


Abstract
Universal bases hybridize with all other natural DNA or RNA bases, and have applications in PCR and sequencing. We have analysed by nuclear magnetic resonance spectroscopy the structure and dynamics of three DNA oligonucleotides containing the universal base analogues 5-nitroindole and 5-nitroindole-3-carboxamide. In all systems studied, both the 5-nitroindole nucleotide and the opposing nucleotide adopt a standard anti conformation and are fully stacked within the DNA duplex. The 5-nitroindole bases do not base pair with the nucleotide opposite them, but intercalate between this base and an adjacent Watson–Crick pair. In spite of their smooth accommodation within the DNA double-helix, the 5-nitroindole-containing duplexes exist as a dynamic mixture of two different stacking configurations exchanging fast on the chemical shift timescale. These configurations depend on the relative intercalating positions of the universal base and the opposing base, and their exchange implies nucleotide opening motions on the millisecond time range. The structure of these nitroindole-containing duplexes explains the mechanism by which these artificial moieties behave as universal bases.
INTRODUCTION
Universal base analogues have the ability to hybridize with all other natural DNA or RNA bases without discrimination. In contrast to their natural counterparts, they generally are hydrophobic aromatic moieties lacking hydrogen-bonding sites. Although there are many other bases described in the literature, the two universal analogues that have received most attention are 3-nitropyrrole (1) and 5-nitroindole (2) (Figure 1A). Based on their enhanced hybridization ability, these bases have been incorporated into DNA to prepare primers for PCR and sequencing because they increase the effective size of the primer without augmenting base-pairing multiplicity (3,4). In PCR, this translates into higher reaction temperatures and better performance. Universal bases have also been used as part of probes to target DNA and RNA sequences (5–7).
Figure 1.

(A) Chemical structure of nitropyrrole and nitroindole base analogues. Note that, by analogy, the atom numbering scheme in NI and NIC is similar to that used for DNA purine bases. (B) Sequence and numbering scheme of the three nitroindole-containing self-complementary DNA oligonucleotides analysed in this study.
As most universal bases lack hydrogen-bonding groups, it has been proposed that their effects derive from their ability to stack within double-helical DNA. In this regard, the structure of a DNA decamer containing 3-nitropyrrole opposite adenine has been determined by NMR, revealing that the nitropyrrole ring is stacked within the duplex (8). However, the amount of stacking with the neighbouring bases was less than expected, and the nitro group was found to protrude into the major groove rather than stack within the duplex. Relative to 3-nitropyrrole, incorporation of 5-nitroindole analogues causes a smaller decrease in the melting temperature of DNA duplexes (2). This is probably the result of better stacking interactions within double-helical DNA due to the larger aromatic surface area of nitroindoles relative to nitropyrroles, and provides an immediate advantage for applications based on hybridization.
Although they have been used for applications where the only requirement is hybridization, the analogues developed so far have serious limitations as substrates for natural enzymes. When acting as templates for polymerases, they direct incorporation of standard nucleotide triphosphates opposite them inefficiently, and preferentially incorporate dATP (9). The rate of incorporation of 5-nitroindole and 3-nitropyrrole 5′-triphosphates by wild-type polymerases is also very slow relative to natural triphosphates. In either case, the artificial bases behave as chain terminators, probably because of enhanced interactions with the enzyme. The efficient processing of artificial bases by polymerases is of importance since it would significantly increase the range of their applications by allowing enzymatic synthesis of modified oligonucleotides containing artificial bases, or the preparation of libraries of oligonucleotides from a single universal base-containing oligonucleotide.
Recently, the effect of introducing a hydrogen bonding carboxamide group on the hybridization ability of the universal base analogue 5-nitroindole (Figure 1A) was examined (10). Combined with an alternative strategy currently being explored and based on the development of engineered polymerases, the presence of this group may enhance the ability of these bases to act as enzyme substrates, providing a way to overcome the above problems. In this regard, we have shown that specifically evolved polymerases obtained by compartmentalized self-replication are capable of processing 5-nitroindole analogues more efficiently than wild-type enzymes ((11) and unpublished data).
Despite the potential advantages of these analogues, the structure of nitroindole-containing DNA has not been described so far. In this article, we describe the conformation and dynamics of three modified DNA oligonucleotides containing 5-nitroindole (NI) or 5-nitroindole-3-carboxamide (NIC) (Figure 1B). In addition to explaining the universal hybridization ability of 5-nitroindoles, the solution structure of these modified DNA duplexes should also help to elucidate the mechanisms by which these base analogues are processed by engineered polymerases.
