

Abstract
The Ag85 family enzymes are responsible for the synthesis of cell wall components in mycobacterial species. Inhibitors to these enzymes are potential antimycobacterial agents. We have carried out the docking of phoshonate and trehalose analog inhibitors into the three dimensional structure of mycolyltransferase enzyme, Ag85C of M. tuberculosis using the GOLD software. The inhibitor binding positions and affinity were evaluated using both the scoring fitness functions- GoldScore and ChemScore. We observed that the inhibitor binding position identified using the GoldScore was marginally better than the ChemScore. A qualitative agreement between the reported experimental biological activities (IC50) and the GoldScore was observed. We identified that amino acid residues Arg541, Trp762 are important for inhibitor recognition via hydrogen bonding interactions. This information can be exploited to design Ag85C specific inhibitors.
Keywords: M. tuberculosis, drug design, docking, mycolyltransferases, trehalose analogs, phosphonate inhibitors
Background
Tuberculosis is an infection caused by the bacterium Mycobacterium tuberculosis. It is a major disease infecting two billion people, or approximately one-third of the world's population. These numbers are rising also in the developed countries due to the compromised immune systems, typically as a result of immunosuppressive drugs. This population is at particular risk of infection and active tuberculosis disease.
In M. tuberculosis, a major secreted protein complex, antigen 85, constitutes three proteins antigen 85A, 85B and 85C [1] that are responsible for the synthesis of cell envelope. These enzymes catalyze the transfer of mycolyl residue from one molecule of α, α' trehalosemonomycolate (TMM) to another TMM leading to the formation of α, α' trehalosedimycolate (TDM) [2] and are hence termed mycolyltransferases. These enzymes have the ability to bind the human fibronectin [3] through which the bacterium gets attached to host cells. Due to their immunodominant and secretory nature, the components of Ag85 complex represent promising protective antigen candidates. Also, the cell envelope of M. tuberculosis has been one of the targets of antimycobacterial agents. Drugs such as isoniazid, ethionamide, isoxyl, thiolactomycin, and triclosan and ethambutol, target the synthesis of mycolic acids in M. tuberculosis. In addition, pyrazinamide was shown to inhibit fatty acid synthase type I that in turn, provides precursors for fatty acid elongation to long-chain mycolic acids by fatty acid synthase II. [4] Mycolic acid biosynthesis is known to be essential for mycobacterium growth, in particular trehalose mycolates aid in virulence of the organism and the structure of mycolates has been found to be important for initial replication and persistence in vivo. [5] A mutated M. tuberculosis strain lacking the functional Ag85C gene showed a 40% decrease in the amount of cell wall linked mycolic acids indicating its role in cell wall synthesis. [6] The importance of mycolyltransferases in pathogenesis and cell wall formation makes them an attractive target for drug design studies. A series of 6,6'-bis(sulfonamido), N,N'-dialkylamino and related derivatives of 6,6'-dideoxytrehalose were designed and synthesized to inhibit the Ag85 complex. [7]
The three dimensional crystal structures of Ag85A (PDB ID: 1SFR) [8] Ag85B (PDB IDs: 1F0N, 1F0P) [9] and Ag85C (PDB IDs: 1DQZ, 1DQY, 1VA5) [10] were determined for both native and substrate bound forms. The protein structure corresponds to a α/β hydrolase fold and the catalytic triad responsible for the mycolyltransferase activity comprise the amino acid residues Ser624, Glu728 and His760 (numbering according to the PDB_ID: 1DQZ, B chain). This active site is typical of a serine protease family. Structural comparison of mycolyltransferases revealed that their backbone superimposes with an overall RMSD (root mean square deviation) of 0.577 Å. The catalytic residues are highly superimposed in these structures indicating that the structure and function of these isozymes are highly similar. Minor deviations are observed in the loop region connecting strand 4 and helix 3, comprising the sequence motif, 85Q-S-N-G-Q-N90 in Ag85C. This region is away from the substrate binding site and therefore will not affect substrate/inhibitor binding to the enzyme. A putative picture of the catalytic mechanism of the mycolyl transfer reaction been proposed by Ronning et al., 2000. [10] In the first step, catalytic serine attacks the carboxyl carbon of TMM molecule to give a mycolylenzyme intermediate and a free trehalose. In the next step, the 6'-OH group of the second TMM molecule attacks the carboxylate carbon of the acyl-enzyme intermediate to yield TDM. Both acylation and deacylation of the enzyme, proceed via a high-energy tetrahedral transition state. It is known that substituted tetrahedral phosphorus (V) species like phosphonates, phosphonamidates and phosphinates represent good tetrahedral transition state analogous of both amide and ester bond cleavage or formation. Incorporation of the phosphorus based transition state mimetics into the substrate or product analogs generally leads to useful enzyme inhibitors. Gobec et al., 2004 [11] synthesized a series of phosphonate inhibitors and reported the inhibition of Ag85C mycolyltransferase activity in the presence of inhibitors. Rose et al., 2002 [7] studied the antimycobacterial activity of trehalose analogs against M. tuberculosis H37Ra and clinical isolates of M. avium.
