

Abstract
Prostaglandin E2 (PGE2) inhibits fibroblast proliferation and collagen production. Its synthesis by fibroblasts is induced by profibrotic mediators including transforming growth factor (TGF)-β1. However, in patients with pulmonary fibrosis, PGE2 levels are decreased. In this study we examined the effect of TGF-β1 on PGE2 synthesis, proliferation, collagen production, and cyclooxygenase (COX) mRNA levels in fibroblasts derived from fibrotic and nonfibrotic human lung. In addition, we examined the effect of bleomycin-induced pulmonary fibrosis in COX-2-deficient mice. We demonstrate that basal and TGF-β1-induced PGE2 synthesis is limited in fibroblasts from fibrotic lung. Functionally, this correlates with a loss of the anti-proliferative response to TGF-β1. This failure to induce PGE2 synthesis is because of an inability to up-regulate COX-2 mRNA levels in these fibroblasts. Furthermore, mice deficient in COX-2 exhibit an enhanced response to bleomycin. We conclude that a decreased capacity to up-regulate COX-2 expression and COX-2-derived PGE2 synthesis in the presence of increasing levels of profibrotic mediators such as TGF-β1 may lead to unopposed fibroblast proliferation and collagen synthesis and contribute to the pathogenesis of pulmonary fibrosis.
Pulmonary fibrosis is characterized by inflammatory cell infiltration, fibroblast proliferation, and excess deposition of extracellular matrix proteins including collagen in the lung parenchyma. This results in the loss of normal alveolar structure and impaired lung function. The disease can occur as an isolated pulmonary disorder in response to known or unknown causes 1 and in association with connective tissue disorders such as systemic sclerosis 2 and rheumatoid arthritis. 3 The pathogenesis of pulmonary fibrosis is incompletely understood. However, fibroblast proliferation and collagen synthesis are known to be regulated, at least in part, by a complex interaction between stimulatory and inhibitory mediators. 4 Several stimulatory mediators including transforming growth factor (TGF)-β1, platelet-derived growth factor (PDGF), insulin-like growth factor-1, and thrombin as well as inhibitory mediators such as interferon, glucocorticoids, epidermal growth factor, and prostaglandin E (PGE)2 have been suggested to play a role in the pathogenesis of pulmonary fibrosis. 4
PGE2 is a potent inhibitor of fibroblast proliferation 5,6 and collagen synthesis. 7,8 It is normally present in the lung at much higher concentrations than in plasma 9 and is the major eicosanoid product of fibroblasts. 10,11 Together, this suggests that PGE2 may play an important role in maintaining normal lung extracellular matrix homeostasis. In addition, a number of pro-inflammatory mediators such as TGF-β1, interleukin (IL)-1β, tumor necrosis factor-α, and PDGF induce fibroblasts to synthesize PGE2. 6,12-16
In pulmonary fibrosis, levels of TGF-β and other profibrotic mediators that induce synthesis of PGE2 are elevated. 17,18 Despite this, PGE2 levels in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis have been shown to be 50% lower than in normal individuals. 19 In addition, fibroblasts cultured from patients with idiopathic pulmonary fibrosis fail to induce PGE2 synthesis on stimulation with IL-1β, tumor necrosis factor-α, or lipopolysaccharide because of aberrant expression of the inducible cyclooxygenase (COX)-2 enzyme, the rate-limiting enzyme in prostanoid biosynthesis 20,21 but the functional effects of this have not been investigated. Furthermore, mice deficient in COX-2 exhibit fibroproliferative disorders of the heart and kidneys 22,23 but there is no data on the fibroproliferative response in the lungs of these animals.