MATERIALS AND METHODS
DNA synthesis
NI phosphoramidites were purchased from Glen Research, and NIC phosphoramidites were prepared as described (10). The DNA oligonucleotides d(GTAG NI CGCGACTAC) (ini), d(GTAG NIC CGCGACTAC) (inic), d(GTAC NIC ACGCGTTGTAC) (trnic), d(GTAGTCGCGACTAC)2 (ini/inic unmodified control) and d(GTACAACGCGTTGTAC) (trnic unmodified control) were synthesized by solid-phase methods (10). The NIC-containing oligonucleotides were deprotected in concentrated ammonia at 55°C for at least 48 h. Deprotection was continued until all traces of the nitroindole methyl ester was converted to the amide as monitored by MALDI-TOF mass spectrometry and NMR spectroscopy. All samples were purified by denaturing polyacrylamide gel electrophoresis followed by ethanol precipitation, dialysis and desalting. To eliminate a minor trnic fraction containing nitroindole-3-carboxylate, this sample was further purified by HPLC on a Dionex DNAPac PA-100 column, using a gradient of 0.1–1.2 M NaCl in 0.025 M NaOH (pH 12.4) over 40 min. After purification, all samples were microdialysed in aqueous solutions containing 100 mM NaCl, 20 mM sodium phosphate (pH 6.9) and 0.2 mM EDTA. The final DNA concentration in the NMR samples was ∼1 mM.
UV melting experiments
DNA thermal denaturation was monitored by measuring the absorbance of UV light at 260 nm in a quartz cuvette with a standard 1-cm path length. Absorbance was measured over a temperature range of 20–90°C using a Perkin Elmer Lambda 40 spectrophotometer fitted with a Peltier cell. Temperature was increased at 0.5°C/min, and then decreased back to the initial value at the same rate. Prior to the experiment, the samples were heated at 70°C for 5 min, then allowed to cool slowly to room temperature. Samples of 0.5 ODU/ml each were used, diluted in H2O containing 100 mM NaCl, 20 mM sodium phosphate (pH 6.9) and 0.2 mM EDTA.
NMR spectroscopy
NMR spectra were acquired on Bruker 500 and 600 MHz spectrometers, processed with Nmrpipe (12), and analysed using Sparky 3.110 (13). For all systems, 2D NMR spectra recorded in D2O included 1H–31P HETCOR and uninterrupted series of dqf-COSY, TOCSY (60 ms), ROESY and several NOESY experiments at different mixing times (typically 80, 120 and 250 ms). Most of the experiments were recorded at 27 and 38°C, but we also carried out measurements at 6 and 16°C, depending on the dynamic characteristics of the system under study. NOESY spectra were also obtained in H2O for all systems at low temperature (typically 14°C), using Watergate solvent suppression and a mixing time of 150 ms. We used a relaxation delay of 2 s for all experiments, and typically collected 2048 points in t2, and 600, 200 and 800 points in the t1 dimension of D2O, HETCOR and H2O experiments, respectively.
Constraints for trnic structure determination
Distance constraints between non-exchangeable protons were estimated from NOESY build-ups at 80, 120 and 250 ms mixing times. Cross-peaks corresponding to covalently constrained intra-base and intra-sugar distances were used as a reference to calibrate the constraints. Lower limit distance constraints were imposed on several proton pairs, based on the absence of NOE interactions. With the exception of hydrogen bonding restraints, all distance restraints had a minimum width of 1 Å. Hydrogen bonding base-pairing restraints were only introduced after observing the expected pattern of NOE interactions and chemical shifts in the H2O-NOESY experiments. No hydrogen bonding restraints were therefore imposed on the NIC, A6, T11 and T12 bases, since the exchangeable protons of these residues were either not detectable or too broad to extract any conclusion about their interactions. Constraints on sugar conformation were deduced from analysis of the dqf-COSY and TOCSY spectra. Sugar–phosphate backbone dihedral angle constraints were introduced based on the observation of cross-peak patterns in the 1H–31P HETCOR and 1H–1H NOESY spectra (14).
trnic structure generation
The NI and NIC bases were parameterized into the ff99 force field of Amber 8.0 using gaff atom types (15). The molecular electrostatic potential around N9-methyl-NIC and N9-methyl-NI (Figure 1A) was calculated at the 6-31G* ab initio level with Gamess 2006 (16), and used to generate resp atomic charges for the NI and NIC bases with R.e.d. II (17). The distance and dihedral constraints specified above were then used to determine the 3D structure of one symmetrical half of the self-complementary trnic duplex (Figure 1B). One hundred structures were iteratively refined by restrained molecular dynamics within the Sander module of Amber 8.0. We set up a protocol in which each structure undergoes two simulated annealing stages of 25 ps. In the first stage, the structure is heated to 1500 K, slowly cooled to 1 K and minimized. During this stage, no force field torsional terms or dihedral constraints are applied, and distance restraints and non-bonded terms are first reduced 100 times and then gradually allowed to return to their initial values, first the distance restraints followed by the non-bonded terms. In the second (refinement) stage, the structure is heated to 1000 K, slowly cooled to 1 K and minimized. Dihedral restraints and torsion terms are imposed at this stage together with non-bonded terms and distance restraints. No cut-off is used for the non-bonded interactions, and the solvent is mimicked with a distance-dependent dielectric constant. For each iteration, the 100 initial structures are disordered, high temperature snapshots obtained from the first stage of the previous iteration. This procedure ensures that any memory that the structures may keep from their initial conformation is erased; this is an important consideration when determining the structure of non-standard nucleic acid systems. After several iterations, a final set of 28 converged structures was selected on the basis of restraint and total energy values, and used to generate the refinement statistics (Table 1).
