


Abstract
Hexameric DnaB type replicative helicases are essential for DNA strand unwinding along with the direction of replication fork movement. These helicases in general contain an amino terminal domain and a carboxy terminal domain separated by a linker region. Due to the lack of crystal structure of a full-length DnaB like helicase, the domain structure and function of these types of helicases are not clear. We have reported recently that Helicobacter pylori DnaB helicase is a replicative helicase in vitro and it can bypass Escherichia coli DnaC activity in vivo. Using biochemical, biophysical and genetic complementation assays, here we show that though the N-terminal region of HpDnaB is required for conformational changes between C6 and C3 rotational symmetry, it is not essential for in vitro helicase activity and in vivo function of the protein. Instead, an extreme carboxy terminal region and an adjacent unique 34 amino acid insertion region were found to be essential for HpDnaB activity suggesting that these regions are important for proper folding and oligomerization of this protein. These results confer great potential in understanding the domain structures of DnaB type helicases and their related function.
INTRODUCTION
DNA helicases are important for various cellular processes like DNA replication, recombination and repair. Hexameric replicative DNA helicases are essential for chromosomal DNA replication. These enzymes unwind the double stranded DNA in the presence of ATP during replication fork movement (1). Among these helicases, the most extensively studied is Escherichia coli DnaB helicase. Although the crystal structures of some of the hexameric replicative helicases like RepA of plasmid RSF1010 (2) and gp4 of E. coli T7 bacteriophage (3) are available, a high-resolution crystal structure of full-length DnaB helicase is not available yet except for the N-terminal region of EcDnaB (4). The structural details of different functional domains of DnaB type helicases are based on biochemical characterization and low-resolution electron microscopic analysis of EcDnaB and a recent study on bacteriophage SPP1 G40P helicase (5,6).
EcDnaB can be divided into two major domains: the N-terminal domain and the C-terminal helicase domain separated by a linker region (7,8). There is very little homology at the amino acid level at the N-terminus among the different DnaB helicases (9). However, it looks like that the N-terminal is essential since a deletion mutant of EcDnaB and an equivalent mutant from B. strearothermophilus containing the C-terminal helicase domain and a part of the linker region shows in vitro ATPase activity but fails to demonstrate in vitro helicase activity (8,10,11). A similar study on bacteriophage SSP1 G40P helicase shows that the deletion of N-terminal region only does not affect in vitro ATPase and helicase activity but another mutant deleting the linker region along with the N-terminal region loses both ATPase and helicase activity (5). Based on these observations, it has been suggested that N-terminal domain including the linker region is essential for DnaB activity in vitro. However, none of these studies include in vivo analysis of DnaB functional domains.
Electron microscopic analysis of wild-type EcDnaB shows C6 or C3 conformational states as well as an intermediate state (12–14). It has been suggested that the N-terminal region is essential for the C6:C3 conformational changes that might be important for the helicase activity of EcDnaB (4,9). Electron microscopic analysis of G40P helicase revealed the presence of similar C3:C6 conformational changes for the wild-type protein whereas the N-terminal domain deletion mutant (including the linker region) was mostly found to be in the C6 conformation at pH 7.0 (6). These results suggest that the N-terminal domain is essential for C6:C3 conformational changes. However, it is not clear whether any such conformational change is important for in vitro and in vivo activities.
Although several deletion mutant studies have been performed for different DnaB helicases at the N-terminal region, such studies have been restricted to only EcDnaB for functional domain mapping at the C-terminal helicase domain. Deletion of 55 amino acid residues from the extreme C-terminal region of the helicase domain does not affect the in vitro ATPase and helicase activity of EcDnaB. Further deletion from the C-terminal region results in the loss of helicase activity retaining the ATPase activity probably due to the loss of leucine heptad repeat regions that might be responsible for oligomerization (15). However, there is no direct evidence that leucine heptad regions form coiled coil domain and are truly responsible for oligomerization.
We have recently characterized Helicobacter pylori DnaB (HpDnaB) helicase both in vitro and in vivo. HpDnaB does not require a DnaC homologue for its function (16). Using two different E. coli dnaC (EcdnaC) temperature-sensitive mutant strains, we showed the complementation of EcDnaC function by HpDnaB suggesting that HpDnaB can bypass EcDnaC activity in vivo (17). HpDnaB shows overall 32% identity and 53% homology with EcDnaB although this homology is more prominent at the C-terminal helicase domain except for the extreme C-terminal region that include a unique 34 amino acid residue insertion region, the function of which is still not known (17).
In order to investigate the functions of different domains of HpDnaB that might allow us to understand the overall structure function relationship of DnaB in general, here we show both in vitro and in vivo analysis of several N-terminal and C-terminal deletion mutants of HpDnaB. We find that an N-terminal deletion mutant (HpDnaBDelN2) equivalent to that of EcDnaB (that shows in vitro ATPase activity but no helicase activity; EcDelN156, Supplementary Figure 1) including the N-terminal region and a part of the linker region shows in vitro helicase activity and it can complement E. coli dnaBts mutant strain at the restrictive temperature suggesting that N-terminal region might not be essential for HpDnaB function. Furthermore, the deletion mutant lacking the N-terminal domain along with the subsequent linker region retains ATPase activity but no helicase activity suggesting that the linker region is important for the overall function of HpDnaB. Finally, we show that the extreme C-terminal region including the 34 amino acids insertion is essential for its structural integrity and function. These results have a great implication in understanding the role of different domains of HpDnaB that might be helpful to understand the domain organization of DnaB type helicases in general.
MATERIALS AND METHODS
Bacterial strains and plasmid construction
Helicobacter pylori (strain 26695) genomic DNA and specific primers (as shown in Table 1) were used for the polymerase chain reaction (PCR) in order to amplify the coding regions of HpdnaBwt (1.5 kB). The amplified products were cloned at the BamHI site of pET28a expression vector as described earlier (16). Different mutant variants (except HpDnaBMut and HpDnaBDel34) as shown in Figure 1A were first amplified by using PCR with the help of respective primers (as shown in Table 1) followed by cloning them at the BamHI site of pET28a expression vector as described earlier (16). For PCR amplification of respective genes for HpDnaBmut and HpDnaBDel34, overlapping primers were used as shown in Table 1. HpDnaBMut (the ATP binding site mutant) was generated by introducing a point mutation (R204 to C) at the conserved WALKER A domain (ARPSMGKT) of HpDnaB essentially following the protocol as described elsewhere (16). All the recombinant clones were sequenced using suitable primers to avoid any error in the reading frames following PCR reactions.
Table 1.
Primers list
| No | Primer name | Sequence |
|---|---|---|
| 1 | HpDnaBWt(Fw) | 5′-GCGGATCCATGGATCATTTAAAGCATTTGC-3′ |
| 2 | HpDnaBWt(Rv) | 5′-GCGGATCCTCAAGTTGTAACTATATCATAA-3′ |
| 3 | HpDnaBmut(Fw) | 5′-GTCATTATAGGGGCATGCCCGTCTATGGGTAAA-3′ |
| 4 | HpDnaBmut(Rv) | 5′-TTTACCCATAGACGGGCATGCCCCTATAATGAC-3′ |
| 5 | HpDnaBDelN1(Fw) | 5′-CGGGATCCATGCGCCAAAAAATGCCTAAAGAC-3′ |
| 6 | HpDnaBDelN1(Rv) | same as HpDnaBWt(Rv) |
| 7 | HpDnaBDelN2(Fw) | 5′-CGGGATCCATGGTGGAGCGAGAAGTCTATG-3′ |
| 8 | HpDnaBDelN2(Rv) | same as HpDnaBWt(Rv) |
| 9 | HpDnaBDelN3(Fw) | 5′-CGGGATCCGAAGTTACTGGCATACCG-3′ |
| 10 | HpDnaBDelN3(Rv) | same as HpDnaBWt(Rv) |
| 11 | HpDnaBDelN4 (Fw) | 5′-CGGGATCCATGGGGAGTTTAGTCATTATAGG-3′ |
| 12 | HpDnaBDelN4 (Rv) | same as HpDnaBWt(Rv) |
| 13 | HpDnaBDelC1(Fw) | same as HpDnaBWt(Fw) |
| 14 | HpDnaBDelC1 (Rv) | 5′-CGGGATCCTCAAGCGTTAAAGCGCGTATAAAC-3′ |
| 15 | HpDNABDel34(Fw) | 5′-CAAATGAGGGCTGAAAGCATTGAAGAGGCT-3′ |
| 16 | HpDnaBDel34(Rv) | 5′-AGCCTCTTCAATGCTTTCAGCCCTCATTTG-3′ |
Figure 1.

