

Abstract
The recognition of palindromic specific DNA sequences by the human papillomavirus (HPV) E2 proteins is responsible for regulation of virus transcription. The dimeric E2 DNA-binding domain of HPV-16 (E2c) dissociates into a partially folded state under high hydrostatic pressure. We show here that pressure-induced monomers of E2c are highly structured, as evidenced by NMR hydrogen–deuterium exchange measurements. On binding to both specific and nonspecific DNA, E2c becomes stable against pressure. Competitive binding studies using fluorescence polarization of fluorescein-labeled DNA demonstrate the reversibility of the specific binding. To assess the thermodynamic parameters for the linkage between protein dissociation and DNA binding, urea denaturation curves were obtained at different pressures in the presence of specific and nonspecific DNA sequences. The change in free energy on denaturation fell linearly with increase in pressure for both protein–DNA complexes, and the measured volume change was similar to that obtained for E2c alone. The data show that the free energy of dissociation increases when E2c binds to a nonspecific DNA sequence but increases even more when the protein binds to the specific DNA sequence. Thus, specific complexes are tighter but do not entail variation in the volume change. The thermodynamic data indicate that DNA-bound E2c dissociates into monomers bound to DNA. The existence of monomeric units of E2c bound to DNA may have implications for the formation of DNA loops, as an additional target for viral and host factors binding to the loosely associated dimer of the N-terminal module of the E2 protein.
Human papillomaviruses (HPVs) cause warts and proliferative lesions in epidermal tissues. Infection of the anogenital tract by this virus is associated with several premalignant and malignant lesions, especially dysplasia and carcinoma of the uterine cervix (1). The HPV strains 16, 18, 31, 33, and 45 are the most dangerous, prone to causing lesions that evolve into tumors. The products of the E2 gene are crucial to the life cycle of the virus because they regulate transcription from all viral promoters (2, 3), which makes E2 protein a potential target for antiviral therapy. The E2 protein is comprised of an N-terminal transactivation domain (E2n) separated from the C-terminal DNA-binding and dimerization domain (E2c) by a flexible region rich in proline residues (3). The solution structure of the E2c module from HPV-31 was determined by NMR spectroscopy (4) and the crystal structure of the E2c from HPV-16 was recently determined (5). The structure of the E2n module of HPV16 (6) and of a proteolytic fragment of HPV 18 E2n (7) also were recently solved. The structural biology of E2, together with folding, dimerization, and DNA-binding data reveal an elegant protein–DNA complex, in which dimerization occurs at both the E2c and E2n domains, giving rise to the potential formation of a DNA loop (6). Earlier studies have shown that intact E2 proteins build into loops on DNA with the E2-binding sites hundreds of bases apart (8). Whereas the binding of cellular and viral proteins may regulate E2n dimerization, the dimerization of E2c is modulated by the specific sequence of DNA.
Protein–nucleic acid interactions frame the basis for the regulation of key biological functions such as transcription, translation, replication, gene regulation, virus assembly, and recombination. Structural data have demonstrated that the reading of a DNA sequence is achieved by a combination of van der Waals, electrostatic, and hydrogen bonds (9–12). Structural studies alone cannot separate the contribution of each of these factors. Cooperative interactions among protein subunits play an important role in the binding to the specific DNA sites (9–13). Deciphering the interactions among the players (different domains, DNA, and binding proteins) requires biophysical studies in addition to structure. Noncovalent interactions can be perturbed reversibly by using high hydrostatic pressure, which allows protein folding, protein–protein, and protein–ligand interactions to be characterized thermodynamically (14–17). Protein folding and protein–protein interactions result in volume increase because of the combined effects of the formation of solvent-excluding cavities and the release of bound solvent (17–19).
In a previous study, we found that E2c dimers dissociate into partially folded monomers as evidenced by fluorescence spectroscopy (20). Here, we study the free-energy linkage between dimerization and DNA binding of E2c by using high pressure and urea to drive the equilibrium toward the dissociated species. We find that the monomers are highly folded as evidenced by hydrogen–deuterium exchange measurements using NMR. We also demonstrate that the free energy of dissociation increases when E2c binds to a nonspecific DNA sequence, but increases even more when the protein binds to the specific DNA sequence. Specific complexes are tighter but do not elicit variation in the volume change, which means that there is no further burying of surface area in moving from a nonspecific to a specific mode of binding. Our data also indicate that DNA-bound E2c dissociates into monomers bound to DNA.
Experimental Procedures
Chemicals.
