

Abstract
Biological pacemakers were recently created by genetic suppression of inward rectifier potassium current, IK1, in guinea pig ventricular cells. We simulated these cells by adjusting IK1 conductance in the Luo-Rudy model of the guinea pig ventricular myocyte. After 81% IK1 suppression, the simulated cell reached steady state with pacemaker period of 594 ms. Pacemaking current is carried by the Na+-Ca2+ exchanger, INaCa, which depends on the intracellular calcium concentration [Ca2+]i. This [Ca2+ ]i dependence suggests responsiveness (increase in rate) to β-adrenergic stimulation (βAS), as observed experimentally. Simulations of βAS demonstrate such responsiveness, which depends on INaCa expression. However, a simultaneous βAS-mediated increase in the slow delayed rectifier, IKs, limits βAS sensitivity.
Keywords: pacemaker, arrhythmias, ion channels, gene therapy
Recent experiments demonstrate that cardiac biological pacemakers (BPs) can be created by genetic suppression of inward rectifier potassium current (IK1) in guinea pig ventricular myocytes.1 A potential advantage of this approach, as a therapeutic alternative to electronic pacemaking, is possible responsiveness to regulatory inputs, eg, β-adrenergic stimulation (βAS).
To advance this technology, it is important to understand the BP pacemaking mechanism. In the present study, we demonstrate that Na+-Ca2+ exchanger (INaCa) is the pacemaker current and explore BP responsiveness to βAS.
Materials and Methods
The Luo-Rudy (LRd) guinea pig ventricular myocyte model2 was used to investigate BP pacemaking. Two IK1 suppression levels (81% and 100%) and INaCa expression levels (control2 and 100% increase) were simulated. βAS effects were simulated3 based on experimental observations. Abbreviations are defined in the Figure legend.
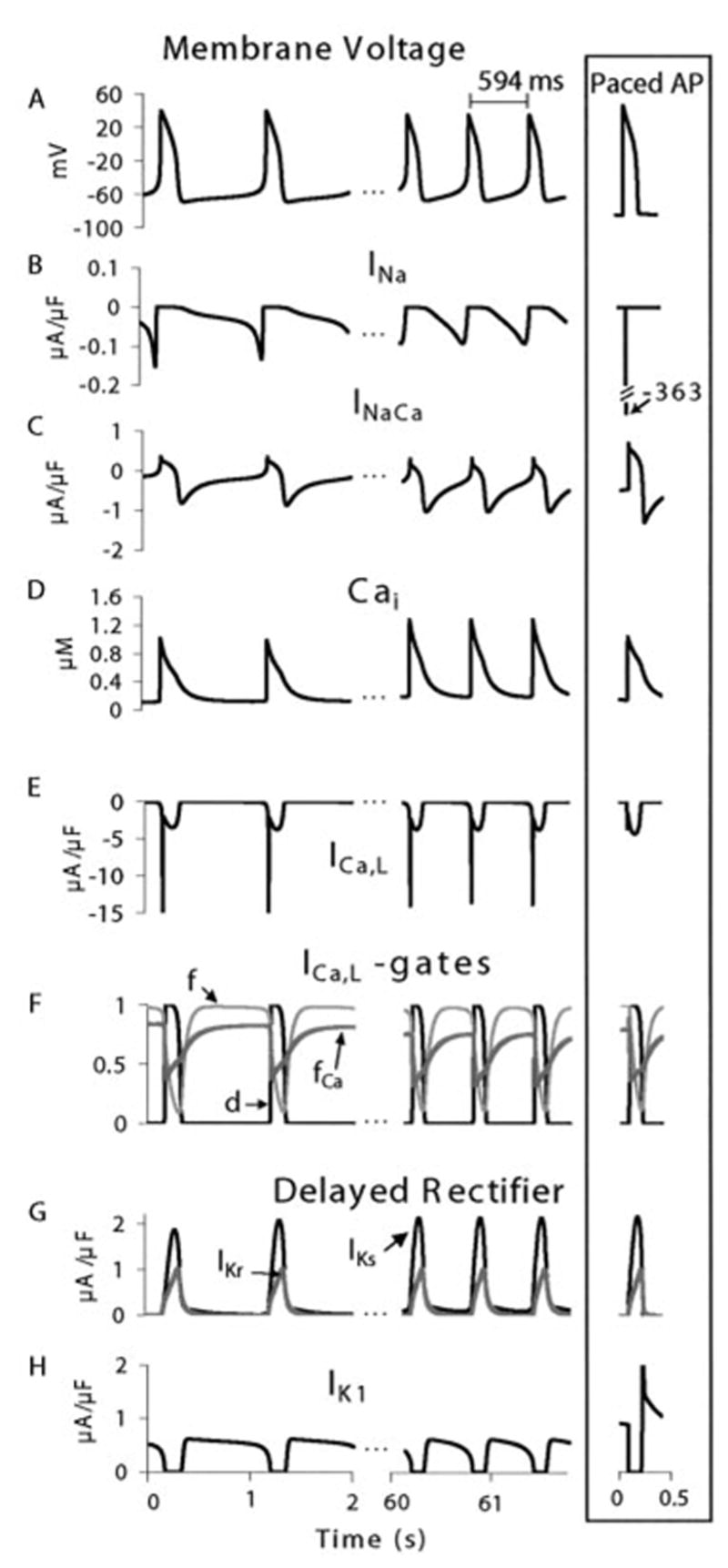
Selected processes during spontaneous initiation (first two APs) and steady-state oscillations in BP cells. IK1 suppressed by 81%. Paced AP (same CL) is shown for reference (framed right column). A, Membrane potential, Vm. B, Sodium current, INa. C, Na+-Ca2+ exchange current, INaCa. D, Intracellular calcium, [Ca2+]i. E, L-type calcium current, ICa,L. F, ICa,L gating: activation gate (d), voltage-dependent inactivation (f), and calcium-dependent inactivation (fCa). G, Fast delayed rectifier K+ current, IKr, and slow delayed rectifier K+ current, IKs. H, Inward rectifier potassium current, IK1.
An expanded Materials and Methods section can be found in the online data supplement available at http://www.circresaha.org.
Results