Several groups have earlier carried out a comparative evaluation of the docking methods and scoring functions. [14,15,16] Results from Verdonk et al., 2003 [13] indicated that for “drug like” and “fragment like” ligands, the docking accuracies obtained from GoldScore and ChemScore are similar, while for larger ligands, GoldScore gave better results. Docking involves the identification of ligand conformation and orientation in the protein binding pockets. The scoring functions are helpful to predict the biological activity of the ligand. The aim of the present work is to study the docking of phosphonate and trehalose analog inhibitors into the active site of Ag85C using the GOLD software. [12,13] In this work, we compared the scoring functions, GoldScore and ChemScore that are available in the Gold docking software. The three mycolyltransferases have highly similar structure, we have therefore chosen the three dimensional structure of Ag85C (PDB ID: 1DQZ) as a representative structure for docking studies. Also, the inhibition of Ag85C by the phosphonate inhibitors by Gobec et al., 2004 has been studied and the experimental IC50 values are reported. Since this enzyme functions as a serine protease, these docking studies will help characterize the inhibitor binding site. The information about the inhibitor binding site can be used to design Ag85C specific inhibitors that would not interfere with the other physiologically important ubiquitous serine proteases in humans.
Methodology
Energy minimization of protein
The crystal structure coordinates of the Ag85C (PDB_ID: 1DQZ) were obtained from the protein data bank ( http://www.rcsb.org/) and the B chain was selected for docking studies. All hydrogen atoms were added to the protein, including those necessary to define the correct ionization and tautomeric states of amino acid residues such as Asp, Ser, Glu, Arg and His using Cache software ( www.cachesoftware.com/cache). A two stage protocol was set up for the energy minimization of protein using Hyperchem7 (Hypercube Inc.), molecular modeling software. In the first stage, all hydrogen atoms in the protein were allowed to optimize. The hydrogen locations are not specified by the X-ray structure but these are necessary to improve the hydrogen bond geometries. In the second stage, all protein atoms were allowed to relax. Minimization in both stages was performed using 100 steps of steepest descent and 2000 steps of conjugate gradient algorithm.
Binding site analysis
The binding site identification of mycolyltransferases, Ag85A, Ag85B and Ag85C was carried out using the Binding Site Analysis module available in Insight II (Acclerys, USA). The default parameters for grid size 1.00 Å and site open size 7.00 Å were used in binding site calculation.
Selection of docking molecules
A set of 17 phosphonate inhibitors is taken from the reported literature [11] for the docking studies. A list of these molecules is provided in Figure 1a and a schematic representation of the molecules with least IC50 values is provided in Figure 1b. Similarly, a set of 9 trehalose analogs is taken from the reported literature [7] for docking studies. A list of these molecules is provided in Figure 5a and a schematic representation of the molecules with least MIC values are provided in Figure 5b. We have adapted the numbering of these inhibitors as represented in the original papers. [7,11]
Figure 1.
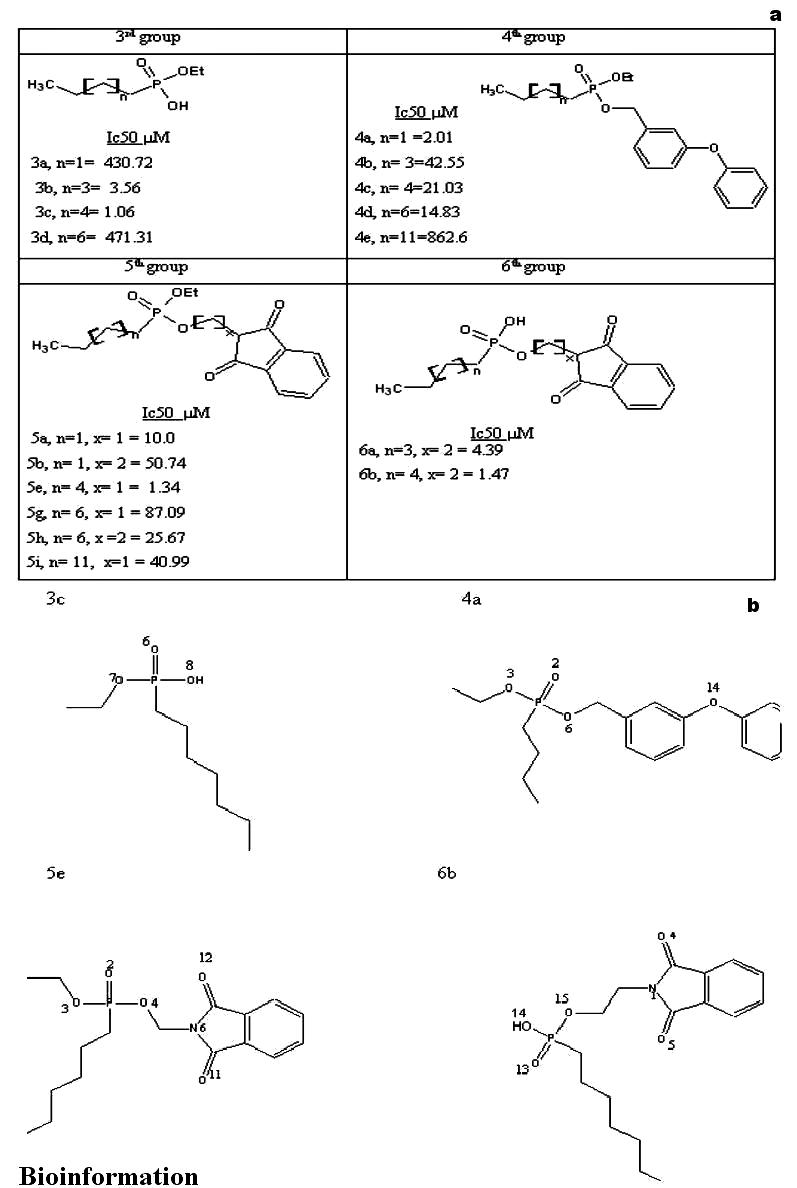
(a) A list of 17 phosphonate inhibitors docked into the active site of mycolyltransferase, Ag85C. The reported Ic50 values of the corresponding molecules are also indicated. (b) A schematic representation of the phosphonate inhibitors with least Ic50 values
Figure 5.