All reagents were of analytical grade. Distilled water was deionized and filtered through a Millipore water purification system before use. Urea was purchased from Sigma and bis-ANS (4,4′-dianilino-1,1′binaphthyl-5,5′-disulfonic acid) from Molecular Probes. The urea stock solution was prepared just before use and its concentration was checked by refractive index (21).
E2c Expression and Purification.
The C-terminal 80-aa DNA-binding domain of HPV-16 E2 protein (E2c) was overexpressed in Escherichia coli and purified according to Mok et al. (22). Protein concentration was determined by using the extinction coefficient of 41,900 M−1⋅cm−1 at 280 nm (23).
Synthetic Oligonucleotides.
The single-stranded synthetic oligonucleotides, HPLC-purified, were purchased from Integrated DNA Technologies (Coralville, IA). Unlabeled and fluorescein-labeled double-stranded 18-bp oligonucleotides containing one E2 recognition sequence were prepared: E2DBSssA (5′-GTAACCGAAATCGGTTGA-3′) and its complementary strand with a fluorescein molecule attached to the 5′ end via a six-carbon linker (F-E2DBS). Complementary oligonucleotides were annealed as previously described (23). The same procedure was used to prepare the nonspecific double-stranded poly(A-T) (5′-ATA TAT ATA TAT ATA TAT-3′).
Fluorescence Measurements.
The high-pressure bomb has been described (24) and was purchased from ISS (Champaign, IL). The urea unfolding of E2c at high pressures was carried out at 25°C in 50 mM Bis-Tris⋅HCl containing 1 mM DTT, pH 5.5, in the presence of the indicated concentrations of urea. After preincubation in urea for 2 h, unfolding under pressure was monitored by using the shift in tryptophan fluorescence spectra recorded on an ISSPC1 spectrofluorometer (ISS), with excitation at 276 nm and emission scanned from 300 nm to 450 nm. Fluorescence polarization was measured in the “L” geometry with the excitation set to 475 nm and emission recorded through a Corning 3–69 filter (50% cut-off at 520 nm).
Tryptophan Fluorescence Lifetime and Rotation Measurements.
Fluorescence lifetime and dynamic depolarization measurements were performed on a multifrequency cross-correlation phase and modulation fluorimeter which uses the harmonic content of a high-repetition-rate, mode-locked Nd–aluminum-garnet laser to pump a dye laser (25). The quality of fits was assessed by using χ2 values and plots of weighted residuals (25, 26).
Analysis of the Data by Using a Two-State Model.
In this model we consider only native dimer (N) and denatured monomer (D) in the equilibrium reaction (N2 ⇋ 2D). The equilibrium constant for denaturation at pressure p (Kp) and at atmospheric pressure (Kdo) (14, 15):
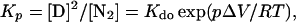 |
1 |
where [N2] and [D] are, respectively, the concentrations of the dimer and the monomer at pressure p and ΔV is the standard volume change on association. The total protein concentration, Pt, is expressed in terms of the monomer. Fluorescence spectra at pressure p were quantified by the center of spectral mass 〈νp〉:
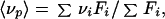 |
2 |
where Fi stands for the fluorescence emitted at wavenumber νi, and the summation is carried out over the range of appreciable values of F. The extent of reaction at pressure p (αp) is related to 〈νp〉 by the expression,
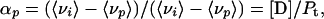 |
3 |
where 〈νi〉 and 〈νf〉 are the initial and final values of the center of spectral mass, respectively, and 〈νp〉 is the center of spectral mass at pressure p.
The Gibbs free energy change at pressure p (ΔGp) for a monomer–dimer association equilibrium can be calculated from Eq. 4.
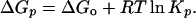 |
4 |
Combination of Eqs. 1 and 3 gives the quadratic equation 2αp2Pt + Kpαp − Kp = 0, which can be solved for αp as:
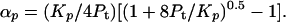 |
5 |
Combining Eqs. 1, 3, 4, and 5 gives:
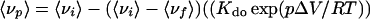 |
6 |
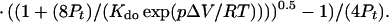 |
Similarly, we can deduce the following general equation for this process by combining the Eqs. 1 and 3,
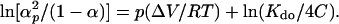 |
7 |
From this equation, one may calculate the standard volume change and the Kdo at total protein concentration C expressed as dimer.
Analysis of the urea-induced denaturation curves was carried out as described in detail (22, 27). All thermodynamic parameters were obtained by fitting the following equation to the data:
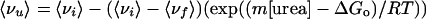 |
8 |
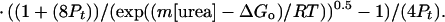 |
in which m is a proportionality constant.
NMR.