Evidence suggests that TGF-β1 plays a key role in the pathogenesis of pulmonary fibrosis. It is a potent stimulator of collagen synthesis and regulator of fibroblast proliferation. 6,13,24 TGF-β1 levels are increased in patients with pulmonary fibrosis 17,18 and in the lungs of animals with experimentally-induced pulmonary fibrosis. 25 TGF-β1 has been localized to sites of extracellular matrix gene expression 26 and increased mRNA expression of TGF-β1 has been reported after bleomycin-induced lung injury. 24,27 Subcutaneous injection of TGF-β1 induces granulation tissue formation 28 and adenoviral transfer of a gene construct that expresses active TGF-β1 to rat lung results in a severe and sustained fibrotic response. 29 Inhibition of TGF-β1 limits the fibroproliferative response in animal models. 30-33 In addition, we have shown that in lung fibroblasts, TGF-β1 stimulates autocrine synthesis of PGE2 that is responsible for the anti-proliferative effects of TGF-β1 6 and limits its stimulation of collagen synthesis. 13
In this study we investigated the effects of TGF-β1 on PGE2 synthesis, proliferation, and collagen production by lung fibroblasts isolated from human fibrotic and nonfibrotic lung. In addition, we have assessed the role of COX-1 and COX-2 in mediating the effects of TGF-β1 on fibroblast PGE2 synthesis by Northern analysis and using selective COX inhibitors. We have also performed in vivo experiments examining the effect of bleomycin-induced pulmonary fibrosis in COX-2-deficient mice. We provide evidence, for the first time, to demonstrate that fibroblasts from patients with pulmonary fibrosis have a limited capacity to up-regulate PGE2 synthesis in response to TGF-β1 and that in control fibroblasts this response is mediated via COX-2. The lack of stimulation of PGE2 synthesis by the fibroblasts derived from fibrotic lung correlates with a loss of the anti-proliferative response to TGF-β1. We also demonstrate that mice deficient in COX-2 are more susceptible to bleomycin-induced lung injury.
Materials and Methods
Fibroblast Cell Lines
Thirty-five lung fibroblast cell lines were studied. The fibrosis group consisted of 17 cell lines derived from patients with pulmonary fibrosis (idiopathic pulmonary fibrosis, n = 9; systemic sclerosis, n = 7). All biopsies from which cell lines were derived showed histological evidence of pulmonary fibrosis. In addition one cell line (CCL-134) established from a patient with idiopathic pulmonary fibrosis was obtained from the American Type Culture Collection (ATCC, Rockville, MD). The control group consisted of 18 cell lines that were established from lung tissue derived from various sources. Thirteen were biopsy samples from patients undergoing lung resection for localized tumor. Tissue was taken from areas of macroscopically normal lung parenchyma, distal to any tumor mass. Two cell lines were established from patients who died of non-lung related causes. In addition to these, another three human lung fibroblast cell lines were obtained from the ATCC (CCI-201, CCI-204, and CCL-200).
Isolation and Culture of Lung Fibroblasts
Fibroblast cell lines were established from explant cultures. 34 Briefly, lung tissue biopsies were cut into 1-mm 3 fragments and placed ∼10 mm apart on the surface of culture dishes with Dulbecco’s modified Eagle’s medium (Life Technologies, Paisley, UK) supplemented with 10% (v/v) newborn calf serum (Imperial Laboratories, Andover, UK), penicillin (100 U/ml), streptomycin (100 μg/ml), and 2.5 μg/ml amphotericin B (all from Life Technologies, Paisley, UK). Fibroblasts were observed growing out of the tissue fragments after 6 to 8 days, developing into a near confluent monolayer of cells after 3 to 4 weeks. Experiments were conducted on cells between passages 3 and 17. Fibroblast cell lines were characterized immunohistochemically to confirm their purity. Staining with antibodies to cytokeratin, von Willebrand factor, and desmin was negative, indicating that the cultures did not contain significant numbers of epithelial, mesothelial, endothelial, or smooth muscle cells. Greater than 95% of the cells stained positively for vimentin, and between 20 and 30% of the cells were also positive for α-smooth muscle actin, confirming the fibroblast/myofibroblast phenotype of the cell lines.
Measurement of Fibroblast Proliferation
Cell proliferation was assessed using either a spectrophotometric assay based on the uptake and subsequent elution of methylene blue as described previously 5 or by measuring the incorporation of 3H-thymidine into DNA. Briefly, 96-well microtiter plates were seeded with 6 × 10 3 cells/well in Dulbecco’s modified Eagle’s medium containing 0.4% newborn calf serum. After a 24-hour preincubation, serum-free media was added containing TGF-β1 (R&D Systems Europe Ltd., Oxon, UK) at concentrations between 0 and 640 pg/ml. The final concentration of newborn calf serum in the media was 0.2% (v/v). Changes in cell number were assessed 72 hours later. Results were expressed as percentage change in mean absorbance compared with cells exposed to medium alone. To measure thymidine incorporation, [3H]-thymidine (Amersham, Buckinghamshire, UK) was added to each well to give a final concentration of 37 KBq/well. Changes in DNA synthesis were assessed at various times up to 72 hours. Thymidine incorporated into DNA was harvested onto glass fiber filters (ICN Flow, Oxfordshire, UK), radioactivity measured, and values expressed as percentage change in mean disintegrations per min (dpm) as compared to cells exposed to medium alone. In experiments to block PGE2 synthesis, indomethacin at a final concentration of 1 μg/ml (Sigma, Poole, England) was added to the cells 30 minutes before the addition of TGF-β1.