Table 1.
Experimental constraints and structural analyses for the trnic structures
| Experimental constraintsa | |
| NOE distance constraints | 440 |
| Intra-residueb | 144 (9) |
| Inter-residue | 296 (19) |
| Hydrogen bonding distance constraints | 16 |
| Dihedral constraintsc | 167 |
| Structure analysisd | |
| NOE violations >0.25 Å | 0 |
| Largest NOE violation (Å) | 0.22 ± 0.04 |
| Dihedral violations >5° | 0 |
| Largest dihedral violation | 2.70 ± 0.57 |
| Distance constraint violation energye (kcal mol−1) | |
| All structures | 10.24 ± 1.18 |
| 5′S structuresf | 10.46 ± 0.67 |
| 3′S structuresf | 9.71 ± 1.82 |
| Dihedral constraint violation energyg (kcal mol−1) | |
| All structures | 1.65 ± 1.96 |
| 5′S structures | 1.53 ± 2.04 |
| 3′S structures | 1.97 ± 1.69 |
| Total ff99 force field energy | |
| All structures | −597.4 ± 9.6 |
| 5′S structures | −599.9 ± 6.9 |
| 3′S structures | −591.4 ± 12.8 |
| Rms deviation from ideal bond lengths (Å × 102) | 0.90 ± 0.01 |
| Rms deviation from ideal bond angles (deg) | 2.41 ± 0.06 |
| Pair-wise rms deviation (Å) | |
| All structures | 0.70 ± 0.34 |
| 5′S structures | 0.48 ± 0.21 |
| 3′S structures | 0.83 ± 0.36 |
| Rms deviation from average structure (Å) | |
| All structures | 0.55 ± 0.22 |
| 5′S structures | 0.41 ± 0.11 |
| 3′S structures | 0.59 ± 0.24 |
aThe average number of NOE distance constraints per residue is also given (in parenthesis). bConstant intra-residue interproton distances were not used or counted as constraints for structure refinement. cIncludes 48 chirality restraints and 16 hydrogen bonding broad (±30°) planarity restraints, introduced to increase convergence. dAverage and standard deviation values obtained from 28 converged structures. eForce constant K = 30 kcal mol−1 Å−2. fThere are 20 5′S structures and 8 3′S structures. gForce constant K = 90 kcal mol−1 rad−2.
RESULTS
ini, inic and trnic NMR spectra analysis
NOESY, COSY and 1H–31P correlation data in D2O and H2O indicate that all three ini, inic and trnic oligonucleotides form anti-parallel duplexes adopting a B conformation in solution. With the exception of the terminal residues (for which there are some indications of partial C3′-endo sugar conformation), anti glycosidic angles, standard backbone angles and predominantly C2′-endo sugars are observed for all nucleotides, including NI and NIC, in all of the systems. Assignments were carried out using 1H–1H and 1H–31P correlation spectra and the sequential and inter-strand NOE connectivities observed in all systems.
NI opposite adenine: the ini duplex
Experiments at 27°C showed extensive broadening of the NI resonances together with the resonances of the neighbouring bases (G4, C6, G9, A10 and C11). This selective broadening increased at lower temperatures (6 and 16°C) but diminished significantly at 40°C (see Figure 2A and Supplementary Data), allowing for spectral analysis and assignments at this temperature. The observed broadening indicates internal mobility of NI and/or its neighbouring nucleotides on the chemical shift (millisecond) timescale: although at 40°C the exchange rate is fast enough for the signals to sharpen, the observed resonances and NOEs are likely to correspond to a fast-exchange average of two or more conformations.
Figure 2.

H6/H8–H1′ region of the NOESY spectra of ini at 40°C and 200 ms mixing time (A), inic at 38°C and 250 ms (B) and trnic at 38°C and 250 ms (C). Intra-residue pyrimidine H6-H1′ and purine/nitroindole H8–H1′ cross-peaks are labelled with residue names and numbers (nitroindole nucleotides are identified as N). Intra-residue pyrimidine H5-H6 cross-peaks are labelled with residue numbers. Classical sequential NOE connectivities are indicated with horizontal arrows. For clarity, only A3-T12 assignments and sequential connectivities are shown in (A) and (B). In (A), cross-peaks (a)–(i) are assigned as follows: (a) NI5 H2-H1′, H3-H1′ and H8-H1′ (overlapped); (b) NI5 H7-H8; (c) NI5 H6-H7; (d) NI5 H8-C6 H5; (e) NI5 H2-C6 H1′ and NI5 H3-C6 H1′ (overlapped); (f) A3 H2-G4 H1′; (g) G7 H8-C8 H5; (h) A10 H8-C11 H5; (i) G4 H8-N5 H7. In (B), cross-peaks (a)–(i) are assigned as follows: (a) NIC5 H3-H1′; (b) NIC5 H2-H1′; (c) NIC5 H8–C6 H5; (d) A10 H2-T11 H1′; (e) A3 H2-A13 H1′; (f) A3 H2-G4 H1′ and A3 H2-T12 H1′ (overlapped); (g) G7 H8-C8 H5; (h) A10 H8-C11 H5; (i) NIC5 H2-C11 H1′. Likewise, in (C), for clarity only A3-T14 assignments and connectivities are indicated. Cross-peaks (a)–(k) are assigned as follows: (a) NIC5 H3-H1′; (b) NIC5 H2-H1′; (c) NIC5 H6–C4 H5; (d) A6 H2-NIC5 H1′; (e) A6 H2-T12 H1′; (f) A6 H2-T11 H1′; (g) A3 H8-C4 H5; (h) A6 H8-C7 H5; (i) A3 H2-A15 H1′; (j) A3 H2-C4 H1′ and A3 H2-T14 H1′ (overlapped); (k) G8 H8-C9 H5.