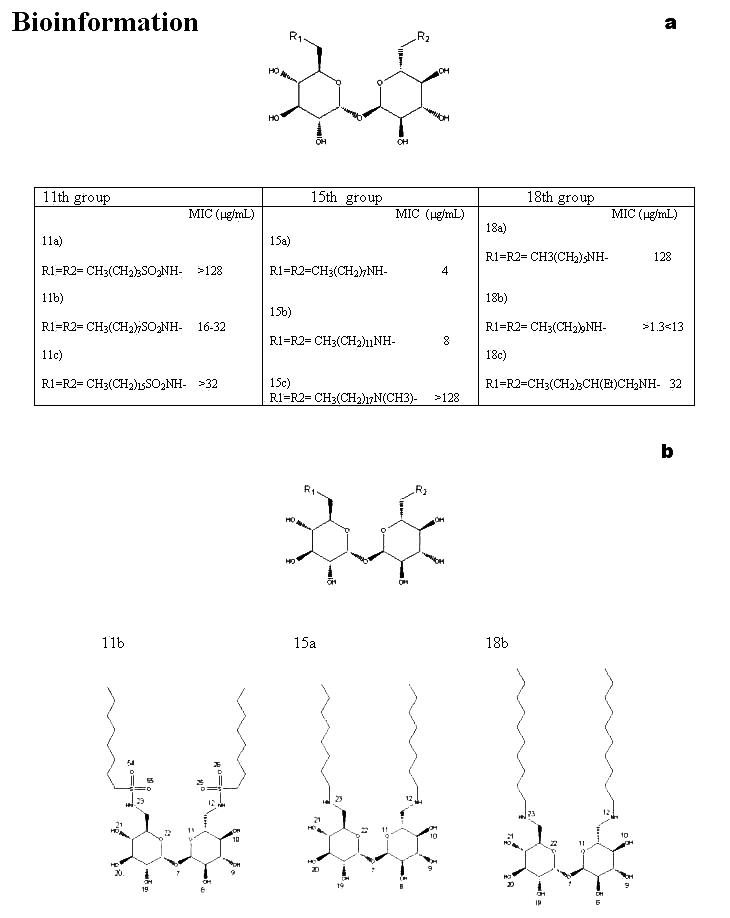
(a) A list of 9 trehalose analog inhibitors docked into the active site of mycolyltransferase, Ag85C. The reported MIC values of the corresponding molecules are also indicated (b) A schematic representation of the trehalose analog inhibitors with least MIC values
Molecular Modeling
The phosphonate and trehalose analog inhibitors were built using the Hyperchem7. The structures were energy minimized using the steepest descent algorithm with a convergence gradient value of 0.001 kcal/mol. Further, the geometry optimization was carried out for each compound using the MOPAC 6 package and the semi empirical AM1 Hamiltonian.
Docking
Docking was carried out using GOLD (Genetic optimization for Ligand Docking) software, that uses the Genetic algorithm (GA). This method allows a partial flexibility of protein and full flexibility of ligand. All water molecules and hetero atoms were removed from the protein to evaluate the two scoring functions in GOLD software. For each of the 25 independent GA runs, a maximum number of 100000 GA operations were performed on a set of five groups with a population size of 100 individuals. Operator weights for crossover, mutation, and migration were set to 95, 95, and 10, respectively. Default cutoff values of 2.5 Å (dH-X) for hydrogen bonds and 4.0 Å for van der Waals distance were employed. When the top three solutions attained RMSD values within 1.5 Å, GA docking was terminated. The RMSD values for the docking calculations are based on the RMSD matrix of the ranked solutions. We observed that the best ranked solutions were always among the first 10 GA runs, and the conformation of molecules based on the best fitness score was further analyzed. All the 17 phosphonate and 9 trehalose analog inhibitors were analyzed using docking studies and the molecules with least IC50 and MIC values respectively in each group are described in detail in the results and discussion section.
The two scoring functions available in the GOLD software to measure the affinity of ligand for protein binding site are
GoldScore fitness function
GoldScore performs a force field based scoring function and is made up of four components: 1. Protein-ligand hydrogen bond energy (external H-bond); 2. Protein-ligand van der Waals energy (external vdw); 3. Ligand internal van der Waals energy (internal vdw); 4. Ligand intramolecular hydrogen bond energy (internal- H- bond). The external vdw score is multiplied by a factor of 1.375 when the total fitness score is computed. This is an empirical correction to encourage protein-ligand hydrophobic contact. The fitness function has been optimized for the prediction of ligand binding positions. GoldScore = S(hb_ext) + S(vdw_ext) + S(hb_int) + S(vdw_int)
Where S(hb_ext) is the protein-ligand hydrogen bond score, S(vdw_ext) is the protein-ligand van der Waals score, S(hb_int) is the score from intramolecular hydrogen bond in the ligand and S(vdw_int) is the score from intramolecular strain in the ligand.