After 81% IK1 suppression, we observe spontaneous action potentials (APs) that, after a 16-second transition, settle into a stable oscillatory pattern (Figure, panel A). Activity is initiated by slow depolarization generated by sodium and calcium leakage (background currents) and INaCa that extrudes calcium to maintain homeostasis at rest.2 In unmodified cells (intact IK1), these inward currents are balanced by outward IK1 and resting Vm is stable.4 In the BP cell, when Vm reaches −60 mV, INa activates and increases depolarization rate. Peak INa is two orders of magnitude smaller than that of a paced AP2 due to inactivation during the slow depolarization (Figure, panel B). INa and initial activation of ICa,L continue depolarizing Vm as INaCa decreases (higher Vm reduces its driving force). T-type calcium current (ICa,T) does not contribute because of inactivation during the slow depolarization (in ventricular myocytes these channels are unavailable at potentials above −65 mV).5 Once ICa,L is fully activated, it supports the subsequent upstroke and plateau of the AP (Figure, panel E). dVm/dtmax corresponds to peak ICa,L and is much smaller than that of INa-dependent paced APs (15 V/s versus 388 V/s).2 As IKr and IKs repolarize Vm, INaCa driving force increases, causing larger inward current (Figure, panel C). At −67.8 mV (maximum diastolic potential, MDP, Figure, panel A), outward IKr, IKs, and the suppressed IK1 (Figure, panel H) do not balance inward INaCa and background currents. This imbalance causes slow phase-4 depolarization (φ4d) that leads to generation of a subsequent AP and continuous pacemaking.
Pacemaking mechanism remains similar during steady state. While removing residual Ca2+ from calcium-induced calcium release (CICR) of the previous AP, INaCa generates inward current that, in absence of balancing IK1, depolarizes Vm to AP threshold. During sustained oscillations, there is higher [Ca2+]i due to loading (Figure, panel D). Increased [Ca2+]i affects rate by augmenting forward-mode INaCa (inward current) during φ4d (Figure, panel C), which accelerates depolarization (Figure, panel A). At the end of φ4d, as INaCa decreases, INa transiently increases and depolarizes Vm to threshold for ICa,L activation, which generates the AP upstroke. At the end of the AP (beginning of φ4d), IKs is still partially activated (Figure, panel G) and is important in determining MDP and rate of early φ4d. IK1 expression also affects the rate of φ4d; 81% IK1 suppression results in oscillations at cycle length (CL) of 594 ms (Figure, panel A), and complete IK1 suppression leads to much faster rate (CL = 366 ms, not shown).