Expression and purification of different domains of HpDnaB. (A) Schematic diagrams of full-length and different deletion mutants of HpDnaB. The top panel shows the N-terminal domain and the helicase domain separated by the linker region with amino acid residues as indicated on top. The unique 34 amino acid residues insertion is shown by a black box. The N-terminal deletions (N1–N4) and the C-terminal deletion (C1) are shown. Asterisk indicates the single amino acid residue change (R204 to C) in the WALKER A domain of wild-type HpDnaB. (B) SDS-PAGE analysis of purified proteins. His6-tagged fusion proteins as indicated in the panel ‘A’ were purified as described in the materials and methods followed by SDS-PAGE analysis and Coomassie staining of the gel.
For complementation assay, plasmid constructs were made by subcloning the respective genes (including the His6-tag) from the pET28a recombinant clones into pBR322 downstream of Bla-P2 promoter (18). The details of the cloning strategies are described elsewhere (16).
Protein purification
His6-tagged proteins were purified as described earlier (16) unless specified otherwise. To get the better yield of soluble proteins (HpDelN4, HpDelC1 and HpDel34), the bacterial culture was induced for 5–6 h at 22°C in the presence of 0.2 mM IPTG followed by usual protein extraction and purification procedures as described earlier (16).
ATP hydrolysis assay: pyruvate kinase/lactate dehydrogenase linked ATP hydrolysis assay
The substrate dependent ATPase activity of HpDnaBWt and its various mutant forms were carried out using coupled pyruvate kinase/lactate dehydrogenase linked assay as described earlier (19). Typically, the reactions (75 μl) were carried out at 22°C in the reaction buffer containing 20 mM Tris–Cl (pH 8.0), 5 mM MgCl2, 100 mM KCl, 8 mM DTT, 4% sucrose, 80 μg/ml BSA. The reaction mixture was further supplemented with 250 μM NADH, 2 mM Phosphoenolpyruvate, 2.5 units of pyruvate kinase, 1.6 units of lactate dehydrogenase and varying concentrations of the ATP in the absence or in the presence of 50-mer single-stranded oligi-dT DNA (1 μM). The above reaction mixture was incubated for 5 min at room temperature prior to the addition of the respective protein (1.2 μM each). After the addition of the protein, the reaction kinetics was followed by measuring the decrease in the absorbance at 340 nm continuously for first 5 min by using the Ultrospec 2100 spectrophotometer (Amersham Biosciences, Piscataway, NJ, USA). The conversion of ATP to ADP is stoichiometrically coupled to the oxidation of NADH and was calculated using the equation:
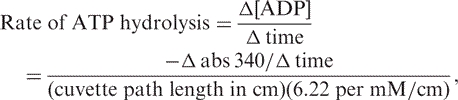 |
where 6.22 per mM/cm is the molar extinction coefficient of NADH at 340 nm.
The rate of ATP hydrolysis against different substrate concentrations and Lineweaver-Burk plot for each enzyme was plotted using GraphPad Prism curve fitting software and subsequently Vmax (maximum velocity) and Kcat (turn over number) values of each enzyme was calculated.
ATP-binding assay
UV-mediated cross-linking reactions were carried out using the protocol as described earlier (16). In brief, HpDnaBWt or other mutant proteins (1 μg each) were incubated in ice for 10 min in the cross-linking buffer containing 20 mM HEPES buffer (pH 7.5), 10% glycerol, 0.1 mM DTT, 10 μCi of ∝-32PATP (3000 Ci/mmol) followed by UV (254 nm) irradiation at a distance of 5 cm (Stratagene) for 30 min at 4°C. The reaction mixtures were further processed and proteins were separated by SDS-PAGE for the detection of labeled proteins using phosphorimager as described elsewhere (16).
Helicase assay
Helicase assays were performed essentially following the protocol as described earlier (16). The substrates for the helicase assays were obtained by annealing a radio-labeled 5′-tailed 29 mer oligo (5′-CCAAAACCCAGTCACGACGTTGTAAAACG-3′) to M13mp18 DNA followed by purification of the annealed products as described elsewhere (16).
The time-course of helicase reaction was performed using a 160 μl reaction mixture containing 500 nM each protein and ∼40 femto moles helicase substrate in helicase buffer as described earlier (16). Immediately after addition of the protein, the mixture was transferred to 4°C and a 20 μl aliquot (‘0’ time point) was taken out followed by the addition of 5 μl 5× stop buffer (1.25% SDS, 75 mM EDTA, 25% glycerol). The reaction mixture was then incubated at 37°C and 20 μl aliquot was taken out at 5, 10, 15, 20, 30, 45 and 60 min followed by the addition of the stop buffer. The reaction products were finally resolved in native PAGE. The released radio-labeled probe can thus be distinguished from the annealed substrate following autoradiography and quantitated by densitometric scanning.
Gel filtration chromatography
Gel filtration chromatography was performed essentially following the protocol described earlier (16). About 400 μg each protein (1 μg/μl concentration; either wild-type or different deletion mutants) was dialyzed against buffer A [50 mM Tris–Cl (pH 7.4), 50 mM NaCl, 1 mM DTT, 1 mM ATP, 2 mM MgCl2] at 4°C. The dialyzed sample was centrifuged at 15 000 rpm for 15 min. The supernatant was transferred to a fresh tube and 200 μl protein mixture (∼200 μg) was subjected to size-exclusion chromatography on a Superdex 200 (Pharmacia) column using buffer A. The column was previously calibrated using Pharmacia low and high molecular weight standards as followed: thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), bovine serum albumin (66 kDa), ovalbumin (43 kDa) and chymotrypsinogen (25 kDa).
Western blot analysis and antibodies
Western blot analysis was carried out following standard procedures (20). Anti-His rabbit polyclonal antibodies were purchased from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA.
Complementation assay
Escherichia coli temperature sensitive mutant DJ 58 (dnaBts) were transformed either with pBR322 or pBR-HpdnaBwt or pBR-HpdnaBmut, pBR-HpdelN2 or pBR-HpdelC1. The transformed cells were grown either at the permissive (30°C) or non-permissive (41°C) temperature.
Electron microscopy
The full-length HpDnaB, HpDnaBDelN2 and HpDnaBDelN3 were analyzed at a concentration of 0.03 mg/ml at pH 8.0 in the presence of 2 mM ADP. About 3 μl of each protein was applied to copper glow discharged carbon-coated grids and was later stained with 2% Uranyl acetate for 30 s. The Grids were examined on Morgagni 268 transmission electron microscope at 80 kV voltage, and 50 000× magnification and finally images were taken on the CCD camera. Single Particles in ring shaped forms were selected manually using the Signature software (21). About 173 single particles of wild-type and 111 single particles of D2 were selected. These particles were classified and processed using IMAGIC software. According to the shape wild-type particles were classified into two groups, in which 106 particles were in C6 symmetry and 67 particles were in C3 symmetry whereas all HpDnaBDelN2 particles were found to be in C6 symmetry.