ChemScore fitness function
ChemScore is an empirical scoring function to estimate the free energy of ligand binding to protein. It uses simple contact terms to estimate lipophilic and metal-ligand binding contributions, including hydrogen bonding interactions. It does not differentiate between different types of hydrogen bonds, depending on the nature and geometry of the interaction. It adds a clash penalty and internal torsion terms, which influence against close contacts in docking and poor internal conformations. Covalent and constraint scores may also be included.
ChemScore estimates the total free energy change that occurs on ligand binding to protein as per the equation given below ΔG binding = ΔG o+ ΔG hbond + ΔG metal + ΔG lipo + ΔG rot .... (1)
Each component of this equation is the product of a term dependent on the magnitude of a particular physical contribution to free energy (e.g. hydrogen bonding) and a scale factor determined by regression. ChemScore = ΔG binding + Pclash +Cinternal Pinternal + (Ccovalent Pcovalent + Pconstraint) .... (2)
Results and Discussion
Binding site analysis of mycolyltransferases, Ag85A, Ag85B and Ag85C identified that, their binding pockets are identical and the largest binding pocket overlaps with the trehalose binding position in the PDB ID: 1F0P (data not shown). This further increased our confidence that the structures of mycolyltransferases are highly similar and we have therefore taken Ag85C as the representative structure for docking studies. The docking of phosphonate and trehalose analog inhibitors into the active site of mycolyltransferase, Ag85C was carried out using the GOLD software and the docking evaluations were made on the basis of two fitness functions, GoldScore and ChemScore. For both the scoring functions, the docking conformation generated with best fitness has been analyzed using two different criteria. These are 1) ligand binding position, and 2) fitness function score comparison.
Phosphonate inhibitors
Criteria-1
Ligand binding position. From the docking of molecule 3c into the active site of Ag85C, we observed that, the hydrogen bond acceptor atoms are 6, 7 and 8, and atom 8 is also a hydrogen bond donor, as indicated in Figure 1b.
GoldScore fitness function
One hydrogen bond was observed when the molecule 3c was docked into the active site of Ag85C, see Figure 2a and Table 1a. The alkyl chain of inhibitor entered into hydrophobic region of the protein comprising amino acid residues indicated in Table 2a. The best RMSD was found to be 0.28 Å.
Figure 2.

GoldScore based interactions of molecules (a) 3c (b) 4a (c) 5e and (d) 6b, docked into the active site of mycolyltransferase, Ag85C
Table 1a. List of hydrogen bonding interactions between the phosphonate inhibitors and mycolyltransferase, Ag85C.
| Molecule | Goldscore | ChemScore | ||||||
|---|---|---|---|---|---|---|---|---|
| Mol Name | No. of H bonds | Protein residue atom | Ligand atom | H bond distance Å | No. of H bonds | Protein residue atom | Ligand atom | H bond distance Å |
| 3c | 1 | Arg-541(NH) | 6(O) | 2.78 | 2 | Arg-541(NH) | 6(O) | 3.04 |
| Ser-624(OG) | 8(O) | 2.94 | ||||||
| 4a | 1 | Arg-541(NH) | 14(O) | 2.71 | 1 | His -760(C=O) | 2(O) | 3.08 |
| 5e | 2 | Arg-541(NH) | 6(O) | 2.81 | 1 | Ser-624(OG) | 12(O) | 2.65 |
| Trp-762(NE2) | 11(O) | 3.12 | ||||||
| 6b | 3 | Arg-541(NH) | 5(O) | 2.64 | 1 | Ser-624(OG) | 5(O) | 2.41 |
| Trp-762(NE1) | 4(O) | 3.11 | ||||||
| Ser-624(OG) | 13(O) | 2.93 | ||||||
Table 1a. List of amino acid residues contributing to the hydrophobic pocket in docking of phosphonate inhibitors into mycolyltransferase, Ag85C.
| Phosphonate inhibitor | GoldScore | ChemScore |
|---|---|---|
| 3c | Gly539, Leu540, Ala665, Phe576, Phe650, Leu727, Leu623 Ser624, and Met625 | Gly539, Leu540, Ala665, Phe576, Phe650, Leu727, Ile662, Ser624, Met625 and Trp658 |
| 4a | Leu540, Met625, Phe576, Leu727, Phe650, Leu661, Ile662, Ala665, Trp658, and Ser624 | Trp762, Ala724, and His760 |
| 5e | Leu540, Leu727, Phe650, Leu661, Ile662, Trp658, and Ser624 | Leu540, Asp538, Gly539, Leu623, Ser624 |
| 6b | Leu727, Phe650, Leu661, Ile662, Trp658, and His760 | Gly548, Gly539, Leu623, Asp538 and Trp762 |
ChemScore fitness function
Two hydrogen bonds were observed when the molecule 3c was docked into active site of Ag85C, see Figure 3a and Table 1a. The alkyl chain of inhibitor entered into hydrophobic region of protein comprising amino acid residues indicated in Table 2a. The best RMSD was found to be 0.62 Å.
Figure 3.

ChemScore based interactions of molecules (a) 3c (b) 4a (c) 5e and (d) 6b, docked into the active site of mycolyltransferase, Ag85C
From the docking of molecule 4a into the active site of Ag85C, we observed that the hydrogen bond acceptor atoms are 2, 3, 6 and 14 as indicated in Figure 1b.
GoldScore fitness function
One hydrogen bond was observed when the molecule 4a was docked into the active site of Ag85C, see Figure 2b and Table 1a. The alkyl chain of inhibitor entered into hydrophobic region of protein comprising amino acid residues indicated in Table 2a. The best RMSD was found to be 0.90 Å.