All NI aromatic resonances are upfield-shifted with respect to the resonances of isolated NI (data not shown), suggesting stacking of the NI ring between neighbouring bases. This is confirmed by aromatic–aromatic and sugar–aromatic sequential NOE interactions between G4 H8, H1′, H2′, H2″ and H3′ and NI H7 and H8, and between NI H1′, H2′, H2″ and H3′ and C6 H6 and H5 (Figure 2A). NOEs between C6 H1′, H5 and H6 and NI H2, H3 and H8 (Figure 2A, peaks d and e) also indicate that the NI base is stacked on the C6:G9 base pair most of the time. In fact, the intensity of the intra-nucleotide NI H7-H1′ and sequential G4 H1′, H2′ and H2″ to NI H7 NOEs (not shown) indicate that the NI nucleotide is anti and is stacked between its neighbouring nucleotides in a manner similar to that observed for standard purine DNA bases. This means that the NI H6, H7 and H8 protons are in the major groove, H2 and H3 are in the minor groove, and the nitro group, located in the Watson–Crick edge of the ring, is oriented towards A10 in the opposite strand (see Figure 1A and B). This is a remarkable observation, because the ‘complementary’ residue A10 is also anti (Figure 2A). Therefore, a steric clash would be expected if both bases occupied the same plane within the DNA duplex. However, broadening of A10 H2, G4 H1 and G9 H1 and overlapping of NI H2, H3 and H8, together with the dynamic nature and the broadening at lower temperatures of the shifts and NOEs emanating from NI and its neighbouring nucleotides, did not allow us to carry out detailed analyses on the structure of this system.
NIC opposite adenine: the inic duplex
Relative to ini, resonance broadening is significantly diminished in inic, but is still apparent at lower temperatures particularly for the H5 and amino resonances of the NIC-neighbouring residues C6 and C11 (Figure 2B), the amino protons of the NIC carboxamide group, and the imino protons of G4 and G9. This indicates that millisecond dynamics is also taking place around the region occupied by NIC.
The NIC aromatic spin system was assigned from COSY and TOCSY spectra at 38°C and found to be well resolved. The relative intensities of the NIC intra-residue H8-H1′ and H3-H1′ cross-peaks (Figure 2B, peaks N5 and a) clearly establish that the NIC nucleotide is anti. NIC is connected to the preceding residue (G4) and to the following one (C6) by many standard sugar–aromatic sequential interactions, including NOEs between G4 H1′, H2′, H2″ and H3′ and NIC H8, and between NIC H1′, H2′, H2″ and H3′ and C6 H6 (see Figure 2B). Aromatic–aromatic interactions are also observed between NIC H8 and C6 H5 (Figure 2B, peak c), C6 H6 and G4 H8. These intra-residue and inter-residue interactions clearly demonstrate that (i) NIC is stacked between G4 and C6; (ii) NIC H8 and H6 (and hence the carboxamide group) are located in the major groove, and NIC H2 and H3 are located in the minor groove. This conformation is similar to that adopted by anti purine DNA bases. As before, this implies that the nitro group is pointing towards A10 in the opposite strand (Figure 1B).
At the other side of the helix, A10 is also anti and shows sequential sugar–aromatic NOEs with G9 and C11 (Figure 2B), indicative of A10 stacking within the DNA duplex. Aromatic–aromatic interactions are likewise seen between G9 H8 and A10 H8, and between A10 H8 and C11 H5 (Figure 2B, peak h) and H6. A10 H2 gives rise to a weak NOE with C11 H1′ (Figure 2B, peak d), but the expected NOE with C6 H1′ is not observed. There are also NOE interactions between NIC H3 and A10 H2, and between NIC H2 and C11 H1′ (Figure 2B, peak i), and weak NOEs between NIC H2 and H6 and G9 H1. Taking these observations together, the observed NOEs indicate that the NIC-‘complementary’ residue A10 is stacked between G9 and C11, and that both NIC and A10 are unpaired and stacked on each other.