Measurement of Hydroxyproline
Hydroxyproline was measured as an index of fibroblast procollagen production using previously described methods. 13 Cells were grown to confluence in 2.4-cm diameter wells in Dulbecco’s modified Eagle’s medium supplemented with 5% newborn calf serum. Once confluent, cells were incubated for a further 24 hours. The media was removed and replaced with 1 ml of preincubation medium containing 4 mmol/L glutamine, 50 μg/ml ascorbic acid (Sigma), 0.2 mmol/L proline (Sigma), and 0.4% newborn calf serum (v/v) and incubated for 24 hours. The media was then replaced with either 0.5 ml of fresh preincubation medium alone or preincubation media containing indomethacin (1 μg/ml) and incubated for 30 minutes. Finally 0.5 ml of media or media containing TGF-β1 (1 ng/ml final concentration) was added and incubated for 24 hours before harvesting. Parallel sets of plates were seeded and treated in the same way to determine cell number. To assess procollagen production, the cell layer and medium were combined and proteins precipitated in 67% (v/v) ethanol at 4°C overnight. The precipitated proteins were recovered by vacuum filtration onto polyvinylidene difluoride filters (pore size, 0.45 μm; Millipore Ltd., Watford, UK) and hydrolyzed in 2 ml of 6 mol/L HCl at 110°C overnight. Hydroxyproline was isolated and quantified by reverse-phase high-pressure liquid chromatography of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD-CI)-derivatized hydrolysates as described previously. 13 Values were corrected for the amount of hydroxyproline present in the cell layer and culture medium at the start of the incubation period as well as cell number and expressed either as pmol of hydroxyproline/10 5 cells/hour or as a percentage increase greater than basal procollagen production.
Measurement of PGE2 Synthesis
PGE2 was measured in the medium from cells cultured in the same way as described for the determination of procollagen production. In addition to indomethacin, cells were also preincubated with the COX-2 selective inhibitor; NS-398 (Biomol Research Labs; Plymouth, UK) at a final concentration of 5 μg/ml 35 or the COX-1 preferential inhibitor, piroxicam (Sigma) at a final concentration of 2.5 ng/ml 36 Cells were exposed to the selective and nonselective inhibitors for 30 minutes before the addition of TGF-β1 (1 ng/ml) or preincubation medium. After a further 24-hour incubation period, PGE2 was measured using a specific enzyme immunoassay (Amersham, Bucks, UK). Results were expressed as pg of PGE2 per 10 5 cells or per ml of media.
RNA Extraction and Analysis
Cells were seeded into 10-cm Petri dishes at a concentration of 5 × 10 5 cells/dish and treated in an identical manner to those cultures used for the procollagen assay. Cells were exposed to TGF-β1 (1 ng/ml) for various times up to 24 hours. Total RNA was extracted from the cells using Trizol reagent (Life Technologies, Paisley, UK) in accordance with the manufacturer’s instructions. Five to 10 μg of RNA was fractionated by electrophoresis through a 1% (w/v) agarose/formaldehyde gel, transferred to a nylon membrane (Hybond N, Amersham, Bucks, UK) by Northern transfer, and fixed by UV crosslinking. To assess COX-1 and COX-2 mRNA levels, membranes were hybridized with a [32P]dCTP-labeled human cDNA probe for COX-1 (Biogenesis, Poole, UK), or a [32P]dCTP-labeled human cDNA probe for COX-2 (kindly donated by T. Hla, Dept. of Molecular Biology, Holland Lab, American Red Cross, Rockville, MD). mRNA levels were quantitated by densitometric laser scanning.