NMR spectra were obtained in a Bruker 600-MHz spectrometer at 25°C. The sample was prepared in 10% D2O by using 10 mM Bis-Tris⋅HCl and 1 mM DTT at pH 5.5. Protein concentration was 100 μM dimer. Water suppression was achieved by using the watergate sequence (28) with the composite pulse 3, 9, 19 and a 1-ms, z-pulsed field gradient at 10 G/cm. The urea peak was presaturated.
Results
Pressure Dissociation of E2c to Partially Folded States.
We have used high hydrostatic pressure to cause the dissociation and denaturation of the E2c protein. In our previous study (20), we were puzzled by the finding that the protein dissociates into a partially folded monomer despite the expected dismantling of the β–barrel interface. We determined pressure isotherms for E2c alone (Fig. 1, circles) and in the presence of specific and nonspecific DNA (Fig. 1, squares and triangles, respectively), in which changes in the center of spectral mass of Trp fluorescence are followed. HPV-16 E2c has three residues per monomer, one exposed to the solvent and the other two located at the interface between subunits, making them excellent probes for association and folding (20). The equilibrium dissociation constant at atmospheric pressure for E2c alone was determined for the two-state approach by adjusting Eq. 6 for these experimental data. We obtained a Kd of 17 nM and a volume change of 87.9 ml/mol (Fig. 1, circles; Table 1), which is in very good agreement with the results previously demonstrated for pressure dissociation (20) and urea denaturation (22, 23).
Figure 1.

Effects of specific and nonspecific DNA binding on pressure-induced dissociation of E2c. Spectral shift was measured as a function of hydrostatic pressure applied to 1.0 μM E2c alone (●), complexed to a specific DNA sequence (▪), E2-DBS (0.25 μM dimer–DNA complex), or to a nonspecific DNA sequence (▴), poly(A-T) 18-mer (0.25 μM dimer–DNA complex). The experiment was performed as described in Experimental Procedures. The solid line for dissociation of E2c alone represents the fitting obtained with Eq. 6. The lines represent second-order linear regressions. 1 atm = 0.101325 MPa.
Table 1.
Effects of DNA binding on pressure-induced dissociation of E2c
We previously found that E2c monomers obtained by pressure were only partially unfolded as evidenced by different fluorescence techniques (20). The use of high-resolution NMR has been important in characterizing pressure-denatured proteins (16, 29–31). To appraise the retention of substantial secondary and tertiary structures in the pressure-induced monomers, proton–deuterium (H/D) exchange experiments under pressure were evaluated by using high-resolution NMR (Fig. 2). Fig. 2 Bottom shows that a high urea concentration resulted in a large extent of proton–deuterium exchange, consistent with substantial unfolding of the monomers. In contrast, the H/D exchange of the amide protons in the pressure-dissociated monomers (Fig. 2 Middle) was similar to that obtained for the control E2c dimer (Fig. 2 Top). Most of the amide protons retained a high protection factor in the pressure-dissociated state, and several of these peaks belong to β-sheet and α-helical regions. In contrast, most of the amide protons became labile when the protein was denatured by high concentrations of urea.
Figure 2.

One-dimensional NMR spectra of E2c in several conditions after solubilization in 100% D2O. Comparison made after 30 min at atmospheric pressure in the absence of urea (Top) and after 30 min at 3 kbar (1 kbar = 100 MPa) in the absence of urea (Middle) shows that at high pressure there is considerable secondary structure as indicated by the slowly exchanging amide resonance peaks between 8 and 10 ppm. The spectrum in the presence of 5 M urea (Bottom) shows that the amide resonances are completely exchanged after 10 min.
According to the rate constant at pH 5.5 (k = 0.53 s−1) for unprotected protons (32), it would take ≈1.3 s to exchange 50% of the amide protons when in an open state (not making hydrogen bonds). We can conclude that the conformational changes that accompany dissociation into monomers are shorter events resulting in dimeric and monomeric structures in fast equilibrium (t ≪ 1.5 s).
Stabilization by DNA.
E2c from different papillomavirus strains binds to a single ACCG-N4-CGGT palindromic consensus with the equilibrium constant values ranging from 7 × 10−11 M to 4 × 10−10 M for gel-shift assays (33, 34) and from 2 × 10−9 M to 5 × 10−7 M for solution measurements (35).