NIC opposite thymine: the trnic duplex
As in the previous two duplexes, the NIC nucleotide is anti (Figure 2C, peaks N5 and a), and is connected to the preceding residue (C4) and to the following one (A6) by many standard sugar–aromatic sequential interactions, including NOEs between C4 H1′, H2′, H2″ and H3′ and NIC H8, and between NIC H1′, H2′, H2″ and H3′ and A6 H8 (see Figure 2C). A weak NOE between A6 H2 and NIC H1′ is also observed (Figure 2C, peak d). Aromatic–aromatic interactions are detected between NIC H8 and C4 H6 and A6 H8, between NIC H6 and C4 H5 (Figure 2C, peak c) and A6 H2, between the NIC carboxamide protons and the C4 H5, H6 and amino resonances, and between NIC H3 and A6 H2. These intra-residue and inter-residue interactions clearly demonstrate that: (i) NIC is stacked between C4 and A6; (ii) NIC H6, H8 and carboxamide group are in the major groove, and NIC H2 and H3 are in the minor groove. This conformation is similar to that observed in ini and inic, and implies that the nitro group of NIC is pointing towards T12 in the opposite strand (Figure 1B).
At the other side of the helix, T12 is anti and is connected by sequential sugar–aromatic and aromatic–aromatic NOEs with T11 and G13 (Figure 2C), including T11 H1′, H3′, H2′ and H2″ to T12 H6, T11 H6 and H1′ to T12 methyl, T11 H6 to T12 H6, T12 H3′ H2′ and H2″ to G13 H8, T12 H6 to G13 H8, and G13 H1 to T12 methyl. There are weak NOEs between the H1′ protons of T11 and T12 and A6 H2 (Figure 2C, peaks e and f), and a very weak NOE between G13 H1 and NIC H2 is also observed. Importantly, NIC H6 gives rise to NOE interactions with the T12 H6 and methyl protons and a weaker one with the T11 methyl protons. These interactions demonstrate that NIC and T12 are not base-paired, but stacked on each other.
Selective broadening also affects the aromatic resonances of NIC and its neighbouring residues at lower temperatures (Supplementary Data), and the imino protons of T11 and T12 are undetectable at all temperatures tested. These observations indicate that, as ini and inic, trnic is also undergoing significant millisecond dynamics in the vicinity of NIC. The carboxamide resonances of NIC are broadened by exchange, and give rise to NOEs of similar intensity with both NIC H6 and NIC H8 (Figure 1A), and to weak NOEs with both C4 H2′, H2″ and H3′ and the T12 methyl group. As these NOE interactions cannot be generated by a single conformation, they must be the result of a fast-exchange average of two possible carboxamide group orientations.
trnic solution structure
A superimposed set of 28 converged structures of one symmetrical half of the trnic self-complementary duplex are shown in Figure 3, and the structure refinement data are summarized in Table 1. In all of the converged structures, the double helix is in the B conformation, all sugars except C16 are C2′-endo, and anti glycosidic angles are observed for all nucleotides including NIC. The nitro group of NIC points towards the opposite strand, whereas its carboxamide group is located in the major groove, in a position equivalent to that of N7 of standard purine bases (Figure 4). The nitro group is not hydrogen-bonded in any of the structures, and is coplanar with the nitroindole aromatic system. This coplanar geometry was observed in the NI and NIC base structures optimized by ab initio methods (see the Material and methods section) and in 83% (±30°) of the nitro-benzenes available in the CSD (18). Nitro groups are relatively poor hydrogen bonding acceptors (19), but they are expected to polarize the indole aromatic system, enhancing in this way the electrostatic component of the stacking interactions with DNA bases (20). Because of the steric constraints imposed by the orientation of the nitro group, the NIC nucleotide is not coplanar with its ‘complementary’ base T12. Instead, both NIC and T12 are stacked on each other and on an adjacent base pair. In this regard, two conformational families are observed in the final set of converged structures (Figure 3). In the first family of structures (identified as 5′S), NIC stacks between A6:T11 and T12 and T12 stacks between NIC and C4:G13 or, in other words, NIC and T12 stack on each other using their 5′ faces, whereas in the second family of structures (designated 3′S), NIC and T12 stack on each other using their 3′ faces (see Figures 4 and 5). Geometrical conversion between each of the stacking configurations can be easily accomplished by modifying the inclination of the NIC and T12 bases, without major changes in the positions of the sugar and backbone atoms (Figure 3). Although the 5′S family is better represented in the final ensemble of structures, both stacking conformers are equally compatible with the trnic NOEs and constraints, and give rise to similar NOE violation energies and force field energies (Table 1). For both stacking families, the NH2 atoms of the carboxamide group of NIC are mostly (82% of the structures) oriented towards the opposite strand and within hydrogen-bonding distance (1.91 ± 0.03 Å) of T12 O4 in the major groove. This is accomplished by a slight (16°) deviation out of the nitroindole plane of the carboxamide group in the 5′S structures, or by a negative rolling of the T12 base in the 3′S structures (Figure 4A and B). Although 20° deviations out of plane are commonly observed in the aromatic carboxamides available in the CSD (18), the NIC-T12 hydrogen bond is exposed to the solvent, and its geometry is not ideal because NIC and T12 are in different planes. In the remaining structures, the NH2 atoms of the carboxamide group of NIC are oriented in the opposite direction, and are found to establish a hydrogen bond with one of the phosphate oxygens of the NIC nucleotide in 4 of the 28 structures (14%). In spite of the absence of hydrogen bonding restraints for A6 base pairing during structure refinement (see the Material and methods section), full A6:T11 pairing is observed in almost all (86%) of the structures (Figure 3). In addition, T12 is found to hydrogen bond to A6 in four 3′S structures (14%).