Animals
COX-2+/+ (wild-type, strain SV129/C57BL/6 F2) and COX-2-deficient mice (COX-2−/−, obtained from the Jackson Laboratory Bar Harbor, ME (stock numbers 101045 and 002476), aged 6 weeks received a single intratracheal instillation of saline (0.9%) or saline containing bleomycin sulfate (1 mg/kg body weight) in a volume of 50 μl and were killed after 14 days by pentobarbitone overdose. Lungs were harvested from between four and six mice of each genotype for histological analysis. The vasculature was perfused with heparinized phosphate-buffered saline (PBS) and the lungs fixed by intratracheal instillation of freshly prepared 4% paraformaldehyde in PBS at a pressure of 25 cm H2O. The trachea was ligated just caudal to the larynx and the thoracic contents were removed and immersed in fixative overnight, transferred to 15% sucrose in PBS before dehydrating and embedding in paraffin wax. Sections (5 μm) were cut and stained with Masson’s trichrome. The extent of fibrosis was scored in a blinded manner by three independent observers based on a previously described method. 37 Each lung lobe was scored on a scale of 0 to 4 and a mean derived from the five lobe scores for each individual mouse.
Statistical Analysis
For comparisons between patient groups, data were expressed as the median (range) and statistical differences were determined using the Mann-Whitney U test. To compare the effects of TGF-β1, indomethacin, and the COX selective inhibitors on individual cell lines, data were expressed as the mean ± SEM and statistical differences were evaluated using the Student’s two-tailed unpaired t-test for single group comparisons and Newman-Keuls one-way analysis of variance (analysis of variance) for multiple group comparisons. Fibrosis scores were evaluated by calculating the mean ± SEM, followed by single comparisons between individual treatment groups using the Student’s two-tailed unpaired t-test. Data were considered to be statistically significant when P < 0.05.
Results
Basal and TGF-β1-Induced PGE2 Synthesis by Fibroblasts
Analysis of the nonfibrotic lung fibroblast cell lines suggested that these fell into two distinct phenotypic groups in terms of their PGE2 production, basally and in response to TGF-β1, as well as their functional responses to TGF-β1 (see below). The groups consisted of those cell lines that produced PGE2 basally that was further stimulated by incubation with TGF-β1, and those that produced little or no PGE2 basally and that were not stimulated further by TGF-β1. We therefore divided this group on the basis of basal and TGF-β1-induced PGE2 production. Group II (n = 4) consisted of cell lines isolated from nonfibrotic lung where basal PGE2 production was lower than the highest level produced by cell lines derived from fibrotic lung and was not stimulated by incubation with TGF-β1. Group I (n = 6) contained all other cell lines derived from nonfibrotic lung and group III (n = 6) contained the cell lines derived from fibrotic lung. Basal and TGF-β1-induced PGE2 levels for cell lines in the three groups are shown in Figure 1 ▶ . Under basal conditions, levels of PGE2 synthesis in group I ranged from 30 to 2,176 pg/10 5 cells (median, 321 pg/10 5 cells) and TGF-β1 (1 ng/ml) enhanced this further by up to 16-fold (median, 798 pg/10 5 cells; range, 446 to 3,077 pg/10 5 cells). Basal levels of PGE2 synthesis by group II cell lines ranged from 10 to 26 pg/10 5 cells (median, 12 pg/10 5 cells) and treatment with TGF-β1 had no effect (median, 29 pg/10 5 cells; range, 13 to 38 pg/10 5 cells). Group III cell lines also synthesized lower levels of PGE2 basally (median, 43 pg/10 5 cells; range, 22 to 153 pg/10 5 cells) and in response to TGF-β1 (median, 87 pg/10 5 cells; range, 47 to 371 pg/10 5 cells) compared with group I cell lines (P < 0.05 and P < 0.01, respectively). None of the cell lines derived from fibrotic lung exhibited a phenotype similar to that of the group I cell lines. Furthermore, a comparison of group II with group III indicated that group II cell lines produced less PGE2 than group III both basally and in response to TGF-β1 (P < 0.05 and P < 0.01, respectively). Basal and TGF-β1-induced PGE2 synthesis was found to be consistent in different experiments conducted with cells at the same passage number and in experiments throughout three passages (data not shown).
Figure 1.