Understanding the way in which a protein recognizes its specific DNA base sequence is crucial for understanding the basis of the molecular mechanism for the action of transcription factors and other molecular events involved in DNA processing. With this goal, we attempted to assess the contribution of specific and nonspecific DNA binding to the stability of the E2c dimer. It has been demonstrated that high hydrostatic pressure can be used to study protein–DNA complexes (13, 36–39). Interestingly, pressures up to 3,000 atm promoted almost no shift in the average energy of E2c intrinsic fluorescence emission, regardless of whether E2c was bound to a highly specific double-stranded DNA sequence, E2-DBS 18-mer, or to a nonspecific double-stranded DNA, poly(A-T) 18-mer (Fig. 1, squares and triangles, respectively). Foguel and Silva (38) have demonstrated that the P-22 Arc repressor–DNA complex can be cold-denatured at subzero temperatures under pressure. However, under the same conditions (−15°C and 2,500 atm) we did not detect any significant change in the fluorescence emission for either of the E2c–DNA complexes (data not shown).
E2c–DNA Complex Formation and Dissociation.
To verify whether DNA can dissociate from E2c in the time frame of our experiments, we performed DNA exchange experiments by using fluorescence polarization. Because F-E2DBS fluorescence polarization varies as a direct function of E2c binding, it can be used to monitor the kinetics of binding and dissociation (36, 40).
A fluorescence polarization baseline was obtained using 10 nM F-E2DBS alone. When E2c (10 nM final concentration) was added, there was an immediate increase in fluorescence polarization, indicating a rapid E2c–DNA complex formation (Fig. 3A). Ten minutes later, dissociation was measured by adding a 100-fold excess (1 μM) of unlabeled E2DBS and monitoring the decrease in polarization due to an exchange between labeled and unlabeled DNA. A decrease in polarization to almost the same as in the beginning of the experiment occurred, indicating the reversibility and relatively fast equilibrium of the E2c–DNA complex formation. The specificity of E2c for the E2DBS was confirmed by performing a similar experiment, using an excess of unlabeled poly(A-T) instead of unlabeled E2DBS (Fig. 3B). As expected, the poly(A-T) was not able to displace the E2c–E2DBS complex. The full exchange observed for the specific sequence indicates that the reaction protein–DNA is at equilibrium. These results indicate that pressure dissociation of the E2c–DNA complex was not detected in the experiment of Fig. 1 because the effects were on the thermodynamic potentials (either ΔG or ΔV) rather then on the kinetics of the reaction.
Figure 3.

Off-rate analysis for E2c–E2-DBS complex. The binding of E2c to fluorescein-labeled F-E2-DBS (10 nM double stranded) was measured as a function of the increasing in fluorescence polarization. After ≈10 min of equilibration, 10 nM E2c was added to DNA. After ≈10 min of incubation, the dissociation process was analyzed as a function of time on a continuous measurement of fluorescence polarization, following the addition of a 100-fold excess (for a final concentration of 1 μM double-stranded DNA) of unlabeled competitor: (A) E2-DBS; (B) poly(A-T) 18-mer. Experiments were carried out in 50 mM Bis-Tris⋅HCl/1 mM DTT/200 mM NaCl (pH 7.0) at 22°C. In all cases, maximal dilution was 1.5%.
Urea Denaturation Under Pressure.
Pressure and urea studies on the stability of E2c provide equivalent thermodynamic parameters (20, 22, 23). Because we could not induce dissociation of E2c–DNA complexes by using pressure alone (Fig. 1, squares and triangles), we repeated the experiments with different amounts of urea added to the buffer (Fig. 4 A and B). Each sample was prepared in buffer containing the indicated concentration of urea and allowed to equilibrate at atmospheric pressure for 2 h and then submitted to increasing variation in pressure. From these curves we were not able to calculate either the Kd or the ΔV for complex dissociation. However, when these results are plotted as urea denaturation curves at different pressures, we obtained the curves in Fig. 4C (specific DNA) and D (nonspecific DNA). We can obtain the free-energy change for dissociation and denaturation of E2c (ΔGo) at each pressure by adjusting Eq. 8 to experimental data. A plot of ΔGo versus pressure (Fig. 5A) is linear for both the E2c complexes of E2c dimer with slopes that provide the volume change (ΔV) that accompanies the dimer dissociation and denaturation, and intercepts that give the free energy at atmospheric pressure. It should be pointed out that the changes produced by pressure and/or urea were completely reversible.
Figure 4.

Urea dependence of E2c spectral changes induced by high pressure.
High hydrostatic pressure was applied to samples containing 0.25 μM
(E2c–DNA) complex in the standard buffer at 25°C in the absence of
urea (●) or in the presence of: (A) 1.5 M
(○), 3 M (▪), 3.8 M (□), 4.5 M
(▴), 6.5 M (▵), or 8.1 M (▾) urea;
(B) 0.5 M (○), 1.0 M
(▪), 2.0 M (□), 3.0 M (▴), 3.8 M
(▵), 6.0 M (▾), or 8.0 M (▿) urea.