CD and fluorescence spectroscopy
Samples of helicase (0.37 mg/ml) subjected to circular dichroism (CD) measurements were dissolved in 10 mM sodium cacodylate buffer, pH 7.4. All measurements were carried out at 25°C. Buffer was used as control. CD spectra were recorded on a Jasco (J-810) spectropolarimeter using cells of 0.2 cm path length. The spectropolarimeter was routinely calibrated with the asymmetric compound, (1S)-(+)-10-camphorsulfonic acid. Spectral data were acquired over the range 250–200 nm for the far-UV CD measurement.
Ellipticity values (θ) in mdeg, obtained from the instrument readings, were expressed in terms of mean residue ellipticity [θ] using the following equation:
where M is the mean residue mass, l the path length (0.2 cm), C the concentration in g/ml and [θ] is the mean residue ellipticity (22). CD data was further processed using K2d software for the prediction of secondary structure (23,24).
For intrinsic tryptophan fluorescence measurements (25), samples of different forms of helicases were dissolved in 10 mM sodium cacodylate buffer, pH 7.0. All measurements were carried out at 25°C. Buffer was used as control. The fluorescence spectra of wild-type and different mutant forms of helicase (0.185 mg/ml protein) were recorded on a Perkein-Elmer LS-55 Luminescence Spectrometer in the wavelength range of 300–400 nm at 25°C. The excitation wavelength was 278 nm. Scan rate of 500 nm/min was used with a slit size of 10 and 7 nm for excitation and emission, respectively. Data were collected at 0.5 nm wavelength resolution.
Homology modeling
The helicase domain of H. pylori DnaB shows 17% sequence identity and 40% homology with the bacteriophage T7 helicase, whose crystal structure has been determined (26). The homology model of H. pylori DnaB helicase domain was generated with MODELLER, a comparative modeling method (27). This method yielded a partial model, and the model was incomplete in those regions where the sequence homology was very poor or where there was no homology. The helicase domain homology model was also generated with the ‘threading’ method with the help of program LOOP (28). Both the methods generated almost similar models (with root-mean-square deviation of 2 Å) although the model generated by MODELLER showed better stereochemistry with respect to bond angle, bond length and allowed residues in Ramachandran plot, etc. Therefore, we combined both the models to generate a complete model with better stereochemistry. The resultant model was viewed on ‘O’ program (29) and the stereochemistry of the model was adjusted and short contacts were removed. This model was taken to Discover, Insight II, the energy minimization of the model was performed using both molecular dynamics and energy minimization. The iterative steps of model building by ‘O’ program and energy minimization using Discover, Insight II yielded a model, displayed by MOLSCRIPT (30). The stereochemistry of the final model was validated by PROCHECK (31).
RESULT
Expression and purification of different amino terminal and carboxy terminal deletion mutants of HpDnaB
Most replicative DnaB like helicases can be divided into two major domains: an N-terminal domain and a C-terminal helicase domain separated by a linker region (7,8,11).
Based on the boundaries of these domains as described earlier (7,8,11), analysis of primary amino acid sequence of HpDnaB following alignment with E. coli DnaB amino acid sequence reveals the presence of these two major domains and the linker region too. The N-terminal region spans from residues 1 to 120 whereas the C-terminus helicase domain lies within the residues 175 and 488 (Figure 1A and Supplementary Figure 1). The linker region is within these two domains. It may be noted that due to the poor homology at the N-terminal region, there is a possibility that these boundaries are more flexible than it has been suggested here. Interestingly, the extreme C-terminal region of HpDnaB shows very poor homology with its other DnaB counterparts. In fact, there is a unique 34 amino acid insertion in this region (400–433), the function of which is still not known (Supplementary Figure 1 and Figure 1A).
In order to investigate the functional domains of HpDnaB both in vitro and in vivo, we decided to generate series of deletion mutants ranging from N-terminus to C-terminus of HpDnaB. These mutants were generated by PCR amplification using cloned HpDnaB as template and respective primers as shown in the primer list. A point mutant having mutation in the ATP binding site was also used (HpDnaBMut). Different deletion mutants and their respective amino acid residue positions are shown in Figure 1A. Four deletion mutants were generated from the N-terminus (DelN1-DelN4) and two deletion mutants were generated from C-terminal region (DelC1 and Del34). It is important to note at this point that in E. coli, the equivalent deletion mutant of DelN2 (without having the N-terminal region and some portion of the linker region) shows ATP hydrolysis rate comparable to that of wild-type protein but it fails to show any detectable helicase activity (EcDelN156, Supplementary Figure 1) (10). Following PCR amplification, the mutant forms of the genes were subsequently cloned into pET28a vector and finally the proteins were expressed and purified as described in the materials and methods. Initially all the proteins were expressed at 37°C. However at this temperature, DelN4 and all the C-terminal deletion mutants mostly went into the inclusion bodies. Later, with several modifications, these proteins were expressed at 22°C with low level of IPTG that yielded reasonably good quantity of protein at the soluble form that were enough for the biochemical assays. All the purified proteins are shown in Figure 1B.
Analysis of biochemical activities of HpDnaBWt and deletion mutants
One of the hallmarks of helicases is their ATPase activity that is either DNA-dependent or DNA-stimulated (32,33). The energy thus obtained is transduced for helicase activity. In order to allow quantitative comparison of activities of wild-type and mutant proteins, ATP hydrolysis rates were calculated at various substrate concentrations in a time-course dependent manner in the absence or presence of 50-mer single-stranded oligo-dT DNA (1 μM) as described in the materials and methods. The DNA stimulated ATPase activity of HpDnaB has been described earlier (16). Wild-type and various mutant proteins were taken at 1.2 μM concentration and ATP hydrolysis reaction rates were plotted against substrate concentrations along with the Lineweaver-Burk plots (Figure 2A–H) that allowed us to get the maximum rate of each reaction (Vmax) and turn over number (Kcat) of each enzyme in the absence or presence of ssDNA as shown in Table 2. These results indicate that the wild-type enzyme is the most efficient one with the highest Vmax and Kcat values both in the absence or presence of DNA followed by DelN1 (that retains ∼80% activity, compared to that of wild-type in the presence of DNA) and DelN2 (that retains ∼70% activity, compared to that of wild-type in the presence of DNA) that show moderate ATPase activities whereas DelN3 shows very poor ATPase activity with a drastic decrease in both Vmax and Kcat values. DelN4, DelC1 and Del34 show almost no ATPase activity comparable to the background activity of control HpDnaBMut protein containing a mutation in the NTP binding motif (data not shown). We find that both Vamx and Kcat values increase around 10-fold or more for HpDnaBWt and HpDnaBDelN1-2 enzymes in the presence of DNA whereas DelN3 shows around 2-fold stimulation of these values under the same experimental conditions. HpDnaBMut does not show any detectable ATPase activity either in the absence or presence of DNA. These results suggest that binding to DNA confers favorable structural conformations to HpDnaBWt and HpDnaBDelN1-2 enzymes resulting such a profound stimulation of ATPase activity.
Figure 2.

ATPase activity of the wild-type and different deletion mutant proteins. ATPase assay using various HpDnaB forms. Enzyme-linked ATPase assays were performed using protocol as described in the materials and methods either in the absence or presence of DNA. The rates of the reactions against different substrate (ATP) concentrations were plotted for HpDnaBWt, HpDnaBDelN1, HpDnaBDelN2 and HpDnaBDelN3 as shown in panels A, C, E and G, respectively. Lineweaver-Burk plots of the same enzymes were also plotted either in the absence or presence of DNA in each case (panels B, D, F and H, respectively). Maximum velocity of the reaction (Vmax) and turn-over number (Kcat) were calculated in each case (as shown in Table 2).