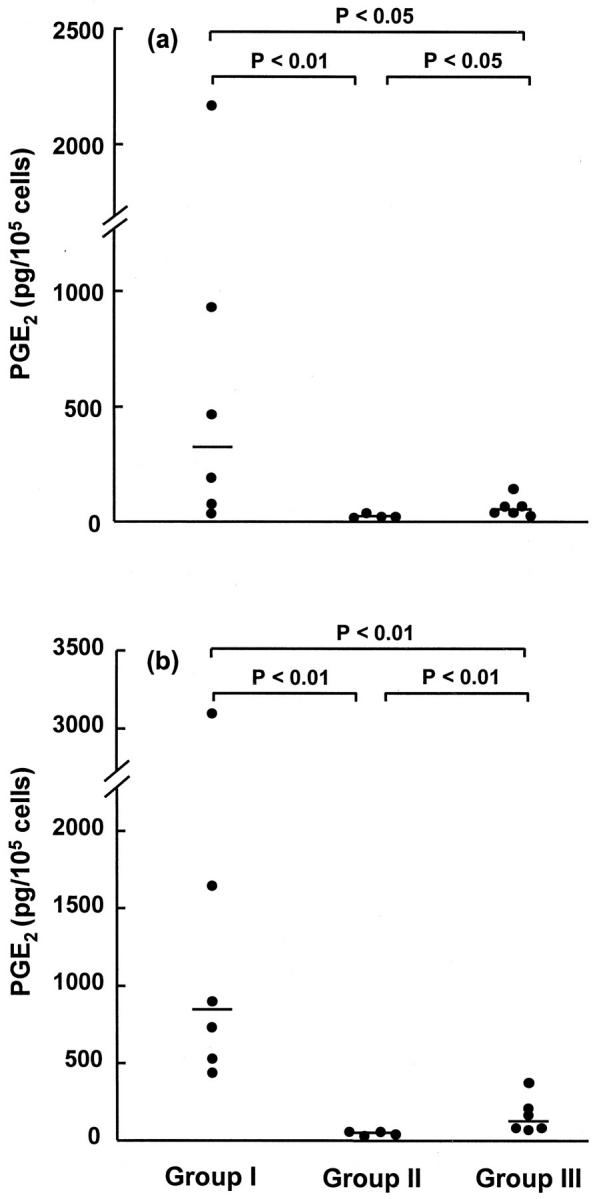
Basal and TGF-β1-induced lung fibroblast PGE2 production. Fibroblasts were grown to confluence and then incubated for 24 hours in media alone (a) or in TGF-β1 at a concentration of 1 ng/ml (b). Cell lines from groups I and II were established from nonfibrotic lung and were categorized according to their ability to synthesize PGE2. Group III cell lines were derived from lung tissue from patients with pulmonary fibrosis. Results are expressed in pg of PGE2 per 10 5 cells. Each point represents an individual cell line and is derived from the mean for six replicate cultures. The median is indicated by the horizontal bar.
Effect of TGF-β1 on Fibroblast Proliferation
To ascertain whether the differences in PGE2 synthesis affected fibroblast function, the effect of TGF-β1 on the proliferation of cell lines from groups I, II, and III was examined. Figure 2 ▶ shows the effect of TGF-β1 on representative cell lines from the three groups. In response to the TGF-β1, group I fibroblasts exhibited a biphasic pattern of response with significant stimulation of proliferation at TGF-β1 concentrations of 5 to 40 pg/ml. With increasing concentrations of TGF-β1, the mitogenic response declined and at concentrations of 160 pg/ml TGF-β1 and greater, proliferation was inhibited. In contrast, group II fibroblasts failed to demonstrate an anti-proliferative response to TGF-β1. Concentrations of TGF-β1 at and less than 80 pg/ml had no significant effect whereas concentrations of 160 pg/ml and above induced proliferation. Fibroblasts from group III responded to TGF-β1 in a similar manner to the group II fibroblasts with a concentration-dependent increase in proliferation. Concentrations of TGF-β1 at and greater than 40 pg/ml induced significant proliferation. However, fibroblasts derived from fibrotic lung were still capable of responding to PGE2. In the presence of exogenous PGE2 (2 to 64 ng/ml), fibroblast proliferation was significantly inhibited in a dose-dependent manner ranging from −20 ± 3% at the lowest concentration of PGE2 (P < 0.05) to −35 ± 1% inhibition with 64 ng/ml PGE2 (P < 0.005). Treatment of cell lines with TGF-β2 and -β3 produced similar results to those observed for TGF-β1 (data not shown).
Figure 2.