(A) E2-DBS; (B) poly(A-T) 18-mer. C
and D are replots of A and B,
respectively, as urea unfolding under different pressures: 408
(●), 610 (○), 815 (█), 1020
(□), 1220 (▴), 1430 (▵), 1630
(▾), 1840 (▿), 2040 (⧫), 2240 (⋄),
2450 ( ),
or 2653
(
),
or 2653
( ) atm.
The curves were fitted for Eq. 8.
) atm.
The curves were fitted for Eq. 8.
Figure 5.

(A) Changes in E2c free energy as a function of pressure. Plot of ΔGo obtained from Fig. 4 (see text) vs. pressure for E2c–E2-DBS (●) and for E2c–poly(A-T) 18-mer (█). The extent of dissociation of E2c alone was calculated as described under Experimental Procedures (Eq. 7) for data from Fig. 1, converted to ΔGo values and plotted as a function of pressure (▴). (B) Volume vs. free energy diagram. Free energy levels of the relation between specific DNA binding and monomer association. D represents the double-stranded specific DNA; E represents an E2c monomer unit, which does not correspond necessarily to the folded monomer in the associated state; (E)2 represents the E2c in the dimer state; (E)2D represents the complex obtained from the association between one E2c dimer and a double-stranded DNA containing one DNA-binding site. (C) Scheme for subunit dissociation of E2c bound to DNA. In the upper situation, the monomers dissociate and remain bound. In the lower situation, there is dissociation from DNA as well. The hatched symbols correspond to void volumes.
The primary effect of the DNA was to stabilize the E2c subunit interaction (Fig. 5 and Table 1). Both specific and nonspecific DNA sequences promoted a large stabilization of E2c dimer when compared with the protein alone, with a higher change in free energy for the specific DNA sequence (ΔΔGo = 6.7 kcal/mol) than for the nonspecific DNA (ΔΔGo = 4.5 kcal/mol).
Dynamics of the E2c–DNA Complex.
The increase in stability of E2c on DNA binding was not accompanied by significant changes in thermodynamic volume (Table 1). To investigate the dynamic properties of the free and DNA-bound proteins at pH 5.5, we carried out Trp lifetime (25, 26, 41) and rotation measurements (Table 2). For E2c alone, the best fit for the data are achieved by using a Lorentzian distribution of lifetimes centered at 1.70 ns rather than a single exponential decay. For the DNA-bound state, the distribution shifts to longer lifetimes, centered at 2.60 ns (Table 2). This difference between the free and DNA-bound states indicates that the phase space explored by the Trp is different. That longer Trp lifetimes are found for the E2c–DNA complex suggests that a conformation-dependent dequenching takes place, likely to reduction in solvent exposure.
Table 2.
Effects of DNA binding on Trp lifetime and rotation
| Lifetime,
ns
|
Rotational correlation time (θ), ns
|
||||||
|---|---|---|---|---|---|---|---|
| Center | Width | Average θ | θ1 | f1* | θ2 | f2* | |
| E2c | 1.70 | 0.85 | 4.4 | 9.2 | 0.14 | 0.18 | 0.16 |
| E2c–DNA | 2.26 | 1.62 | 15.8 | 36.9 | 0.13 | 0.32 | 0.17 |
Excitation wavelength was 295 nm and the emission was observed through a long-wavelength pass filter (WG335) with a cutoff at 335 nm. All measurements were performed in 50 mM Bis-Tris⋅HCl/1 mM DTT (pH 5.5), at 25°C. The protein and DNA concentrations were both 5 μM. The χ2 values for the fittings of lifetime distribution and rotations were typically less than 2.0.
f is the fraction of each rotational correlation time.
Rotations of the Trp residues in E2c dimer (free or bound to DNA) were also evaluated (25, 26, 42). For both free and DNA-bound E2c, the best fit to the data were obtained by using a model that assigns two rotational motions (Table 2), a longer one (θ1) corresponding to the tumbling of the protein or protein–DNA complex and another one (θ2) related to the local motions of the Trp side chains. The longer component of the rotational correlation time increases about 4-fold on formation of the complex and its value (37 ns) is reasonable for a particle containing the E2c dimer (18.9 kDa) tightly bound to the 18-bp DNA.
Discussion and Conclusions
We demonstrate in this article the remarkable stabilization conferred by DNA on the dimerization of E2c and we provide evidence that the difference between the effects produced by specific and nonspecific DNAs is likely the basis for the sequence discrimination. In some DNA-binding proteins, dimerization occurs only on DNA binding (43, 44). However, in most cases, the subunits associate with high affinity in solution in the absence of DNA (9, 10) and a free energy linkage between folding/oligomerization and DNA binding seems to be crucial for the recognition (9–13, 36–38, 45, 46). The stabilization of E2c dimer by specific binding of DNA results in an almost 2-fold increase in the value for the free energy of the protein–protein interactions. This increase in stability is one of the largest ever measured for protein–ligand interaction (47, 48).