Figure 3.

View of the superimposed 28 converged structures of one symmetrical half of the trnic duplex. The 5′S and 3′S stacking families can be clearly distinguished in the central part of the helix.
Figure 4.

View of 3′S (A and C) and 5′S (B and D) trnic representative conformers. For clarity, only C4, NIC5, A6, T11, T12 and G13 are shown. The views are towards the major groove (A and B), and down the helix axis, showing the stacking interactions between the bases (C and D). The C4:G13 pair is coloured magenta, NIC5 is yellow, T12 is green and the A6:T11 pair is shown in blue.
Figure 5.

Scheme showing the two possible 3′S and 5′S stacking configurations of nitroindoles (coloured yellow and black) in a DNA duplex. Note that these two conformations are not equivalent.
Thermal stability
The ini and inic duplexes containing NI and NIC give rise to similar UV melting temperatures (Table 2), in agreement with previous findings (10). Relative to the unmodified controls, the destabilization caused by the presence of two universal bases in the ini and inic duplexes is greater relative to trnic (Table 2), which shows the melting temperature variation (6°C decrease per artificial nucleotide) typically observed in nitroindole-substituted duplexes (2). This is probably caused by the sequence difference between the two systems (Figure 1B).
Table 2.
UV melting temperatures (°C) of the DNA duplexes analysed in this studya,b. Experimental standard deviations are 0.5°C
| Duplex | Tm(°C) |
|---|---|
| ini | 41 |
| inic | 42 |
| trnic | 50 |
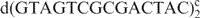 |
60 |
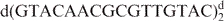 |
62 |
aNote that the sequence of ini and inic is different from that of trnic (Figure 1B). bUV melting temperatures, obtained at micromolar concentrations, are substantially lower than melting temperatures at millimolar (NMR) concentrations. cini- and inic-unmodified control. dtrnic unmodified control.
DISCUSSION
The most important conclusion of this work is the observation that nitroindole universal bases are fully stacked within the DNA duplexes that contain them. They adopt an anti conformation and give rise to very good overlap with the adjacent bases. Relative to 3-nitropyrrole, the stacking is more extensive, and the nitro group is stacked within the helix rather than protruding out into the major groove (Figure 4C and D). This explains why, relative to 3-nitropyrrole, substitution of a natural base with 5-nitroindole analogues causes a smaller decrease in the melting temperature of DNA duplexes (2), providing the nitroindole family of universal bases with an important advantage for applications based on hybridization.
In all duplexes studied, both NI and NIC nucleotides are anti and in this conformation the nitro group (located in the hydrogen bonding position 1 of purines and blocking the Watson–Crick edge of the base, Figure 1A) is directed towards the complementary strand without giving rise to any hydrogen bonding interactions. Related to this fact, a second important conclusion is that NI and NIC do not base pair with the base opposite them in the duplex, but intercalate between this base and an adjacent Watson–Crick base pair. At the other side of the helix, the opposing base is also anti. However, this nucleotide is not bulged out but is smoothly accommodated within the duplex with minimal distortions, keeping stacking interactions with the nitroindole base and the adjacent Watson–Crick base pair (Figures 3 and 4). This observation explains the mechanism by which nitroindoles behave as universal base analogues, since they do not base pair but stack on the opposing natural base. The DNA duplex accommodates the presence of the nitroindole nucleotide while maintaining a B conformation with full stacking and standard nucleotide and backbone dihedrals. This is not surprising, as an artificial base causing significant distortions in the duplex would not be expected to behave as a universal analogue.
Despite causing minimal structural distortions, the insertion of NI or NIC nucleotides in double helical DNA causes significant line broadening in all of the three duplexes studied here, indicative of dynamic processes taking place on the millisecond timescale. These dynamics are most apparent for the NI-containing duplex ini, for which extensive broadening is observed at temperatures below 40°C, but the phenomenon is present in all three systems (Supplementary Data). The broadening selectively affects the resonances of nitroindole and its neighbouring nucleotides, and is specifically associated to the insertion of these artificial nucleotides, since the NMR spectra of an unmodified d(GTAGTCGCGACTAC)2 control duplex containing thymine instead of nitroindole does not show any sign of these dynamics (Supplementary Data), and no broadening was detected in a d(CATGAGTAC).d(GTACXCATG) duplex where X is 3-nitropyrrole (8).