Effect of TGF-β1 on lung fibroblast proliferation. Proliferation was assessed 72 hours after the addition of TGF-β1 at the indicated concentrations using a spectrophotometric assay in representative fibroblast lines from group I (a), group II (b), and group III (c). Results are expressed as percentage change compared to cells exposed to media alone. Mean absorbance at 650 nm for cells grown in media alone were 0.149 ± 0.004, 0.180 ± 0.012, and 0.349 ± 0.011 in groups I, II, and III, respectively. Each point represents the mean ± SEM for six replicate cultures. *, P < 0.05; **, P < 0.005; and ***, P < 0.000,5 respectively.
The effect of TGF-β1 on the proliferation of fibroblasts derived from seven group I cell lines, four group II cell lines, and 11 group III cell lines is shown in Figure 3 ▶ . A similar pattern of response was observed on treatment of these cell lines with a low (5 pg/ml) and high (160 pg/ml) concentration of TGF-β1. At 5 pg/ml TGF-β1, six of seven group I cell lines were stimulated to proliferate (median, 21%; range, −22 to 58%) and in all seven cell lines, proliferation was inhibited at 160 pg/ml TGF-β1 (median, −19%; range, −11 to −44%). Of the group II cell lines, three of four cell lines did not respond significantly to 5 pg/ml TGF-β1 (median, 2%; range, −7 to 26%). Moreover, all four group II cell lines failed to demonstrate any inhibitory response to TGF-β1 at 160 pg/ml (median, 26%; range, 10 to 41%) and in three cell lines this concentration was mitogenic (P < 0.05). A similar response was observed with group III, in which at 5 pg/ml TGF-β1, all 11 cell lines displayed a trend toward stimulation of proliferation (median, 10%; range, 4 to 38%). Again, at 160 pg/ml TGF-β1, all group III cell lines failed to show any inhibitory response (median, 15%; range, 1 to 40%). In eight of 11 cell lines, TGF-β1 at 160 pg/ml caused a further stimulation of proliferation. Further increasing the concentration of TGF-β1 up to 640 pg/ml did not induce an inhibitory response (data not shown).
Figure 3.

Effect of a low and high concentration of TGF-β1 on lung fibroblast proliferation. Fibroblast proliferation was assessed after the addition of TGF-β1 at 5 pg/ml or 160 pg/ml. Results are expressed as percentage change compared to cells exposed to media alone. Each point represents the mean for six replicate cultures.
Effect of TGF-β1 on Fibroblast Procollagen Production
Median basal procollagen production in cell lines from group I was 86 pmol hyp/10 5 cells/hour (range, 18 to 162 pmol hydroxyproline (hyp)/10 5 cells/hour) compared with 46 pmol hyp/10 5 cells/hour (range, 17 to 83 pmol hyp/10 5 cells/hour) in group II fibroblasts, which were not significantly different. In group III cells, derived from fibrotic lung, median procollagen production was approximately twofold to fourfold higher than for group I and II cell lines, respectively (median, 179 pmol hyp/10 5 cells/hour; range 27 to 323 pmol hyp/10 5 cells/hour; P < 0.02 in both cases). Treatment with TGF-β1 increased procollagen production further in all cell lines studied although the magnitude of stimulation varied (Figure 4) ▶ . In group I cell lines, a median stimulation of 54% greater than basal levels was observed with TGF-β1 (range, 7.1 to 94%). In group II cell lines, the median increase in procollagen production was almost threefold higher than that demonstrated for group I (median, 154%; range, 142 to 201%). The group III cell lines also displayed a greater increase in procollagen production on stimulation with TGF-β1, with median values twofold greater than those for group 1 (median, 100%; range, 19 to 224%).
Figure 4.

Effect of TGF-β1 on lung fibroblast procollagen production. Fibroblasts were grown to confluence and procollagen production was assessed 24 hours after the addition of TGF-β1 (1 ng/ml). Values were corrected for cell number and then expressed as percentage change compared with cells exposed to media alone. Each point represents an individual cell line and is derived from the mean of six replicate cultures. The median is indicated by the horizontal bar.
Effect of Indomethacin on TGF-β1-Induced Fibroblast Proliferation and Procollagen Production
To confirm that PGE2 was responsible for the anti-proliferative effects of TGF-β1, proliferation studies were performed in the presence and absence of indomethacin (Figure 5) ▶ . In group I fibroblasts indomethacin reversed the growth inhibition obtained with 160 pg/ml TGF-β1 alone and a mitogenic response was restored. In contrast to this, indomethacin had no effect on group II or group III fibroblasts in which 160 pg/ml TGF-β1 alone induced a proliferative response. Indomethacin had no effect on basal proliferation in any of the cell lines studied (data not shown). Indomethacin blocked the anti-proliferative response to TGF-β1 in three other group I cell lines (data not shown).