Because we measure the dissociation from E2c–DNA to monomers, two possible situations can be visualized (Fig. 5C). The first is that E2c dimers bound to DNA dissociate into free monomers and DNA. The second is the dissociation of E2c dimers into monomers still bound to DNA. The thermodynamic diagrams depicted in Fig. 5B do seem to indicate that the latter case is more likely. Whereas the free energy change increases dramatically on DNA binding, the volume change is not altered. The volume change obtained for dissociation of E2c corresponds to the exposure of surface area in the range 2,800–3,000 Å2. These values correlate well with the calculated values for buried surface area on dimerization determined from the structure of the E2c from bovine papillomavirus-1 (2,567 Å2; ref. 49) and from HPV-16 (1,553 Å2; ref. 5). In fact, the latter value is more realistic because ΔV should encompass the release of cavity (ΔVc; Fig. 5C) as well as the electrostriction effects of the solvent on binding to the exposed surface (ΔVs; ref. 18). The structural calculation takes into account only ΔVc.
The binding of DNA should bury additional surface area as the protein–DNA interface is formed, as well as the formation of electrostatic interactions should also result in an increase of the volume change. However, our data (Fig. 5) show that the presence of DNA does not affect the volume change at all, which indicates that no extra surface area is involved when E2c dissociates on the DNA.
Altogether, our results can be better explained by the dissociation of dimers bound to DNA into monomers still bound to DNA. The finding that monomeric E2c obtained by high pressure is highly structured as evidenced by the hydrogen–deuterium exchange experiment (Fig. 2) reinforces the idea of a monomer competent to bind DNA. Antson et al. (6) recently proposed a model where the dimerization of the E2n modules contributes to the stabilization of DNA loops, serving to relocate distal DNA-binding transcription factors to the site of HPV transcription initiation. Their model explains previous observations that support the formation of DNA loops. In addition to the proposed state of two dimers forming a loop, other states can be envisioned. A multiplicity of situations with movement along the DNA is possible if protein–protein dissociation occurs at the E2n module or at the E2c module. In both cases, the E2c remains bound to DNA either as dimer or monomer. The existence of monomeric units of E2c bound to DNA may have implications for the formation of DNA loops providing an additional source of modulation by viral and host factors binding to the N-terminal module of the E2 protein. The retention of a large amount of tertiary structure by the dissociated monomer as evidenced here corroborates the hypothesis of a functional monomeric unit of E2c.
The change in dimer–monomer dissociation constant on binding to specific DNA corresponds to a decrease of 6.7 kcal/mol in free energy of E2c dimer. In the case of bacteriophage (Arc repressor) and bacterial (LexA) transcription factors, there were large differences between nonspecific and specific DNAs (13, 37, 38). In contrast, the differences between specific and nonspecific DNAs were much smaller for E2c dimer–ΔΔG values equal to 6.7 and 4.5 kcal/mol, respectively (Table 1). The differences between prokaryotic and eukaryotic transcription factors may be related to the more elaborate action of the latter. The difference in free energy between the nonspecific and specific DNA binding is still high (2.2 kcal/mol), and more dramatic is the difference between the free and bound form. The high stability of the DNA-bound form practically precludes the existence of free protein, and the protein has an intrinsic way to distinguish between the nonspecific and specific binding sites. This strategy has the potential for DNA binding to be regulated by other ligands. Equal volume changes obtained for the specific and nonspecific complexes also suggest that there are no changes in hydration states when the protein goes from DNA-bound dimer to DNA-bound monomers.
The overall characteristics of the pressure-dissociated E2c state bound to DNA strongly suggest a folded monomer. The demonstration that a “structured” monomer exists in rapid equilibrium with the folded dimer contributes to the understanding of the switches necessary to turn on and off the transcription functions of the E2 protein. High-resolution NMR studies should reveal the atomic structure of the monomer, opening new avenues to the development of drugs targeted to the monomer that will make it possible to prevent or treat HPV infection.