ChemScore fitness function
One hydrogen bond was observed when the molecule 4a was docked into active site of Ag85C, see Figure 3b and Table 1a. The alkyl chain of inhibitor entered into hydrophobic region of protein comprising amino acid residues, as shown in Table 2a. The best RMSD was found to be 0.80 Å.
From the docking of molecule 5e into the active site of Ag85C, we observed that, the hydrogen bond acceptor atoms are 2, 3, 4, 6, 11, and 12 as indicated in Figure 1b.
GoldScore fitness function
Two hydrogen bonds were observed when the molecule 5e was docked into active site of Ag85C, see Figure 2c and Table 1a. The alkyl chain of inhibitor entered into hydrophobic region of protein comprising amino acid residues, as indicated in Table 2a. The best RMSD was found 0.61 Å.
ChemScore fitness function
One hydrogen bonding interaction was observed when the molecule 5e was docked into the active site of Ag85C, see Figure 3c and Table 1a. The inhibitor alkyl chain entered into hydrophobic region of protein comprising amino acid residues as indicated in Table 2a. The best RMSD was found to be 0.82 Å.
From the docking of molecule 6b into the active site of Ag85C, we observed that, the hydrogen bond acceptor atoms are 1, 4, 5, 13, 14 and 15, and the atom 14 is also a hydrogen bond donor, as indicated in Figure 1b.
GoldScore fitness function
Three hydrogen bonds were observed when molecule 6b was docked into the active site of Ag85C, see Figure 2d and Table 1a. The alkyl chain of ligand entered into hydrophobic region of protein comprising residues as indicated in Table 2a. The best RMSD was found to be 1.04 Å.
ChemScore fitness function
One hydrogen bonding interaction was observed when molecule 6b was docked into the active site of Ag85C with a distance of 2.41 Å, see Figure 3d and Table 1a. The alkyl chain entered into hydrophobic region of protein comprising amino acid residues as indicated in Table 2a. The best RMSD was found to be 1.01 Å.
Criteria-2
Fitness function score comparison. The phosphonate inhibitors were docked into the active site of Ag85C and evaluated based on both the fitness scoring functions- GoldScore and ChemScore available in the GOLD software. We observed that the GoldScore correlated well with the reported IC50 values as shown in Figure 4a. There are no compounds with high GoldScore and low activity (high IC50 values). The exceptions are molecules 3b and 3c that bind the Ag85C with a GoldScore of 50.30 and 51.18, while the IC50 values are 3.5 μM and 1.06 μM, respectively. We hypothesize that the low GoldScore for these molecules is due to the presence of OH substitution alone on the phosphate head group. When the docked molecules were evaluated based on the ChemScore, there is no such correlation with the IC50 values, see Figure 4b. These results indicate that the GoldScore is a better parameter to assess the binding of phosphonate inhibitors to Ag85C and there is an overall 70-80 % correlation between the GoldScore and IC50 values.
Figure 4.
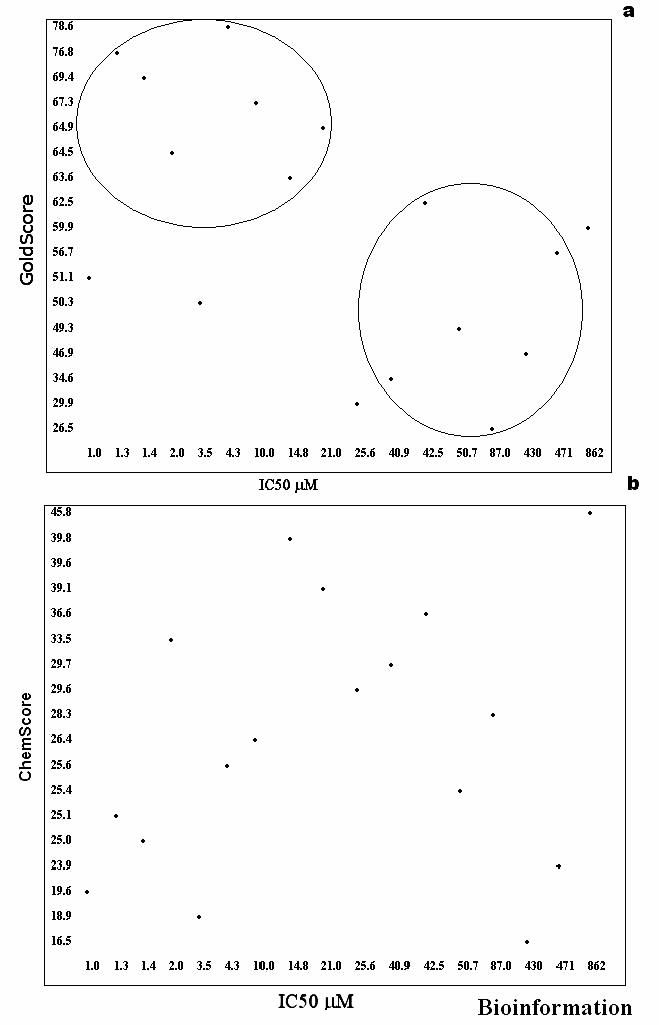
(a) GoldScore based docking of phosphonate inhibitors into the mycolyltransferase, Ag85C (b) ChemScore based docking of phosphonate inhibitors into the mycolyltransferase, Ag85C
Trehalose analogs
Ligand binding position: From the docking of molecule 11b into the active site of Ag85C, we observed that, the hydrogen bond acceptor atoms are 7-12, 19-23, 25, 26, 54 and 55. The atoms 8-10, 12, 19-21 23 are also hydrogen bond donors, as indicated in Figure 5b.