Figure 5.

Effect of indomethacin on TGF-β1-induced lung fibroblast proliferation. Fibroblasts were pretreated for 30 minutes with indomethacin (1 μg/ml) before the addition of TGF-β1 (160 pg/ml) and proliferation was assessed 48 hours later in representative cell lines from groups I, II, and III. Results are expressed as percentage change compared with cells exposed to media alone. The mean absorbance at 650 nm for cells grown in media alone was 0.149 ± 0.004, 0.349 ± 0.011, and 0.154 ± 0.004 in groups I, II, and III, respectively. Each bar represents the mean ± SEM for six replicate cultures.
The effect of indomethacin on basal and TGF-β1-induced procollagen production in representative fibroblast lines from groups I, II, and III is shown in Figure 6 ▶ . In fibroblasts from group I, indomethacin increased basal and TGF-β1-induced procollagen production by 24 ± 13% and 24 ± 7%, respectively, but this just failed to achieve statistical significance (P = 0.087 and P = 0.061, respectively). In fibroblasts from groups II and III, indomethacin had minimal effect on either basal or TGF-β1-induced procollagen production. Similar results were obtained for a further three group I cell lines, three group II cell lines, and two group III cell lines (data not shown).
Figure 6.

Effect of indomethacin on basal and TGF-β1-induced lung fibroblast procollagen production. Representative fibroblast cell lines from groups I, II, and III were grown to confluence and pretreated with indomethacin (1 μg/ml) for 30 minutes before the addition of TGF-β1 (1 ng/ml). Procollagen synthesis was assessed 24 hours later. Values for basal and TGF-β1-induced procollagen production (pmol hyp/10 5 cells/hour) from which percentage changes were calculated are as follows: group I: basal 86 ± 4, TGF-β1 146 ± 9; group II: basal 83 ± 4, TGF-β1 207 ± 16; group III: basal 284 ± 10, TGF-β1 922 ± 74. Each value represents the mean ± SEM for six replicate cultures.
Effect of Selective COX-1 and COX-2 Inhibitors on TGF-β1-Induced PGE2 Synthesis by Fibroblasts
To determine the mechanism of TGF-β1-induced PGE2 synthesis, COX isoforms were selectively inhibited using a selective COX-2 inhibitor, NS-398, and a preferential COX-1 inhibitor, piroxicam (Figure 7) ▶ . NS-398 (5 μg/ml) inhibited TGF-β1-induced PGE2 synthesis in representative cell lines from groups I, II, and III. Similar results were obtained with another three group I cell lines and two group III cell lines (data not shown). In contrast, the COX-1 preferential inhibitor, piroxicam (2.5 ng/ml), had no significant effect on TGF-β1-mediated PGE2 synthesis by group I or group III fibroblasts. Similar results were observed in another two group I and group III cell lines (data not shown). However, partial inhibition of TGF-β1-induced PGE2 synthesis did occur in group II fibroblasts in the presence of piroxicam.
Figure 7.