Acknowledgments
We thank Emerson R. Gonçalves for competent technical assistance; Martha M. Sorenson for critical reading of the manuscript; Fabio Almeida for kind assistance with NMR measurements; Ronaldo Mohana-Borges for lifetime and rotation measurements; and Gonzalo Prat-Gay for early discussion and suggestions. This work was supported in part by an International Grant from the Howard Hughes Medical Institute and by grants from Programa de Núcleos de Excelência (PRONEX), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Fundação de Apoio à Pesquisa do Estado do Rio de Janeiro (FAPERJ) of Brazil (all to J.L.S.). J.L.S. is a Howard Hughes Medical Institute International Researcher.
Abbreviations
- HPV
human papillomavirus
- E2c
E2 C-terminal DNA-binding domain
- E2n
E2 N-terminal transactivation domain
- E2DBS
E2 DNA-binding sequence
- F-E2DBS
fluorescein-labeled E2 DNA-binding sequence
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.250352197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.250352197
References
- 1.Villa L L. Adv Cancer Res. 1997;71:321–341. doi: 10.1016/s0065-230x(08)60102-5. [DOI] [PubMed] [Google Scholar]
- 2.DiMaio D, Neary K. In: Papillomaviruses and Human Cancer. Pfister H, editor. Boca Raton, FL: CRC; 1990. pp. 113–114. [Google Scholar]
- 3.McBride A A, Romanczuk H, Howley P M. J Biol Chem. 1991;266:18411–18414. [PubMed] [Google Scholar]
- 4.Liang H, Petros A M, Meadows R P, Yoon H S, Egan D A, Walter K, Holzman T F, Robins R, Fesik S W. Biochemistry. 1996;35:2095–2103. doi: 10.1021/bi951932w. [DOI] [PubMed] [Google Scholar]
- 5.Hegde R S, Androphy E J. J Mol Biol. 1998;284:1479–1489. doi: 10.1006/jmbi.1998.2260. [DOI] [PubMed] [Google Scholar]
- 6.Antson A A, Burns J E, Moroz O V, Scott D J, Sanders C M, Bronstein I B, Dodson G G, Wilson K S, Maitland N J. Nature (London) 2000;403:805–809. doi: 10.1038/35001638. [DOI] [PubMed] [Google Scholar]
- 7.Harris S F, Botchan M R. Science. 1999;284:1673–1677. doi: 10.1126/science.284.5420.1673. [DOI] [PubMed] [Google Scholar]
- 8.Knight J D, Li R, Botchan M. Proc Natl Acad Sci USA. 1991;88:3204–3208. doi: 10.1073/pnas.88.8.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pabo C O, Sauer R T. Annu Rev Biochem. 1992;61:1053–1095. doi: 10.1146/annurev.bi.61.070192.005201. [DOI] [PubMed] [Google Scholar]
- 10.Luisi B. In: DNA-Protein: Structural Interactions. Lilley D M J, editor. Oxford: IRL; 1995. pp. 1–48. [Google Scholar]
- 11.Smith T L, Sauer R T. J Mol Biol. 1995;249:729–742. doi: 10.1006/jmbi.1995.0332. [DOI] [PubMed] [Google Scholar]
- 12.Holmbeck S M A, Dyson H J, Wright P E. J Mol Biol. 1998;284:533–539. doi: 10.1006/jmbi.1998.2207. [DOI] [PubMed] [Google Scholar]
- 13.Mohana-Borges R, Pacheco A B, Sousa F J, Foguel D, Almeida D F, Silva J L. J Biol Chem. 2000;275:4708–4712. doi: 10.1074/jbc.275.7.4708. [DOI] [PubMed] [Google Scholar]
- 14.Silva J L, Weber G. Annu Rev Phys Chem. 1993;44:89–113. doi: 10.1146/annurev.pc.44.100193.000513. [DOI] [PubMed] [Google Scholar]
- 15.Heremans K, Smeller L. Biochim Biophys Acta. 1998;1386:353–370. doi: 10.1016/s0167-4838(98)00102-2. [DOI] [PubMed] [Google Scholar]
- 16.Jonas J, Jonas A. Annu Rev Biophys Biomol Struct. 1994;23:287–318. doi: 10.1146/annurev.bb.23.060194.001443. [DOI] [PubMed] [Google Scholar]
- 17.Silva J L, Foguel D, Da Poian A T, Prevelige P E. Curr Opin Struct Biol. 1996;6:166–176. doi: 10.1016/s0959-440x(96)80071-6. [DOI] [PubMed] [Google Scholar]
- 18.Frye K J, Royer C A. Protein Sci. 1998;10:2217–2222. doi: 10.1002/pro.5560071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hummer G, Garde S, Garcia A E, Paulaitis M E, Pratt L R. Proc Natl Acad Sci USA. 1998;95:1552–1555. doi: 10.1073/pnas.95.4.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foguel D, Silva J L, Prat-Gay G. J Biol Chem. 1998;273:9050–9057. doi: 10.1074/jbc.273.15.9050. [DOI] [PubMed] [Google Scholar]