Table 2.
Comparison of the ATP hydrolysis activity of HpDnaB wild-type and mutant proteins in the absence and presence of DNA
| Protein | Vmax (μM/s) | Kcat (per s) |
|---|---|---|
| HpDnaBWt (−DNA) | 0.120 | 0.100 |
| HpDnaBWt (+DNA) | 1.50 | 1.25 |
| HpDnaBDelN1 (−DNA) | 0.09 | 0.075 |
| HpDnaBDelN1 (+DNA) | 1.19 | 0.99 |
| HpDnaBDelN2 (−DNA) | 0.083 | 0.069 |
| HpDnaBDelN2 (+DNA) | 1.06 | 0.88 |
| HpDnaBDelN3 (−DNA) | 0.014 | 0.012 |
| HpDnaBDelN3 (+DNA) | 0.032 | 0.026 |
| HpDnaBMut (−DNA) | – | – |
| HpDnaBMut (+DNA) | – | – |
Each enzyme was taken at 1.2 μM concentration and the corresponding maximum rate of the reaction (Vmax) at varying substrate concentration was calculated to get the turn over number (Kcat) of each enzyme either in the absence or presence of 50-mer single stranded oligo-dT DNA (1 μM).
These results also suggest that linker region and the extreme C-terminal region including the unique 34 amino acid residues are important for the activity of the protein. Interestingly, the Michaelis-Menten constant (Km) for the wild-type enzyme is found to be 64 μM in the absence of DNA that is comparable to the Km value of E. coli DnaB helicase (50 μM) as it has been reported earlier (10). The Km values of DelN1-3 deletion mutants were found to be higher than the wild-type enzyme both in the absence and presence of DNA (data not shown).
Earlier we have shown that HpDnaB binds to radio-labeled ∝-32PATP (16). The inability to hydrolyze ATP by the mutant proteins (DelN4, DelC1 and Del34) can be attributed to their inability to bind to ATP. Alternatively, these mutants can bind to ATP but they are unable to hydrolyze ATP. To test which of the above hypothesis is true, we performed ATP binding assay using wild-type and some of the mutant proteins. For this purpose, wild-type and some other deletion mutants were first incubated in the presence of ∝-32PATP followed by cross-linking of the proteins with the radio-labeled ATP in the presence of ultra violet light as described in the materials and methods. The reaction mixtures containing the proteins were then resolved in SDS-PAGE followed by auto-radiography. Wild-type, DelN2 and DelN3 showed ATP binding whereas DelN4 and DelC1 failed to bind radio-labeled ATP (Figure 3A). Coomassie brilliant blue staining of the same gel indicate that comparable amount of proteins were loaded in each lane. Del34 also did not bind to ATP under the same experimental conditions (data not shown). These results suggest that the inability of DelN4 and DelC1 to hydrolyze ATP is truly due to the failure of these proteins to bind to ATP. However, it seems that both DelN2 and DelN3 binds to ATP less efficiently compared to wild-type protein. This effect may explain the reduced ATP hydrolysis activity of DelN2 and DelN3 over the wild-type protein.
Figure 3.

ATP-binding and helicase activity of HpDnaBWt and mutant proteins. (A) ATP binding activity of various HpDnaB forms. Total 1 μg of each protein as indicated on the top was incubated in the presence of radio-labeled α-32P ATP followed by SDS-PAGE analysis of ATP bound protein by autoradiography. The right panel shows that only wild-type, DelN2 and DelN3 bind to ATP whereas DelN4 and DelC1 fail to bind ATP. The left panel shows the coomassie-stained image of the same gel. (B) Helicase activity of various HpDnaB forms. Approximately 500 nM of each protein as indicated on the top was incubated in a reaction mixture containing end-labeled 29 nucleotides probe hybridized to M13 single-stranded DNA for 30 min and the release of the unannealed probe was monitored following PAGE analysis. Annealed and unwound substrates are marked. The sample marked as ‘heated’ was boiled for 3 min before loading. Deletion mutants DelN1-N2 show the release of unwound oligo whereas DelN3-4, DelC1 and HpDnaBMut do not show any activity. Control lane does not contain any protein. (C) Time-course of helicase activity. The helicase activity of each protein as indicated in the figure was monitored every 5 min interval up to 1 h and the intensity of released unwound oligo in each case was quantified using densitometry scanning and subsequently plotted. Helicase reactions were repeated at least three times for each time point.
Further, we were interested to investigate whether the energy obtained from ATPase activity can be transduced to helicase activity that is the major function of the HpDnaB helicase. Equal amounts of wild-type and other deletion mutant proteins (in molar ratio) were incubated in the presence of labeled substrates and ATP as energy source for 30 min followed by resolving the reaction products in native PAGE. The released radio-labeled probe can thus be separated and distinguished from the annealed substrate following autoradiography. The results indicate that wild-type, DelN1 and DelN2 contain helicase activity whereas DelN3, DelC1 and HpDnaBMut (with a point mutation in the ATP binding site) do not show any such activity under these experimental conditions (Figure 3B). The inactivity of these proteins also ensures that the helicase activity of other N-terminal mutants is not due to the presence of any contaminating proteins since all these proteins were purified under the same experimental conditions. The helicase activity of HpDnaBDelN2 is particularly of great interest since the equivalent deletion in E. coli DnaB protein (EcDelN156) has been reported to contain the ATPase activity but not the helicase activity. DelN3 that shows some ATPase activity fails to demonstrate helicase activity. The C-terminal deletion DelC1 does not show helicase activity. This result is consistent with the inability of this protein to hydrolyze ATP.
In order to demonstrate the helicase activity of the wild-type protein and some of the mutants in a more quantitative manner, percent helicase activities of these proteins (500 nM each) were calculated at every 5 min interval until they reach saturation level as indicated in the materials and methods. These values were plotted against various time-points and the relative helicase activities were compared based on three independent experiments for each time point (Figure 3C). The results indicate that both wild-type and DelN1 are efficient helicases that show conversion of more than half of the substrate into products within 5 min of the start of the reaction. The helicase activity of these enzymes reaches a plateau at around 30 min. Although the helicase activity of DelN2 follows a slower kinetics than the wild-type enzyme, overall, after 60 min, it shows more than 70% of the wild-type activity under these experimental conditions suggesting that DelN2 contains significant amount of helicase activity. On the other hand, neither DelN3 nor DelC1 shows any helicase activity above the background level. These results together suggest that N-terminal region might not be essential for in vitro helicase activity whereas the linker region and the extreme C-terminal region are essential for its activity. It is important to note that Del34 mutant also did not show any helicase activity under the same experimental conditions (data not shown). Comparison of biochemical activities of HpDnaBWt and different deletion mutants is summarised in Table 7.
Table 7.