To help explain the origin of the observed dynamics, which is in apparent contradiction with the smooth accommodation of the nitroindole nucleotides within the DNA duplexes, it is important to realise that since they are unpaired, there are two possible stacking or intercalation positions for the nitroindole and the opposing natural base in the complementary strand. In the first conformation, which we will identify as 5′S (5′ stack), the nitroindole base and the opposing unpaired base stack on each other using their 5′ faces, and on the adjacent Watson–Crick base pairs using their 3′ faces. In the alternative 3′S conformation, the nitroindole and its unpaired counterpart stack on each other using their 3′ faces, and on the adjacent Watson–Crick base pairs using their 5′ faces (see Figures 4 and 5).
How do the above observations relate to the observed millisecond dynamics in nitroindole-containing duplexes? DNA base pairs open with millisecond frequencies, as indicated by the exchange of imino protons with solvent. Mismatched or unpaired bases have been shown to open more frequently, since they do not have a hydrogen-bonded complement (21). We propose that the observed millisecond dynamics of NI and NIC in their DNA duplexes are due to the opening of both the nitroindole and the natural base complement towards the groove, and to the resulting exchange between 5′S and 3′S conformations. In fact, the analysis of the ini and inic spectra indicates that these duplexes are a dynamic mixture of 5′S and 3′S stacking configurations exchanging fast on the chemical shift timescale: NOEs exclusive of only one of the two exchanging conformations are broader and weaker than the rest, and NOE interactions consistent with both conformations are observed in the same spectra. For example, the NIC H2-C11 H1′ NOE in inic (Figure 2B, peak i) indicates a 3′S conformation, yet the expected A10 H2-C6 H1′ interaction is not observed, and there are weak A10 H2-C11 H1′ (Figure 2B, peak d), NIC H2-G9 H1 and NIC H6-G9 H1 NOEs that indicate a 5′S conformation. trnic is probably a fast-exchange mixture of 5′S and 3′S conformations as well. This is indicated by the selective broadening of the resonances of NIC and its neighbouring residues at lower temperatures (Supplementary Data), and by the observation of both stacking configurations in the final set of trnic solution structures (Figure 3). In this regard, note that the energy difference between the 5′S and 3′S conformations is probably small. The stacking interactions are similar in the 5′S and 3′S trnic conformations (compare Figure 4C and D) and in ini and inic, the nitroindole base would stack between a G:C pair and A10 in both conformers (see Figures 1B and 5). Thus, the ini, inic and trnic resonances, sharpening at high temperatures when the exchange rate increases, and broadening at low temperatures when the exchange rate decreases and the coalescence point is approached, are likely to correspond to an average of 5′S and 3′S conformations, exchanging fast on the chemical shift (millisecond) timescale.
Because the nitroindoles intercalate rather than base pair with the opposing base, the stacking interactions of NI and NIC with the neighbouring bases are of major importance. These interactions may explain why adenine is preferentially incorporated as the ‘complementary’ base of nitroindoles by DNA polymerases. Adenine contributes more surface area for stacking interactions with the nitroindole base relative to thymine or cytosine, and as it contains two hydrogen bonding groups instead of three, it would be less penalized than guanine for the absence of a hydrogen bonding partner.
What is the role of the carboxamide group of NIC? The NMR results indicate that NIC is anti and in this conformation the carboxamide group is located in the major groove, far away from the hydrogen bonding edge of the base (Figures 1A and 4). The amide protons of the NIC carboxamide are broadened by exchange in the inic spectra, or give rise to NOEs indicating an average between two carboxamide group orientations in the trnic spectra. In agreement with these observations, a weak hydrogen bond is formed between the NIC carboxamide and T12 O4 in the opposite strand in a group of trnic solution structures, but a intra-strand hydrogen-bond with the NIC phosphate group is also established in a smaller number of structures. Modelling of the inic duplex indicates that a similar interstrand hydrogen bond cannot be formed when the base opposing NIC is a purine, but in this case more favourable stacking interactions must compensate for the absence of this hydrogen bond, because the melting temperatures of NIC:purine and NIC:pyrimidine duplexes are similar (10). The fact that, in contrast to NI, NIC generally shows more negative melting enthalpies when opposed to pyrimidines relative to purines (10) supports the inter-strand NIC-T12 interaction observed in trnic, but further work will be needed to clarify the role of the carboxamide group of NIC in relation to engineered polymerase recognition.
The coordinates of the final set of trnic structures have been deposited in the PDB (ID code 2O4Y).
Supplementary Material
ACKNOWLEDGEMENTS
J.G. carried out the first stages of this work at the MRC LMB, UK. He gratefully acknowledges the financial support of UK's MRC and Spain's CIPF, MEC (grant BFU2004-07274/BMC) and Generalitat Valenciana (ACOMP06/139). The authors thank Gabriele Varani for his contributions during the early stages of this project. Funding to pay the Open Access publication charge was provided by MEC.