- 21.Wolf A V, Brown M G, Prentiss P G. In: CRC Handbook of Chemistry and Physics. 67th Ed. Weast R T, editor. Boca Raton, FL: CRC; 1986–1987. p. D266. [Google Scholar]
- 22.Mok Y-K, Prat-Gay G, Butler P J, Bycroft M. Protein Sci. 1996;5:310–319. doi: 10.1002/pro.5560050215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lima L M T R, Prat-Gay G. J Biol Chem. 1997;272:19295–19303. doi: 10.1074/jbc.272.31.19295. [DOI] [PubMed] [Google Scholar]
- 24.Paladini A A, Weber G. Biochemistry. 1981;20:2587–2593. doi: 10.1021/bi00512a034. [DOI] [PubMed] [Google Scholar]
- 25.Alcala J R, Gratton E, Jameson D M A. Anal Instrum NY. 1985;14:225–250. [Google Scholar]
- 26.Beechem J M, Gratton E, Prendergast F G. In: Topics in Fluorescence Spectroscopy. Lackowicz J R, editor. New York: Plenum; 1991. pp. 241–305. [Google Scholar]
- 27.Pace C N. Methods Enzymol. 1986;131:266–279. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 28.Piotto M, Saudek V, Slenar V. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 29.Peng X, Jonas J, Silva J L. Proc Natl Acad Sci USA. 1993;90:1776–1780. doi: 10.1073/pnas.90.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jonas J, Ballard L, Nash D. Biophys J. 1998;75:445–452. doi: 10.1016/S0006-3495(98)77532-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lassale M W, Yamada H, Akasaka K. J Mol Biol. 2000;298:293–302. doi: 10.1006/jmbi.2000.3659. [DOI] [PubMed] [Google Scholar]
- 32.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. p. 24. [Google Scholar]
- 33.Sanders C, Maitland J. Nucleic Acids Res. 1994;22:4890–4897. doi: 10.1093/nar/22.23.4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monini P, Grossman S R, Pepinsky B, Androphy E J, Laimins L. J Virol. 1991;65:2124–2130. doi: 10.1128/jvi.65.4.2124-2130.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexander K A, Phelps W C. Biochemistry. 1996;35:9864–9872. doi: 10.1021/bi960447d. [DOI] [PubMed] [Google Scholar]
- 36.Royer C A, Chakerian A E, Matthews K S. Biochemistry. 1990;29:4959–4966. doi: 10.1021/bi00472a028. [DOI] [PubMed] [Google Scholar]
- 37.Silva J L, Silveira C F. Protein Sci. 1993;2:945–950. doi: 10.1002/pro.5560020608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foguel D, Silva J L. Proc Natl Acad Sci USA. 1994;91:8244–8247. doi: 10.1073/pnas.91.17.8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson C R, Sligar S G. Proc Natl Acad Sci USA. 1998;95:2186–2191. doi: 10.1073/pnas.95.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lackowicz J R. Principles of Fluorescence Spectroscopy. New York: Plenum; 1999. [Google Scholar]
- 41.Prevelige P, King J, Silva J L. Biophys J. 1994;66:1631–1641. doi: 10.1016/S0006-3495(94)80955-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gratton E, Alcala R J, Marriot G. Biochem Soc Trans. 1986;14:835–838. doi: 10.1042/bst0140835. [DOI] [PubMed] [Google Scholar]
- 43.Luisi B F, Xu W, Otwinowski Z, Freedman L P, Yamamoto K R. Nature (London) 1991;352:497–505. doi: 10.1038/352497a0. [DOI] [PubMed] [Google Scholar]
- 44.Kohler J J, Metallo S J, Schneider T L, Schepartz A. Proc Natl Acad Sci USA. 1999;96:11735–11739. doi: 10.1073/pnas.96.21.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Hippel P H. Science. 1994;263:769–770. doi: 10.1126/science.8303292. [DOI] [PubMed] [Google Scholar]
- 46.Spolar R S, Record M T., Jr Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 47.Weber G. Protein Interactions. New York: Chapman & Hall; 1992. [Google Scholar]
- 48.Van Holde K, Johnson W C, Ho P S. Principles of Physical Biochemistry. Englewood Cliffs, NJ: Prentice–Hall; 1998. [Google Scholar]
- 49.Hegde R S, Grossman S R, Laimins L A, Sigler P B. Nature (London) 1992;359:505–512. doi: 10.1038/359505a0. [DOI] [PubMed] [Google Scholar]