Summary of Biochemical activities of HpDnaBWt and di.erent mutant variants as shown in Figures 2 and 3
| Protein | ATP-binding | ATPase | ATPase DNA-stimulated | Helicase |
|---|---|---|---|---|
| HpDnaBWt | +++ | +++ | +++ | +++ |
| HpDnaBMut | − | − | − | − |
| DelN1 | ND | ++ | ++ | +++ |
| DelN2 | ++ | ++ | ++ | ++ |
| DelN3 | ++ | + | + | − |
| DelN4 | − | − | − | − |
| DelC1 | − | − | − | − |
| Del34 | ND | − | ND | − |
‘+++’, ‘++’ and ‘+’ signs stand for maximum, moderate and lowcatalytic activity, respectively, whereas ‘−’ stands for no activity. ‘ND’ stands for experiment not done
N-terminal region is not essential for hexamer formation but required for C6:C3 conformational changes
In E. coli, an N-terminal deletion mutant of EcDnaB (equivalent to HpDnaBDelN2) forms hexamer in solution and shows in vitro ATPase activity but fails to show in vitro helicase activity (10). In contrary, the equivalent HpDelN2 deletion mutant shows both ATPase activity and helicase activity. It has been proposed that the N-terminal region is essential for conversion of EcDnaB molecules between C3 and C6 structural symmetry that is essential for helicase activity. Direct electron microscopy analysis of EcDnaB molecules supports the above hypothesis (12,13). In vitro helicase activity of DelN2 raises the issue whether N-terminal region of HpDnaB has any effect on C3:C6 conformational changes that have been implicated as an essential factor for helicase activity. To address this issue, we performed direct electron microscopic analysis of HpDnaBWt, DelN2, DelN3 and DelC1 proteins as described in the materials and methods. Over hundred images were taken in each case and the images were further processed and superimposed using IMAGIC software to get the final images as shown in Figure 4. Both wild-type and DelN2 proteins form hexamer with a central channel that might be used for passing the DNA through during the replication fork movement. Interestingly, the wild-type proteins were found in both C6 and C3 structural symmetry forms whereas DelN2 proteins were found in C6 symmetry forms only. Statistically, one-third of the total molecules scanned were present in the C3 form in case of HpDnaBWt whereas all DelN2 molecules were present in the C6 form only (Table 3) suggesting that the N-terminal region would be essential for the structural conversion. Neither hexameric nor any other distinct form could be identified in case of DelN3 or DelC1 under the same experimental conditions. We reasoned that these deletion mutants are structurally unstable to confer any stable conformation under electron microscopic observation. A closure examination of the images in C6 symmetry in case of HpDnaBWt molecules reveal an extended portion protruded from the each unit. No such extended portion could be detected in DelN2 molecules that lack the N-terminal region. The electron microscopic results suggest that the N-terminal region is essential for the C3:C6 conformational changes although these conformational changes are not essential for helicase activity, as it has been proposed earlier for E. coli DnaB proteins.
Figure 4.

Electron microscopic observation and analysis of HpDnaBWt, DelN2, DelN3 and DelC1. Above four proteins were processed for electron microscopy as described in the materials and methods and individual sample was scanned under a Morgagni 268 transmission electron microscope at 80 kV voltage. More than hundred images were captured in each case and the raw images were processed using IMAGIC software. The left panel in each pair shows the unprocessed image and the right panel shows the processed image. HpDnaBWt was found in both C6 and C3 conformations whereas DelN2 was found only in C3 conformations. Bars in the panels are equivalent to 10 nm.
Table 3.
Electron microscopic analysis of wild-type and HpDelN2 proteins
| Protein | Number of Molecules scanned | C6 symmetry | C3 Symmetry |
|---|---|---|---|
| HpDnaBWt | 173 | 106 | 67 |
| HpDnaBDelN2 | 111 | 111 |
The table shows the total number of molecules scanned and the represntation of number of molecules at a specific form of rotational symmetry.
Most replicative helicases are hexameric in nature since this is the preferred conformation for the helicases that will allow them to translocate through the fork structure while unwinding the DNA during DNA replication. We have earlier reported that wild-type HpDnaB forms hexamer in solution (16). In order to investigate whether deletion of the N-terminal and C-terminal regions from the wild-type proteins could affect the oligomeric status of the proteins, we performed gel filtration analysis using wild-type and other deletion mutant proteins. For this purpose, wild-type and different mutant variants of these proteins were passed through Superdex 200 gel filtration column along with several molecular mass standards as described in the materials and methods. All the fractions containing the proteins in each case were further resolved in SDS-PAGE for confirmation. The elution pattern of the individual protein was compared with the elution pattern of the molecular mass standards. A distinct pool of hexameric forms of proteins could be detected in case of HpDnaB, HpDnaBMut, DelN1 and DelN2. DelN3 eluted as dimers whereas DelC1 and Del34 eluted mostly as monomers. The results obtained following gel filtration analysis are summarized in Table 4. It is interesting to note that the electron microscopic studies also reveal the presence of hexameric forms of the helicases for HpDnaBWt and HpDelN1-2. These results together confirm that the hexameric status of the protein is essential for the helicase activity. Further, these results indicate that the linker region and the extreme C-terminal region including the 34 amino acid insertion region might be responsible for the stability of the oligomeric forms of HpDnaB.
Table 4.
Oligomeric status of HpDnaBWt and different mutant variants following gel filtration analysis and comparison of the elution pattern of these proteins with those of standard protein markers as described in the materials and methods
| Protein | Oligomeric status |
|---|---|
| HpDnaBWt | Hexamer |
| HpDnaBMut | Hexamer |
| HpDnaBDelN1 | Hexamer |
| HpDnaBDelN2 | Hexamer |
| HpDnaBDelN3 | Dimer |
| HpDnaBDelN4 | Monomer |
| HpDnaBDelC1 | Monomer |
| HpDnaBDel34 | Monomer |
HpDnaBDelN2 can complement E. coli DnaB temperature sensitive strain in vivo
In vitro helicase activity is measured by the release of a short oigonucleotide probe that is hybridized to M13 ssDNA. Therefore, the in vitro helicase activity cannot be correlated to the in vivo activity since at the chromosomal context this activity needs to be more efficient to unwind the two strands of the DNA. In order to investigate the efficacies of the different deletion mutants in vivo, we decided to perform complementation assay using DJ58 E. coli DnaB temperature sensitive mutant strain. This will also help us to test the hypothesis whether a deletion mutant restricted to the C6 conformational state will be able to support helicase activity in vivo. For this purpose, different deletion mutant forms of HpDnaB were cloned into pBR322 under BlaP2 promoter and DJ58 strain was transformed using these recombinant clones along with pBR322 control plasmid. We showed earlier that HpDnaBWt could complement DJ58 temperature sensitive strain at the restrictive temperature suggesting that HpDnaB is a true replicative helicase in vivo (16). Bacterial colonies were streaked on Luria broth containing agar plates and they were incubated at the permissive (30°C) and restricted temperature (41°C). All of them grew nicely at the permissive temperature whereas only the HpDnaBWt and HpDelN2 grew at the restrictive (41°C) temperature (Figure 5A). Neither HpDnaBMut, nor HpDnaBDelC1 or pBR322 could support the growth of DJ58 mutant cells at the restrictive temperature suggesting that the conditions for the in vivo complementation assay are ideal. HpDelN2 result is of particular interest since it suggests that N-terminal region of HpDnaB helicase is not essential for DnaB helicase function. Neither the C6:C3 conformation changes are required for the in vivo activity. This is in sharp contrast to the EcDnaB where an equivalent deletion mutant is defective in helicase activity.
Figure 5.

Complementation analysis. (A) Complementation of E. coli DJ58ts with plasmids expressing different variants of HpDnaB. Escherichia coli DJ58ts cells were transformed with either pBR322 (1) or pBR322 + HpDnaBMut (2) or pBR322 + HpDnaWt (3), pBR322 + HpDnaBDelN2 (4) or pBR322 + HpDnaBDelC1 (5). Cells were plated on LB agar plates and incubated either at the permissive temperature (30°C) or at the non-permissive temperature (41°C). (B) Detection of HpDnaB (lanes 1–3) or HpDnaBDelN2 (lanes 4–6) expression in E. coli DJ58ts cells by immunoblotting using anti-His antibodies. Bacterial cells (grown at indicated temperature) were resuspended in SDS-PAGE loading buffer followed by SDS-PAGE and western blot analysis. Respective recombinant proteins were also loaded as control.