GoldScore fitness function
Seven hydrogen bonds were observed when the molecule 11b was docked into the active site of Ag85C, see Figure 6a and Table 1b. The alkyl chain of the inhibitor entered into the hydrophobic region of the protein comprising amino acid residues indicated in Table 2b. The best RMSD was found to be 2.16 Å.
Figure 6.
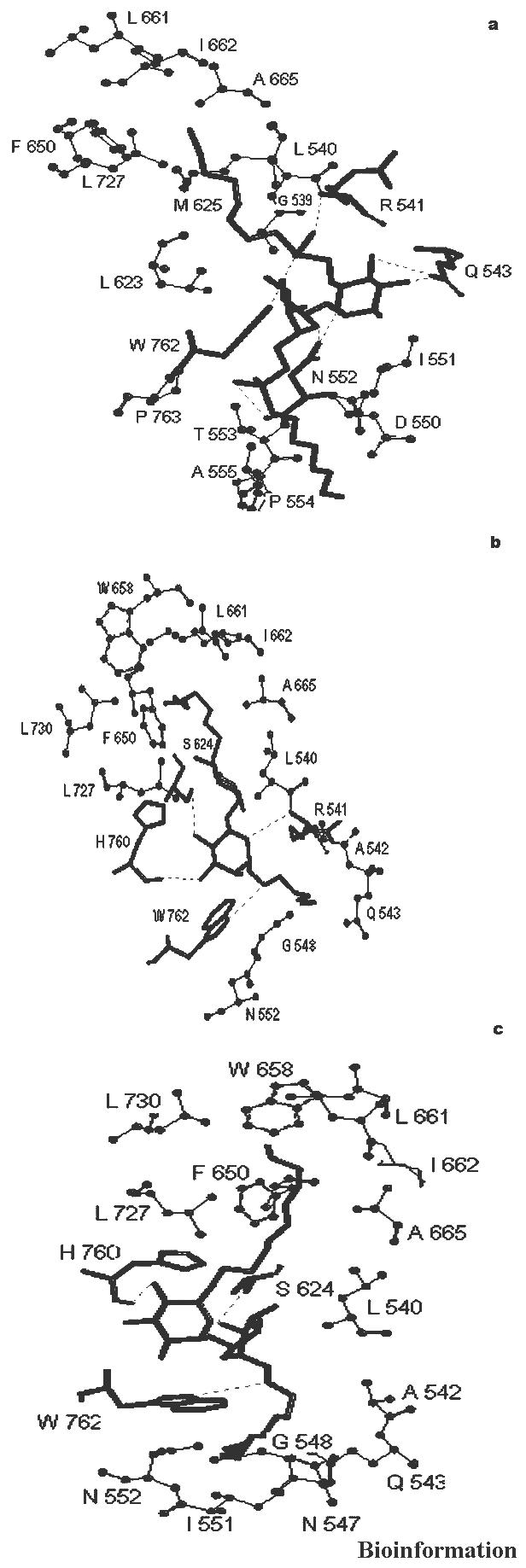
GoldScore based interactions of molecules (a) 11b (b) 15a and (c) 18b docked into the active site of mycolyltransferase, Ag85C
Table 1b. List of hydrogen bonding interactions between the phosphonate inhibitors and mycolyltransferase, Ag85C.
| Goldscore | ChemScore | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mol Name | No. of H bonds | Protein residue atom | Ligand atom | H bond distance Å | No. of H bonds | Protein residue atom | Ligand atom | H bond distance Å | |
| 11b | 7 | Arg-541(N) | 25 (O) | 2.51 | 4 | Arg-541(O) | 25 (O) | 3.13 | |
| Trp-762(NE1) | 26 (O) | 3.63 | |||||||
| Gln-543(NE2) | 9 (O) | 3.04 | Trp-762(NE1) | 26 (O) | 2.80 | ||||
| Gln-543(NE2) | 10 (O) | 2.52 | Gln-543(NE2) | 11 (O) | 2.95 | ||||
| Asn-552(OD1) | 23 (N) | 3.28 | Asn-552(O) | 19 (O) | 3.07 | ||||
| Asn-552(O) | 54 (O) | 3.21 | |||||||
| 15a | 4 | Arg-541(N) | 22 (O) | 3.85 | 3 | Arg-541(N) | 20 (O) | 3.19 | |
| Ser-624(OG) | 19 (O) | 3.21 | |||||||
| His-760(O) | 20 (O) | 3.02 | Ser-624(O) | 12 (N) | 3.34 | ||||
| Trp-762(NE1) | 23 (N) | 3.43 | His-760(O) | 23 (N) | 3.03 | ||||
| 18b | 3 | Ser-624(OG) | 20 (O) | 3.30 | 2 | His-760(NE2) | 19 (O) | 3.30 | |
| His-760(O) | 10 (O) | 3.19 | |||||||
| Trp-762(NE1) | 23 (N) | 2.94 | His-760(NE2) | 20 (O) | 2.76 | ||||
Table 1b. List of amino acid residues contributing to the hydrophobic pockets in docking of trehalose analog inhibitors into mycolyltransferase, Ag85C.