Conflict of interest statement. None declared.
REFERENCES
- 1.Nichols R., Andrews P.C., Zhang P., Bergstrom D.E. Synthesis, structure and biochemical applications of a nucleic-acid spacer: 1-(2′-deoxy-β-D-ribofuranosyl)-3-nitropyrrole. Nature. 1994;369:492–493. [Google Scholar]
- 2.Loakes D., Brown D.M. 5-Nitroindole as an universal base analogue. Nucleic Acids Res. 1994;22:4039–4043. doi: 10.1093/nar/22.20.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loakes D., Brown D.M., Linde S., Hill F. 3-Nitropyrrole and 5-nitroindole as universal bases in primers for DNA sequencing and PCR. Nucleic Acids Res. 1995;23:2361–2366. doi: 10.1093/nar/23.13.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ball S., Reeve M.A., Robinson P.S., Hill F., Brown D.M., Loakes D. The use of tailed octamer primers for cycle sequencing. Nucleic Acids Res. 1998;26:5225–5227. doi: 10.1093/nar/26.22.5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng D., Raskin L. Quantification of Methanosaeta species in anaerobic bioreactors using genus- and species-specific hybridisation probes. Microb. Ecol. 2000;39:246–262. doi: 10.1007/s002480000003. [DOI] [PubMed] [Google Scholar]
- 6.Burgner D., D’Amato M., Kwiatowski D.P., Loakes D. Improved allelic differentiation using sequence-specific oligonucleotide hybridization incorporating an additional base-analogue mismatch. Nucleosides Nucleotides Nucleic Acids. 2004;23:755–765. doi: 10.1081/NCN-120039216. [DOI] [PubMed] [Google Scholar]
- 7.Ganova-Raeva L., Smith A.W., Fields H., Khudyakov Y. New calicivirus isolated from walrus. Virus Res. 2004;102:207–213. doi: 10.1016/j.virusres.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 8.Klewer D.A., Hoskins A., Zhang P., Davisson V.J., Bergstrom D.E., LiWang A.C. NMR structure of a DNA duplex containing nucleoside analogue 1-(2′-deoxy-b-D-ribofuranosyl)-3-nitropyrrole and the structure of the unmodified control. Nucleic Acids Res. 2000;28:4514–4522. doi: 10.1093/nar/28.22.4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith C.L., Simmonds A.C., Felix I.R., Hamilton A.L., Kumar S., Nampalli S., Loakes D., Hill F., Brown D.M. DNA polymerase incorporation of universal base triphosphates. Nucleosides Nucleotides. 1998;17:541–554. [Google Scholar]
- 10.Too K., Brown D.M., Holliger P., Loakes D. Effect of a hydrogen bonding carboxamide group on universal bases. Coll. Czech. Chem. Commun. 2006;71:899–911. [Google Scholar]
- 11.Ghadessy F.J., Ramsay N., Boudsocq F., Loakes D., Brown A., Iwai S., Vaisman A., Woodgate R., Holliger P. Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution. Nat. Biotechnol. 2004;22:755–759. doi: 10.1038/nbt974. [DOI] [PubMed] [Google Scholar]
- 12.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 13.Goddard TD, Kneller DG. San Francisco, CA, USA: University of California; 2001. Sparky 3. [Google Scholar]
- 14.Kim S.G., Lin L.J., Reid B.R. Determination of nucleic acid backbone conformation by 1H NMR. Biochemistry. 1992;31:3564–3574. doi: 10.1021/bi00129a003. [DOI] [PubMed] [Google Scholar]
- 15.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, et al. San Francisco, CA, USA: University of California; 2004. AMBER 8. [Google Scholar]
- 16.Schmidt M.W., Baldridge K.K., Boatz J.A., Elbert S.T., Gordon M.S., Jensen J.H., Koseki S., Matsunaga N., Nguyen K.A., et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993;14:1347–1363. [Google Scholar]
- 17.Pigache A, Cieplak P, Dupradeau F-Y. 227th ACS National Meeting, Anaheim, CA. 2004. Automatic and highly reproducible RESP and ESP charge derivation: application to the development of programs RED and X RED. [Google Scholar]
- 18.Allen F.H. The cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr. 2002;B58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 19.Robinson J.M.A., Philp D., Harris K.D.M., Kariuki B.M. Weak interactions in crystal engineering – understanding the recognition properties of the nitro group. New J. Chem. 2000;24:799–806. [Google Scholar]
- 20.Wheaton C.A., Dobrowolski S.L., Millen A.L., Wetmore S.D. Nitrosubstituted aromatic molecules as universal nucleobases: computational analysis of stacking interactions. Chem. Phys. Lett. 2006;428:157–166. [Google Scholar]
- 21.Bhattacharya P.K., Cha J., Barton J.K. H-1 NMR determination of base-pair lifetimes in oligonucleotides containing single base mismatches. Nucleic Acids Res. 2002;30:4740–4750. doi: 10.1093/nar/gkf601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.