To further rule out the possibility that the successful complementation by HpDnaBWt and HpDnaBDelN2 is not due to recombination of the HpDnaB gene with the E. coli counterpart in the chromosome, western blot analysis was performed using bacterial cell lysate from HpDnaBWt and HpDnaBDelN2 transformed DJ58 cells grown at 41°C. Anti-His6 polyclonal antibodies were used to detect the expression of these proteins since these proteins were expressed as His6–tagged fusion protein (Figure 5B). Both the wild-type and DelN2 proteins were expressed at the restrictive temperature suggesting that the complementation of the DJ58 cells at the restrictive temperature were due to the expression of these proteins and not due to the recombination effect or natural reversion of the mutant cells.
The extreme C-terminal region is essential for the structural integrity of HpDnaB
The inability of HpDelN4 and HpDelC1 to show any in vitro activity raises the issue whether these proteins are folded properly. As an initial indication, we observed that these proteins were mostly going into the inclusion bodies at 37°C whereas the wild-type protein was readily soluble under the same experimental conditions (data not shown). In order to get these proteins in the soluble form we had to express them at the room temperature with low IPTG concentration and several other modifications.
Circular dichroism is a method that would account for the helix content in a polypeptide based on the presence of alpha helix, beta sheet and random coil (22). We prepared five different mutants HpDnaB (HpDnaBMut, DelN2, DelN3 DelN4 and DelC1) and wild-type protein as described in the materials and methods for CD spectra analysis. We probed the secondary structure of wild-type and mutant helicases by far-UV CD. For HpDnaBWt, HpDnaBMut, DelN2, DelN3 and DelN4 proteins, far-UV CD spectra clearly reflect characteristics of (α + β) type of structure (Figure 6A). The first three proteins have nearly same composition of α and β elements. For the DelN3 mutant, although there is sharp decrease in α helical content and increase in β-sheeted structure, the nature of the CD spectrum has not changed suggesting some changes in its overall conformation. DelN4 shows similar CD spectra as seen in case of DelN3 suggesting that these two deletion mutants may contain similar structural conformation. For DelC1 mutant, molar residue ellipticity values reach to a minimum and the protein has lost the helical structure almost completely and there is a parallel increase of β-sheeted structure suggesting that it may be the denatured state of the protein (Figure 6A). These spectra were quantitatively analyzed for percent alpha helix, beta sheet and random coil content using the K2d online program. The estimated levels of secondary structure are listed in Table 5.
Figure 6.

(A) Far-UV circular dichroism spectra of wild-type and different mutant variants of the helicase. CD spectra were measured for each protein in a 2 mm path length quartz cell and data were collected at 1.0 nm wavelength resolution. The arrowheads indicate the CD spectra of the respective protein. (B) Intrinsic fluorescence spectra of wild-type and different deletion mutants. The fluorescence emission spectra of wild-type and different mutant forms of HpDnaB (as indicated) were recorded from 300 to 400 nm at 25°C. The excitation wavelength was 278 nm and data were collected at 0.5 nm wavelengths resolution. The arrowheads indicate the CD spectra of the respective protein.
Table 5.
Secondary structural elements of wild-type and four mutant forms of helicases following CD spectra analysis using K2D online software as described in the materials and methods
| Protein | α-helix (%) | β-sheet (%) | Random coil |
|---|---|---|---|
| HpDnaBWt | 35 | 18 | 46 |
| HpDnaBMut | 31 | 16 | 52 |
| DelN2 | 29 | 18 | 52 |
| DelN3 | 14 | 25 | 51 |
| DelN4 | 9 | 35 | 56 |
| DelC1 | 4 | 48 | 48 |
Del34 also show almost complete loss of alpha helices under the same experimental conditions (data not shown). These results suggest that a major structural change affecting the balance between the alpha helicity and beta sheet content normally present in the wild-type protein has taken place in HpDelC1.
To reconfirm the structural change that has taken place in some of the deletion mutants, intrinsic fluorescence was measured using a spectrofluorometer. There is only one tryptophan residue at the 265th position of HpDnaB. We argued that if there was a gross change in the overall structure of the helicase molecule due to the deletion, the hydrophobic/hydrophilic environment around the trypotophan residue would be changed that would in turn affect the intrinsic fluorescence of these proteins.
For this purpose, intrinsic tryptophan fluorescence spectra of wild-type and different mutant forms of helicase (HpDnaBMut, DelN2 and DelC1) were measured that would allow us to compare the conformational status among the proteins. The emission maxima for all the proteins are in the range of 345–350 nm. Emission intensities for HpDnaBMut and DelN2 are higher than that of the wild-type helicase (Figure 6B). This observation reveals that there might be some conformational differences between wild-type and HpDnaBMut and DelN2. It is possible that in the wild-type protein, tryptophan fluorescence was quenched by neighboring groups, however in the mutant forms those quenching groups got apart from the tryptophan residues and hence the intensity of emitted fluorescence is becoming higher. The intensity of tryptophan emission for the DelC1 was very much reduced compared to the wild-type protein (Figure 6B). There is a possibility that in this mutant form of helicase, a gross conformational change has taken place and the neighboring groups quench the tryptophan fluorescence. The other possibility is that, due to the major conformational change, the mutant protein cannot form native like structure and in the non-native form; the tryptophan residue got exposed to the polar environment, which can explain the reduction of fluorescence intensity of the DelC1 mutant. Different parameters used for the measurement of intrinsic tryptophan fluorescence have been presented in Table 6.
Table 6.
Intrinsic fluorescence measurement parameters of wild-type and mutant helicase proteins
| Protein | Concentration (mg/ml) | Excitation (nm) | Emission (nm) | Slit Ex/Em (nm) |
|---|---|---|---|---|
| HpDnaBWt | 0.185 | 278 | 345 | 10/7 |
| HpDnaBMut | 0.185 | 348 | 10/7 | |
| HpDelN2 | 0.185 | 351 | 10/7 | |
| HpDelC1 | 0.185 | 345 | 10/7 |
Homology model of HpDnaB
Biochemical and biophysical experiments show that the deletion of 133 amino acid residues from the N terminus (DelN2) does not affect overall HpDnaB activity whereas deletion of 174 amino acid residues from the N-terminal region (DelN3) confers some structural changes leading to the loss of helicase activity while retaining ATPase activity. However, deletion of 195 amino acid residues from N-terminal (DelN4) or only 29 amino acid residues (DelC1) from C-terminal region or adjacent 34 amino acid insertion region (Del34) compromised HpDnaB activity completely. These results suggest that though the extreme N-terminal domain can be dispensable for HpDnaB activity, the following linker region and C-terminal region might have an essential function. It is interesting to note at this point that the deletion of 55 residues from the extreme C-terminal region does not have any effect on EcDnaB function. Indeed the extreme C-terminal region of HpDnaB shows very poor homology with EcDnaB and there is a unique 34 amino acid residue insertion in this region that is not present in any other DnaB. Therefore, it is extremely important to get an overview of the three-dimensional structure of the protein in order to explain the roles of the C-terminal and the N-terminal regions of HpDnaB. We made several attempts to crystallize the full-length and different domains of the protein without success. In the absence of a crystal structure, we decided to get a homology model of HpDnaB based on the helicase structures available. This might lead to the explanation of some of the unique HpDanB results presented here.
The helicase domain of HpDnaB (residues 174–488) shows 17% sequence identity and 40% homology with the bacteriophage T7 helicase. The crystal structure of the later helicase has been determined (26). The homology model of HpDnaB helicase domain was generated with MODELLER, a comparative modeling method (27) and also with the ‘threading’ method by the help of program LOOP (28). These two models were superimposed with each other and finally processed as described in the materials and methods.