| Trehalose inhibitors | GoldScore | ChemScore | ||
|---|---|---|---|---|
| R1 | R2 | R1 | R2 | |
| 11b | Ala665, Leu661, Ile662, Leu540, Gly539, Met625, Leu727, Phe650, Leu623 | Pro763, Thr553, Ala555, Pro554, Ile551, Asp550 | Ala542, Gle539, Leu540, Leu727, Leu727 Trp765 | Tyr546,Gln525,Tyr510, Asp550, Ile551 |
| 15a | Leu540, Ala665, Ile662, Leu661, Trp658, Leu730, Phe650, Leu727 | Ala542, Gln543, Gly548, Asn552 | Leu540, Phe576, Ala665, Ile662, Leu661, Trp658, Phe650, Leu730 | Gly548, Asn552, Pro763, Gly719, Trp762 |
| 18b | Leu540, Ala665, Ile662, Leu661, Trp658, Leu730, Phe650, Leu727 | Ala542, Gln543, Gly548, Asn547, Ile551, Asn552 | Ala665, Ile662, Leu661,Trp658, Leu730, Phe650, Leu727 | Ala542,Gln543, Asp545, Ile551, Gly548 |
ChemScore fitness function
Four hydrogen bonds were observed when the molecule 11b was docked into active site of Ag85C, see Figure 7a and Table 1b. The alkyl chain of the inhibitor entered into hydrophobic region of protein comprising amino acid residues, as indicated in Table 2b. The best RMSD was found to be 2.64 Å.
Figure 7.
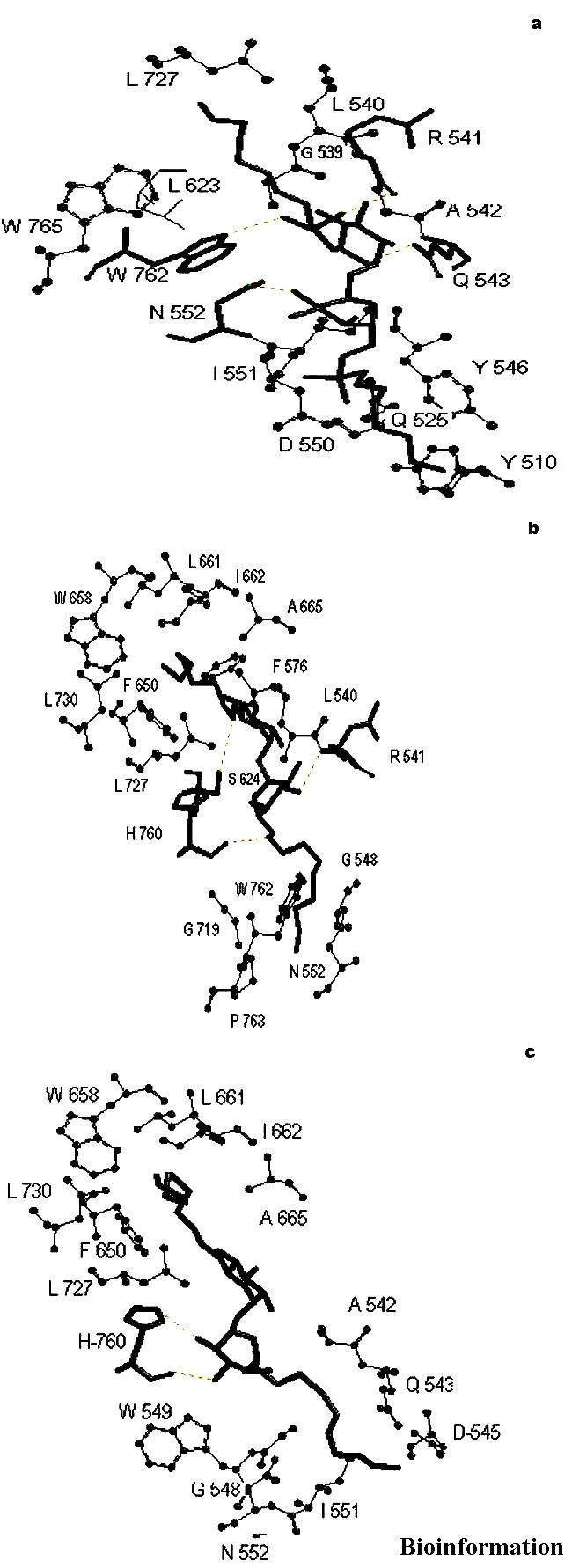
ChemScore based interactions of molecules (a) 11b (b) 15a and (c) 18b docked into the active site of mycolyltransferase, Ag85C
From the docking of molecule 15a into the active site of Ag85C, we observed that, the hydrogen bond acceptor atoms are 7-12, 19-23. The hydrogen bond donor atoms are 8-10, 12, 19-21 and 23, as indicated in Figure 5b.
GoldScore fitness function
Four hydrogen bonds were observed when the molecule 15a was docked into the active site of Ag85C, see Figure 6b and Table 1b. The alkyl chain of the inhibitor entered into the hydrophobic region of the protein comprising amino acid residues as indicated in Table 2b. The best RMSD was found to be 0.78 Å.
ChemScore fitness function
Three hydrogen bonds were observed when the molecule 15a was docked into active site of Ag85C, see Figure 7b and Table 1b. The alkyl chain of inhibitor entered into hydrophobic region of protein comprising amino acid residues, as indicated in Table 2b. The best RMSD was found to be 1.60 Å.