Changes in pacemaker rate under βAS are investigated by modifying AP currents according to their experimental response to β-agonists (see Reference 3 in the online data supplement). Enhanced Iup (Ca2+ uptake by the sarcoplasmic reticulum, SR), ICa,L, or INaK (the Na+-K+ pump) accelerates rate. Increased IKs or negative shift of INa inactivation decreases rate. The Table provides data for individual protocols and their combined effect. Increasing Iup (110%) in the control BP cell (81% IK1 suppression) results in a 24% rate increase. This increase corresponds to SR loading and increased [Ca2+]i that augments forward INaCa. Similarly, increasing ICa,L (300%) increases rate (11%) by augmenting [Ca2+]i and INaCa. This increase occurs despite ICa,L-mediated increase in AP duration (APD), which decreases rate. Contrary to expectation, increasing INaK (an outward current) also increases rate. Augmenting INaK increases the sodium gradient, which increases INa and INaCa by increasing their driving force, accelerating φ4d. Because of INa participation during φ4d, negative shift of its inactivation decreases rate. This effect abolishes INa in BP cells, in contrast to cells with intact IK1 where INa is only reduced by 14%. IKs increase by βAS decreases APD, which accelerates rate. However, it also hyperpolarizes Vm to a more negative MDP, which prolongs φ4d to the next AP threshold. The net effect is slowing of rate (13%), indicating that effects on MDP dominate APD changes. The overall effect of βAS on rate (only 4% increase) is very small, indicating low BP sensitivity to βAS. However, the simulated control BP cell is epicardial,2 which expresses relatively low INaCa density (average midmyocardial is 50% higher).6 When INaCa density is increased 100% (estimated upper limit), βAS causes a 24% rate increase (Table). Note that all other model parameters were kept constant, to study the isolated effect of INaCa expression. We conclude that the βAS sensitivity of BP cells depends strongly on INaCa expression levels.
BP cells show increased [Ca2+]i at steady state compared with paced cells at the same CL (1.22 and 0.94 μmol/L, respectively, Figure, panel D). [Ca2+]i is further increased by βAS and by rate increases, which could cause calcium overload. We test this possibility by applying βAS to a rapidly paced cell (IK1 fully suppressed; control INaCa) and comparing [Ca2+]i to that of a slower BP cell (81% IK1 suppression; control INaCa) without βAS, finding 85% increase in peak [Ca2+]i (from 1.22 to 2.25 μmol/L). This result suggests that, from this perspective, increasing INaCa expression would be a preferred method of increasing βAS sensitivity, because of enhanced calcium removal capacity and protection against calcium overload.
The modulatory role of calcium in pacemaking suggests that ICa,L antagonists may overly suppress pacemaking in BP cells. We simulate this effect by 50% ICa,L block and observe 18.7% decrease in CL, in the range observed for similar block in sinoatrial node (SAN) cells.7
Discussion
In a recent study,1 viral gene transfer was used to convert quiescent myocardial cells into pacemaker cells. With ~80% of IK1 channels suppressed, these cells generated a rhythmic excitation at an intrinsic CL of 600 ms. The spontaneous APs were initiated by slow φ4d from MDP of −60.7 ± 2.1 mV.
Similar behavior is observed in the computer simulations; when IK1 is suppressed by 81%, stable oscillatory behavior is attained. Slow φ4d from MDP of −67.3 mV sustains rhythmic excitation at a CL of 594 ms. Complete IK1 suppression increases the rate to a CL of 366 ms, implying that altering IK1 expression levels could be used to set intrinsic BP cell pacemaker rate.
The simulations identify INaCa as the regulated membrane process responsible for φ4d and pacemaking. Large IK1 conductance determines resting Vm, which is close to K+ reversal potential. When IK1 is suppressed, the steady-state balance between inward and outward currents shifts in the inward direction. The most important currents at this phase are involved with [Ca2+]i homeostasis: calcium leakage that brings calcium into the cell and INaCa that extrudes calcium. These inward currents depolarize Vm to initiate a spontaneous AP. After this AP, residual [Ca2+]i from CICR determines the magnitude of INaCa and consequently the rate of diastolic depolarization. At steady state, CICR (triggered by ICa,L) generates similar [Ca2+]i transients every beat, resulting in a similar φ4d rate between APs and stable pacemaking at constant rate. This mechanism differs from spontaneous activity where spontaneous SR calcium release, an irregular process, underlies AP generation.8 This distinction is essential to the regular rhythm generated by BP cells, a prerequisite for any functional pacemaker.
An important determinant of INaCa is [Ca2+]i, which enhances its forward mode. This property links BP rhythm to βAS and may explain why it accelerates with isoprenaline.1 However, because of simultaneous βAS-mediated IKs increase and INa reduction, our simulations suggest that BP responsiveness to βAS is very limited (quantitative data are not provided in Reference 1) and strongly depends on INaCa expression. For simulated high INaCa density, βAS increases the rate by 24%. For comparison, 115% increase is observed in isolated SAN cells,9 indicating much greater responsiveness to βAS.