The helicase domain of HpDnaB forms hexamer and looks similar to the T7 helicase structure (Figure 7A). However, due to the 34 residues (from 400 to 433) insert near the C-terminal, the nucleotide base-binding region is completely different. According to this model, nucleotide base interacts with the 34-residue insert loop and stabilizes the nucleotide-binding pocket. This model further suggests that this loop (400–433) interacts both with the helicase domain N-terminal (182–192) and C-terminal regions (466–471) of the neighboring molecule (Figure 7B). These interactions might be important for hexamer formation and stabilization of the large loop.
Figure 7.

Homology modeling of HpDnaB. (A) Homology model of HpDnaB based on the T7 helicase domain as template. Six subunits are shown in different colors. The nucleotide (ATP) coupled to each monomer is also shown. (B) Close-up view of interface between two monomers. Two monomers are shown in different colors (cyan and green) and nucleotide (ATP) is shown in ball & stick model. The 34 residues insert from one molecule (brown) is interacting with N-terminal (182–192, light pink) and C-terminal (466–471, dark blue) regions of the next molecule are also shown.
Formation of hexamer is essential for helicase activity. From the homology model, it is revealed that DelN4, DelC1 and Del34 may not be able to form hexamers as these regions are involved in inter-domain interactions. The homology model therefore supports our biochemical experiments that show loss of oligomerization and activity for the above deletion mutants.
DISCUSSION
One of the main observations of this paper is the in vitro helicase activity of HpDelN2 that does not contain the N-terminal region and a part of the linker region. The equivalent deletions in the E. coli and B. stearothermophilus DnaBs have resulted in proteins that can form hexamers and hydrolyze ATP but unable to exhibit helicase activity (10,11). It is believed that this is a consequence of the importance of the N-terminal domain in determining the C6:C3 conformational changes of the DnaB hexamer. While HpDnaBWt showed C6:C3 conformational changes, DelN2 was found to be present only in C6 conformation following electron microscopic analysis of these proteins. In vivo complementation of dnaBts mutant cells with HpDelN2 at the restrictive temperature further confirms that the N-terminal domain is not essential for helicase activity although it might be required for C6:C3 structural conformational changes, the functional significance of which is yet to be determined. It is important to note at this point that HpDelN2 shows less ATPase and helicase activity (as shown in Table 2 and Figure 3B–C) compared to that of the wild-type protein. It is possible that the N-terminal region that is important for C6:C3 conformational changes may likely be required for the full activity of HpDnaB. Alternatively, the N-terminal region would be required for strand discrimination and polarity of translocation as it has been shown in case of bacteriophage SPP1 G40P helicase recently where a deletion mutant lacking the N-terminal domain but having the linker region shows both 5′–3′ and 3′–5′ polarity in contrast to the wild-type protein that shows only 5′–3′ polarity (5).
The difference between EcDnaB and HpDnaB with respect to C6:C3 conformational changes and their activities raises the issue whether these conformational changes are truly the characteristics for DnaB type of helicases. The functional significance of these forms is still not clear for helicase unwinding and translocation. A recent crystal structure analysis of papiloma virus E1 helicase along with single-stranded DNA and nucleotides suggests that only one strand passes through the hexameric channel and each subunit sequentially tracks the oligonucleotide backbone with a straightforward DNA translocation mechanism model (34). It will be very interesting to see whether all hexameric helicases follow this general model for DNA unwinding and translocation. The crystal structure data suggest that E1 heicase forms a rigid collar with 6-fold rotational symmetry. Although it is tempting to speculate that all helicases will follow a general model for DNA unwinding and translocation, there are fundamental differences between E1 helicase and DnaB type helicases. E1 helicase belong to SF3 helicase family that contains three domains out of seven conserved domains found in majority of the helicase proteins whereas DnaB type of helicases contain five conserved domains (35). These subtle differences may play important roles among various helicases.
It seems that the linker region is important for the overall conformation of the HpDnaB helicase and its function. The deletion of the linker region (DelN3) shows significantly reduced ATPase activity both in the absence and presence of DNA compared to that of wild-type protein (Table 2), fails to retain helicase activity (Figure 3B–C) and it shows lower alpha helix content compared to the wild-type protein (Table 5) under our experimental conditions. Electron microscopic analysis of DelN3 also fails to identify any definite structural conformation as seen in HpDelN2. Gel filtration analysis shows that HpDelN3 forms mostly dimers. These results indicate that the linker region is important for intermolecular interaction and structural flexibility required for the stability of the hexameric forms that are key to the helicase activity. As the nucleotide-binding site exists between two molecules, DelN3 is able to bind ATP and it shows some ATPase activity.
Deletion of further 22 amino acid residues into the helicase domain (DelN4) results in monomeric non-functional protein suggesting that these 22 amino acid residues must be very crucial for primary protein–protein interaction interfaces essential for the oligomerization of HpDnaB.
It is interesting to note that both DelN3 and DelN4 show similar trends and poor alpha helix content in their CD spectra although DelN3 shows ATPase activity whereas DelN4 is inactive. The poor alpha helix content of DelN3-4 mutants compared to that of wild-type suggests that the hexameric form of HpDnaB is the most stable form that contribute to the maximum alpha helix content of these molecules. However, the loss of activity of DelN4 can be attributed to the structural instability of the WALKER A domain of the NTP binding motif that is at the extreme N-terminal region in this deletion mutant. In fact, this hypothesis is strengthened with the finding of ATP binding experiments where DelN3 shows ATP binding activity whereas DelN4 fails to show any such activity. Probably the deletion site in DelN3 is too far to interfere with the organization of the ATP binding motif.
Alternatively, the inactivity of HpDelN4 can be explained using the homology model that has been generated using T7 helicase as a template. The homology model suggests that the residues around 182–192 of one molecule interact with and stabilize the 34 amino acid residues from another molecule to stabilize the nucletide-binding site of HpDnaB.
Why do DelC1 and Del34 lose activity? The homology model suggests that the extreme C-terminal residues and the adjoining unique 34 amino acid insertion region stabilizes the ATP binding pocket that gets affected with the deletion of these regions compromising the overall structure and conformation of these proteins yielding unfolded proteins. This hypothesis is substantiated by our experimental results as shown in Figure 3A that clearly indicates that DelN4 and DelC1 fail to bind to radio-labeled ATP.
The 34-insertion region is unique to HpDnaB and not present in any known DnaB helicase. This raises the issue why does HpDnaB selectively possess this region. The explanation might find its root in our previous finding that HpDnaB does not need a helicase loader for its function. It needs to be explored further whether this extra region can account for the independent loading of HpDnaB.
It is now clear from this study and other related studies that the linker region is essential for oligomeric status of the protein and helicase activity. The presence of the unique C-terminal region might provide better flexibility to the oligomeric structure of HpDnaB that would enable HpDelN2 to perform its helicase activity whereas the counterpart deletion mutants in EcDnaB and BsDnaB lacking this region fail to show any such activity since the loss of linker region (either partial or full) cannot be compensated.