From the docking of molecule 18b into the active site of Ag85C, we observed that, the hydrogen bond acceptor atoms are 7-12, 19-23, the hydrogen bond donor atoms are 8-10, 12, 19-21, 23, as indicated in Figure 5b.
GoldScore fitness function
Three hydrogen bonds were observed when the molecule 18b was docked into the active site of Ag85C, see Figure 6c and Table 1b. The alkyl chain of the inhibitor entered into the hydrophobic region of the protein comprising amino acid residues, as indicated in Table 2b. The best RMSD was found to be 2.34 Å.
ChemScore fitness function
Two hydrogen bonds were observed when the molecule 18b was docked into active site of Ag85C, see Figure 7c and Table 1b. The alkyl chain of inhibitor entered into hydrophobic region of protein comprising amino acid residues, as indicated in Table 2b. The best RMSD was found to be 2.28 Å.
FFor the trehalose analog inhibitors, Rose et al., 2002 [7] have reported the MIC values in order to estimate their activity as antimycobacterial agents, therefore, we have not correlated them with the GoldScore and ChemScore.
In the crystal structure of Ag85B complexed with trehalose substrate (PDB ID: 1F0P), it has been shown that Ser124 (equivalent of Ser624 in Ag85C, B chain) forms a hydrogen bonding interaction with atom O6 of trehalose. [9] From our docking studies, we have shown that this serine forms a hydrogen bonding interactions with phosphonate and trehalose analog inhibitors (see Tables 1a and 1b). Further, we also identified additional amino acid residues, Arg541 and Trp762 important for inhibitor recognition mediated via non-bonding interactions. Sequence analysis identified that Arg541, Ser624 and Trp762 are highly conserved in mycolyltransferases [17] of M. tuberculosis. Current docking studies using the GoldScore fitness function reported a hydrophobic tunnel to accommodate the hydrocarbon alkyl chain of phosphonate and R1 alkyl chain of trehalose analog inhibitors. This hydrophobic tunnel comprises the residues Leu540, Phe576, Phe650, Trp658, Leu661, Ile662, Leu623, Met625, Ala665 and Leu727. In the reported crystal structures of Ag85B and Ag85C [9,10], the authors have described a hydrophobic tunnel comprising these residues to accommodate α- alkyl chain of mycolic acids. It was also proposed that β- alkyl chain of mycolic acids would fit the trough on the surface of the protein. Docking of trehalose analog inhibitors identified the R2 alkyl chain to bind the region comprising amino acid residues Tyr510, Gln525, Ala542, Gln543, Asp545, Gly548, Asp550, Ile551, Asn552, Pro554 that are located on the surface of the protein. These results indicate that the predicted nonbonding interactions and hydrophobic region to accommodate the alkyl chains are similar to the experimental data obtained from crystal structures.
Conclusion
A detailed docking analysis of phosphonate and trehalose analog inhibitors into the active site of Ag85C has been studied in the present work, to identify the inhibitor binding position and affinity to Ag85C using the Gold software. We report that the GoldScore fitness function is marginally better than the ChemScore fitness function, to understand the binding conformation. Also, GoldScore provides a qualitative agreement with the reported IC50 values of phosphonate inhibitors.
Acknowledgments
KKI thanks CSIR New Delhi for a SRF fellowship. LGP thanks ILS-UH MOU for research funding.
Footnotes
Citation:Doppelt et al., Bioinformation 1(9): 357-359 (2007)
References
- 1.Wiker HG, Harboe M. Microbiol Rev. 1992;56:648. doi: 10.1128/mr.56.4.648-661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belisle JT, et al. Science. 1997;276:1420. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 3.Abou Zeid C, et al. Infect and Immun. 1988;56:3046. doi: 10.1128/iai.56.12.3046-3051.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schroeder EK, et al. Curr Pharm Biotechnol. 2002;3:197. doi: 10.2174/1389201023378328. [DOI] [PubMed] [Google Scholar]
- 5.Glickman MS, et al. Molec Cell. 2000;5:717. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 6.Jackson M, et al. Mol Microbiol. 1999;31:1573. doi: 10.1046/j.1365-2958.1999.01310.x. [DOI] [PubMed] [Google Scholar]
- 7.Rose JD, et al. Carbohyd Res. 2002;337:105. doi: 10.1016/s0008-6215(01)00288-9. [DOI] [PubMed] [Google Scholar]
- 8.Ronning DR, et al. J Biol Chem. 2004;279:36771. doi: 10.1074/jbc.M400811200. [DOI] [PubMed] [Google Scholar]
- 9.Anderson DH, et al. J Mol Bio. 2001;307:671. doi: 10.1006/jmbi.2001.4461. [DOI] [PubMed] [Google Scholar]
- 10.Ronning DR, et al. Nat Struc Bio. 2000;7:141. doi: 10.1038/72413. [DOI] [PubMed] [Google Scholar]
- 11.Gobec S, et al. Bio & Med Chem Lett. 2004;14:3559. doi: 10.1016/j.bmcl.2004.04.052. [DOI] [PubMed] [Google Scholar]
- 12.Jones G, et al. J Mol Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 13.Verdonk ML, et al. Proteins. 2003;52:609. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 14.Perola E, et al. Proteins. 2004;56:235. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, et al. J Med Chem. 46:2287. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 16.Wang R, et al. J Chem Inf Comput Sci. 44:2114. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 17.Adindla S, et al. Int J Biol Macromol. 2004;34:181. doi: 10.1016/j.ijbiomac.2004.03.008. [DOI] [PubMed] [Google Scholar]