The role of [Ca2+]i in modulating CL suggests further experimental investigation. βAS can cause excessive Ca2+ SR loading and spontaneous release during φ4d, interrupting the regular rhythm. At fast rates with strong βAS, simulated peak [Ca2+]i increases 85% compared with only 50% for SAN cells.9 Therefore, experiments examining a range of βAS are required to determine [Ca2+]i overload levels and likelihood of arrhythmic APs. Ca2+ overload is also likely to induce long-term electrophysiological remodeling, which should be considered. Additionally, the model prediction that ICa,L antagonists will have similar effects in BP and SAN cells should be confirmed.
There are other mechanistic differences between BP and SAN cells. BP cells rely on a single dominant membrane process, INaCa, as the carrier of pacemaker current causing φ4d. Nodal cells rely on several depolarizing currents for φ4d and pacemaking. These include ICa,T, If (the hyperpolarization-activated current), INaCa, ICa,L, and possibly Ist (a sustained inward current, see review10). This multiplicity provides many control points for pacemaking regulation by various (neural and other) inputs. As suggested,10 this multiplicity underlies spatial heterogeneity within the SAN structure, which may be important for its function. In addition, IK,Ach, an acetylcholine-sensitive current not detected in ventricular myocytes, provides vagal control of SAN rate. Finally, SAN ability to drive the heart depends on its architecture (gap-junction distribution; branching fibers), which facilitates optimization of its electrical loading by the surrounding atrial tissue. Therefore, it should be recognized that the engineering of single BP cells is only a first step toward creation of functional BP complexes.
Supplementary Material
Relationship Between Pacemaking Rate and Currents Affected by βAS
| CL, ms | Frequency, bpm | Percent Change | |
|---|---|---|---|
| Control (81% IK1 suppression) | 594 | 101 | 0 |
| Iup increase (110%) | 480 | 125 | 24 |
| ICa,L increase (300%) | 548 | 109 | 8 |
| INaK pump increase (20%) | 549 | 109 | 8 |
| IKs increase (60%) | 682 | 88 | −13 |
| INa shift of inactivation (33.4 mV) | 636 | 94 | −7 |
| Total βAS (all of the above) | 570 | 104 | 4 |
| INaCa increase (100%) | 567 | 106 | 5 |
| βAS with INaCa increase (100%) | 481 | 125 | 24 |
Results of modulating each individual process are shown followed by their cumulative effect (total βAS). Total βAS is also simulated with increased INaCa expression (bottom row), showing dramatic increase in sensitivity. Changes are relative to control.
Acknowledgments
This work was supported by NIH grants R01-HL49054 and R37-HL33343 (to Y.R.) and F31-HL68318 (to J.S.).
Footnotes
This manuscript was sent to Michael R. Rosen, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
References
- 1.Miake J, Marbán E, Nuss HB. Gene therapy: biological pacemaker created by gene transfer. Nature. 2002;419:132–133. doi: 10.1038/419132b. [DOI] [PubMed] [Google Scholar]
- 2.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential, I: simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 3.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hund TJ, Kucera JP, Otani NF, Rudy Y. Ionic charge conservation and long-term steady state in the Luo-Rudy dynamic cell model. Biophys J. 2001;81:3324–3331. doi: 10.1016/S0006-3495(01)75965-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heubach JF, Kohler A, Wettwer E, Ravens U. T-type and tetrodotoxin-sensitive Ca2+ currents coexist in guinea pig ventricular myocytes and are both blocked by mibefradil. Circ Res. 2000;86:628–635. doi: 10.1161/01.res.86.6.628. [DOI] [PubMed] [Google Scholar]
- 6.Zygmunt AC, Goodrow RJ, Antzelevitch C. INaCa contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]
- 7.Masumiya H, Tanaka H, Shigenobu K. Effects of Ca2+ channel antagonists on sinus node: prolongation of late phase 4 depolarization by efonidipine. Eur J Pharmacol. 1997;335:15–21. doi: 10.1016/s0014-2999(97)01150-3. [DOI] [PubMed] [Google Scholar]
- 8.Capogrossi MC, Houser SR, Bahinski A, Lakatta EG. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res. 1987;61:498–503. doi: 10.1161/01.res.61.4.498. [DOI] [PubMed] [Google Scholar]
- 9.Rigg L, Heath BM, Cui Y, Terrar DA. Localisation and functional significance of ryanodine receptors during β-adrenoceptor stimulation in the guinea-pig sinoatrial node. Cardiovasc Res. 2000;48:254–264. doi: 10.1016/s0008-6363(00)00153-x. [DOI] [PubMed] [Google Scholar]
- 10.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. 2000;47:658–687. doi: 10.1016/s0008-6363(00)00135-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.