It is possible that this unique region also serves as a strong interface for protein–protein interaction that stabilizes the oligomerization of HpDnaB and/or other possible interacting partners of HpDnaB. A closure examination of the 34 amino acid insertion reveals the presence of highly charged residues within this region. Distribution of these charges over the interface spanning the insertion region will be the key to understand the functional relevance of this region (Supplementary Figure 1). A detailed point mutation analysis of this region is underway at present. It is possible that some of the positively charged residues present in this region might be oriented in such a way that it might support greater affinity to DNA that might be essential for the loader independent function of HpDnaB.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work is supported by a grant from University grant commission, India [UPOE] to SKD. RGN and AD acknowledge UGC for fellowships. The authors also acknowledge the Swedish Research council for an International Asian-Swedish Research Link grant on Helicobacter pylori replication and control. We thank Olga and Chen for their help with EM studies. SG thanks DBT, India for financial support. The authors also thank specially Dr Neelima Alam for her help with ATPase assay. The authors thank Prof. Sudha Bhattacharya (Jawaharlal Nehru University) and Prof. Anindya Dutta (University of Virginia), Dr Santanu Dasgupta (Upsala University, Sweden) for critically reviewing the manuscript. The Open Access publication charges for this article were waived by Oxford University Press.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 2.Röleke D, Hoier H, Bartsch C, Umbach P, Scherzinger E, Lurz R, Saenger W. Crystallization and preliminary X-ray crystallographic and electron microscopic study of a bacterial DNA helicase (RSF1010 RepA) Acta Crystallogr. D Biol. Crystallogr. 1997;D53:213–216. doi: 10.1107/S0907444996012565. [DOI] [PubMed] [Google Scholar]
- 3.Bird LE, Hakansson K, Pan H, Wigley DB. Characterization and crystallization of the helicase domain of bacteriophage T7 gene 4 protein. Nucleic Acids Res. 1997;25:2620–2626. doi: 10.1093/nar/25.13.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fass D, Bogden CE, Berger JM. Crystal structure of the N-terminal domain of the DnaB hexameric helicase. Structure. 1999;7:691–698. doi: 10.1016/s0969-2126(99)80090-2. [DOI] [PubMed] [Google Scholar]
- 5.Mesa P, Alonso JC, Ayora S. Bacillus subtilis bacteriophage SPP1 G40P helicase lacking the N-terminal domain unwinds DNA bidirectionally. J. Mol. Biol. 2005;357:1077–1088. doi: 10.1016/j.jmb.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 6.Nunez-Ramirez R, Robledo Y, Mesa P, Ayora S, Alonso JC, Carazo JM, Donate LE. Quaternary polymorphism of replicative helicase G40P: structural mapping and domain rearrangement. J. Mol. Biol. 2005;357:1063–1076. doi: 10.1016/j.jmb.2006.01.091. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama N, Arai N, Kaziro Y, Arai K. Structural and functional studies of the dnaB protein using limited proteolysis. Characterization of domains for DNA-dependent ATP hydrolysis and for protein association in the primosome. J. Biol. Chem. 1984;259:88–96. [PubMed] [Google Scholar]
- 8.Biswas SB, Chen PH, Biswas EE. Structure and function of Escherichia coli DnaB protein: role of the N-terminal domain in helicase activity. Biochemistry. 1994;33:11307–11314. doi: 10.1021/bi00203a028. [DOI] [PubMed] [Google Scholar]
- 9.Weigelt J, Brown SE, Miles CS, Dixon NE, Otting G. NMR structure of the N-terminal domain of E. coli DnaB helicase: implications for structure rearrangements in the helicase hexamer. Structure. 1999;7:681–690. doi: 10.1016/s0969-2126(99)80089-6. [DOI] [PubMed] [Google Scholar]
- 10.Biswas EE, Biswas SB. Mechanism of DnaB helicase of Escherichia coli: structural domains involved in ATP hydrolysis, DNA binding, and oligomerization. Biochemistry. 1999;38:10919–10928. doi: 10.1021/bi990048t. [DOI] [PubMed] [Google Scholar]
- 11.Bird LE, Pan H, Soultanas P, Wigley DB. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000;39:171–182. doi: 10.1021/bi9918801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.San Martin MC, Stamford NP, Dammerova N, Dixon NE, Carazo JM. A structural model for the Escherichia coli DnaB helicase based on electron microscopy data. J. Struct. Biol. 1995;114:167–176. doi: 10.1006/jsbi.1995.1016. [DOI] [PubMed] [Google Scholar]
- 13.Yu X, Jezewska MJ, Bujalowski W, Egelman EH. The hexameric E. coli DnaB helicase can exist in different Quaternary states. J. Mol. Biol. 1996;259:7–14. doi: 10.1006/jmbi.1996.0297. [DOI] [PubMed] [Google Scholar]
- 14.San Martin C, Radermacher M, Wolpensinger B, Engel A, Miles CS, Dixon NE, Carazo JM. Three-dimensional reconstructions from cryoelectron microscopy images reveal an intimate complex between helicase DnaB and its loading partner DnaC. Structure. 1998;6:501–509. doi: 10.1016/s0969-2126(98)00051-3. [DOI] [PubMed] [Google Scholar]
- 15.Biswas EE, Biswas SB. Mechanism of DNA binding by the DnaB helicase of Escherichia coli: analysis of the roles of domain gamma in DNA binding. Biochemistry. 1999b;38:10929–10939. doi: 10.1021/bi990049l. [DOI] [PubMed] [Google Scholar]
- 16.Soni RK, Mehra P, Choudhury NR, Mukhopadhyay G, Dhar SK. Functional characterization of Helicobacter pylori DnaB helicase. Nucleic Acids Res. 2003;31:6828–6840. doi: 10.1093/nar/gkg895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soni RK, Mehra P, Mukhopadhyay G, Dhar SK. Helicobacter pylori DnaB helicase can bypass Escherichia coli DnaC function in vivo. Biochem. J. 2005;389(Pt 2):541–548. doi: 10.1042/BJ20050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene. 1977;2:95–113. [PubMed] [Google Scholar]
- 19.Dar MA, Sharma A, Mondal N, Dhar SK. Molecular cloning of apicoplast targeted Plasmodium falciparum DNA gyrase genes: unique intrinsic ATPase activity and ATP-independent dimerisation of PfGyrB subunit. Eukaryot. Cell. 2007;6:398–412. doi: 10.1128/EC.00357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook JF, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 21.Chen JZ, Grigorieff N. SIGNATURE: a single-particle selection system for molecular electron microscopy. J. Struct. Biol. 2007;157:168–173. doi: 10.1016/j.jsb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Keideiling TA. Vibrational circular dichroism application to conformational analysis of biomolecules. In: Fasman GD, editor. Circular Dichroism and the Conformational Analysis of Biomolecules. New York: Plenum Press; 1996. pp. 555–598. [Google Scholar]
- 23.Andrade MA, Chacón P, Merelo JJ, Morán F. Evaluation of secondary structure of proteins from UV circular dichroism using an unsupervised learning neural network. Prot. Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 24.Merelo JJ, Andrade MA, Prieto A, Morán F. Proteinotopic feature maps. Neurocomputing. 1994;6:443–454. [Google Scholar]
- 25.Lackowicz JR. Principles of Fluorescence Spectroscopy. Plenum press: New York; 1983. [Google Scholar]
- 26.Sawaya MR, Guo S, Tabor S, Richardson CC, Ellenberger T. Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell. 1999;99:167–177. doi: 10.1016/s0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- 27.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 28.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods of building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 30.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 31.Teodorescu O, Galor T, Pillardy J, Elber R. Enriching the sequence substitution matrix by structural information. Proteins, Struct. Funct. Genet. 2004;54:41–48. doi: 10.1002/prot.10474. [DOI] [PubMed] [Google Scholar]
- 32.Soultanas P, Wigley DB. Unwinding the ‘Gordian knot’ of helicase action. Trends Biochem. Sci. 2001;26:47–54. doi: 10.1016/s0968-0004(00)01734-5. [DOI] [PubMed] [Google Scholar]
- 33.Konieczny I. Strategies for helicase recruitment and loading in bacteria. EMBO Rep. 2003;4:37–41. doi: 10.1038/sj.embor.embor703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 35.Caruthers JM, Mckay DB. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.