regulate PKR. These conformational changes could also be important determinants of other RNA-folding and RNA-protein assembly processes.
Supplementary Material
Acknowledgments
We thank David Proctor for help with structure analysis and members of the Bevilacqua group for critical reading of the manuscript. This work was supported by a grant from the National Institutes of Health (GM58709).
Abbreviations
- dsRNA
double-stranded RNA
- dsRBD
dsRNA-binding domain
- dsRBM
dsRNA-binding motif
- PKR
dsRNA-activated protein kinase
- DRD
DNA-RNA-DNA
- DDR
DNA-DNA-RNA
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.011355798.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.011355798
References
- 1.Tan S L, Katze M G. J Interferon Cytokine Res. 1999;19:543–554. doi: 10.1089/107999099313677. [DOI] [PubMed] [Google Scholar]
- 2.Williams B R. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 3.St Johnston D, Brown N H, Gall J G, Jantsch M. Proc Natl Acad Sci USA. 1992;89:10979–10983. doi: 10.1073/pnas.89.22.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fierro-Monti I, Mathews M B. Trends Biochem Sci. 2000;25:241–246. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- 5.Kharrat A, Macias M J, Gibson T J, Nilges M, Pastore A. EMBO J. 1995;14:3572–3584. doi: 10.1002/j.1460-2075.1995.tb07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bass B L. Cell. 2000;101:235–238. doi: 10.1016/s0092-8674(02)71133-1. [DOI] [PubMed] [Google Scholar]
- 7.Bycroft M, Grünert S, Murzin A G, Proctor M, St Johnston D. EMBO J. 1995;14:3563–3571. doi: 10.1002/j.1460-2075.1995.tb07362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramos A, Grunert S, Adams J, Micklem D R, Proctor M R, Freund S, Bycroft M, St Johnston D, Varani G. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nanduri S, Carpick B W, Yang Y, Williams B R, Qin J. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryter J M, Schultz S C. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manche L, Green S R, Schmedt C, Mathews M B. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bevilacqua P C, Cech T R. Biochemistry. 1996;35:9983–9994. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- 13.Robertson H D, Manche L, Mathews M B. J Virol. 1996;70:5611–5617. doi: 10.1128/jvi.70.8.5611-5617.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y, Mathews M B. RNA. 1996;2:937–951. [PMC free article] [PubMed] [Google Scholar]
- 15.Clemens M J, Laing K G, Jeffrey I W, Schofield A, Sharp T V, Elia A, Matys V, James M C, Tilleray V J. Biochimie. 1994;76:770–778. doi: 10.1016/0300-9084(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 16.Gunnery S, Rice A P, Robertson H D, Mathews M B. Proc Natl Acad Sci USA. 1990;87:8687–8691. doi: 10.1073/pnas.87.22.8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy S, Katze M G, Parkin N T, Edery I, Hovanessian A G, Sonenberg N. Science. 1990;247:1216–1219. doi: 10.1126/science.2180064. [DOI] [PubMed] [Google Scholar]
- 18.Glickman J N, Howe J G, Steitz J A. J Virol. 1988;62:902–911. doi: 10.1128/jvi.62.3.902-911.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bevilacqua P C, George C X, Samuel C E, Cech T R. Biochemistry. 1998;37:6303–6016. doi: 10.1021/bi980113j. [DOI] [PubMed] [Google Scholar]
- 20.Turner D H. Curr Opin Struct Biol. 1992;2:334–337. [Google Scholar]
- 21.Dingwall C, Ernberg I, Gait M J, Green S M, Heaphy S, Karn J, Lowe A D, Singh M, Skinner M A. EMBO J. 1990;9:4145–4153. doi: 10.1002/j.1460-2075.1990.tb07637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharyya A, Murchie A I, Lilley D M. Nature (London) 1990;343:484–487. doi: 10.1038/343484a0. [DOI] [PubMed] [Google Scholar]
- 23.Tang R S, Draper D E. Biochemistry. 1990;29:5232–5237. doi: 10.1021/bi00474a003. [DOI] [PubMed] [Google Scholar]
- 24.Riordan F A, Bhattacharyya A, McAteer S, Lilley D M. J Mol Biol. 1992;226:305–310. doi: 10.1016/0022-2836(92)90947-i. [DOI] [PubMed] [Google Scholar]
- 25.Zacharias M, Hagerman P J. J Mol Biol. 1995;247:486–500. doi: 10.1006/jmbi.1995.0155. [DOI] [PubMed] [Google Scholar]
- 26.Zacharias M, Hagerman P J. J Mol Biol. 1996;257:276–289. doi: 10.1006/jmbi.1996.0162. [DOI] [PubMed] [Google Scholar]
- 27.Tang R S, Draper D E. Nucleic Acids Res. 1994;22:835–841. doi: 10.1093/nar/22.5.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milligan J F, Uhlenbeck O C. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 29.Moore M J, Sharp P A. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 30.Bevilacqua J M, Bevilacqua P C. Biochemistry. 1998;37:15877–15884. doi: 10.1021/bi981732v. [DOI] [PubMed] [Google Scholar]
- 31.Bevington P R. Data Reduction and Error Analysis for the Physical Sciences. New York: McGraw–Hill; 1969. [Google Scholar]
- 32.Wu H M, Crothers D M. Nature (London) 1984;308:509–513. doi: 10.1038/308509a0. [DOI] [PubMed] [Google Scholar]
- 33.Crothers D M, Gartenberg M R, Shrader T E. Methods Enzymol. 1991;208:118–146. doi: 10.1016/0076-6879(91)08011-6. [DOI] [PubMed] [Google Scholar]
- 34.Schultz S C, Shields G C, Steitz T A. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 35.Serra M J, Turner D H. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 36.Weeks K M, Crothers D M. Science. 1993;261:1574–1577. doi: 10.1126/science.7690496. [DOI] [PubMed] [Google Scholar]
- 37.Dornberger U, Hillisch A, Gollmick F A, Fritzsche H, Diekmann S. Biochemistry. 1999;38:12860–12868. doi: 10.1021/bi9906874. [DOI] [PubMed] [Google Scholar]
- 38.Zacharias M, Hagerman P J. Proc Natl Acad Sci USA. 1995;92:6052–6066. doi: 10.1073/pnas.92.13.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puglisi J D, Chen L, Blanchard S, Frankel A D. Science. 1995;270:1200–1203. doi: 10.1126/science.270.5239.1200. [DOI] [PubMed] [Google Scholar]
- 40.Agalarov S C, Sridhar Prasad G, Funke P M, Stout C D, Williamson J R. Science. 2000;288:107–113. doi: 10.1126/science.288.5463.107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.