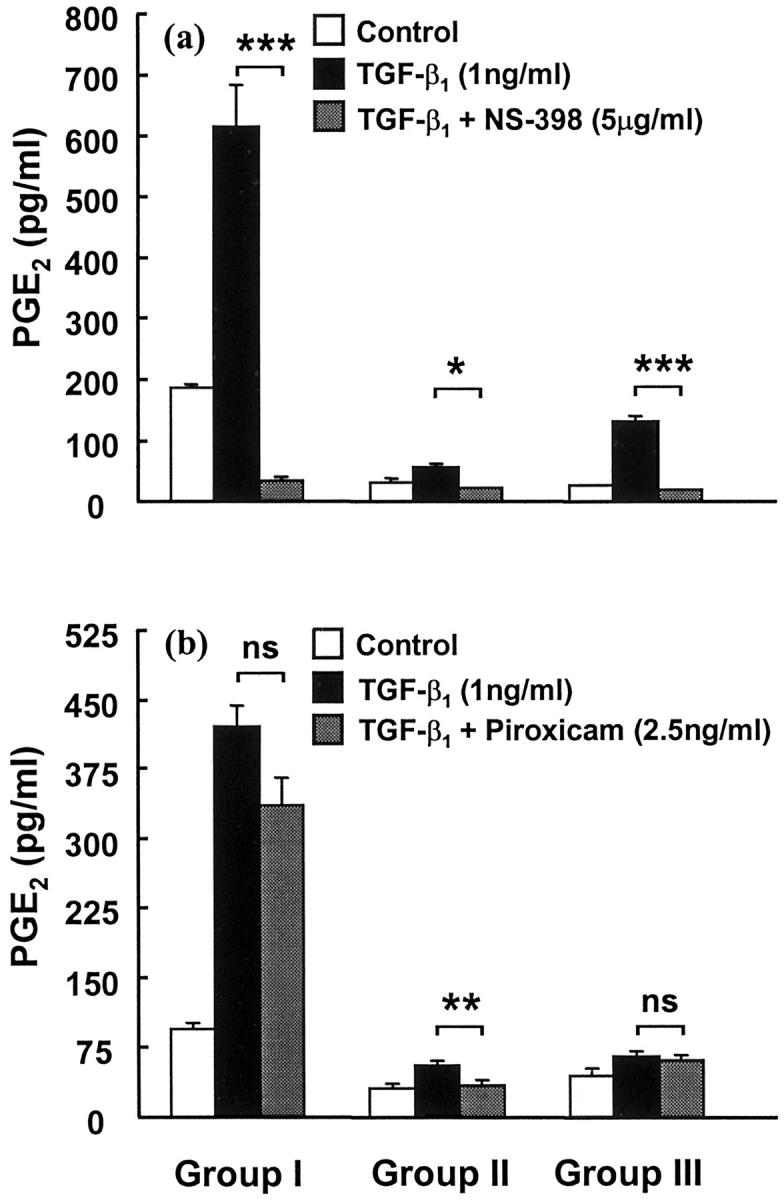
Effect of COX selective inhibitors on TGF-β1-induced lung fibroblast PGE2 production. Fibroblasts were grown to confluence and then pretreated for 30 minutes with either NS-398 (a) or piroxicam (b) before the addition of TGF-β1 (1 ng/ml) for 24 hours in representative cell lines from groups I, II, and III. Results are expressed in pg of PGE2 per ml of media. Each bar represents the mean ± SEM for six replicate cultures. *, P < 0.05; **, P < 0.005; and ***, P < 0.0005, respectively.
TGF-β1-Induced Changes in COX-1 and COX-2 mRNA in Fibroblasts
To examine whether the failure by group II and III fibroblasts to synthesize PGE2 was determined by pretranslational events, Northern analysis was performed on representative cell lines from each group (Figure 8) ▶ . A feint signal corresponding to basal COX-2 expression was detected as a 4.4-kb mRNA species in all three cell lines. A 6-hour treatment with TGF-β1 (1 ng/ml) increased steady-state levels of COX-2 mRNA by approximately fourfold in the group I control fibroblasts and this declined to baseline levels by 24 hours (data not shown). In contrast, levels of COX-2 mRNA were increased by ∼35 and 25% in response to TGF-β1 in group II and group III fibroblasts, respectively. In cells exposed to media alone, abundant COX-1 message was detected as a 2.7-kb transcript in all three groups. Levels of this transcript did not increase further in the presence of TGF-β1 in any of the cell lines studied. COX-1 mRNA was also readily detectable at 24 hours in all three groups (data not shown).
Figure 8.

Effect of TGF-β1 on COX-1 and COX-2 mRNA levels in lung fibroblasts. Representative fibroblast cell lines from groups I, II, and III were grown to confluence and incubated with media alone or media containing TGF-β1 (1 ng/ml) for 6 hours before extraction of total RNA. Hybridized probe was detected by autoradiography and quantitated by densitometric laser scanning. mRNA levels for COX-2 (a) and COX-1 (b) are expressed in arbitrary densitometry units after correcting for levels of 28S RNA.
Bleomycin-Induced Pulmonary Fibrosis in COX-2-Deficient Mice
To determine whether COX-2 and its induction of PGE2 synthesis modulates the fibrotic response, bleomycin was administered to COX-2+/+ and COX-2−/− mice. Figure 9 ▶ shows Masson’s trichrome-stained lung sections from COX-2+/+ and COX-2−/− mice, 14 days after instillation with either saline (0.9%) or bleomycin (1 mg/kg body weight). Alveolar architecture was preserved in saline-treated animals, with no apparent differences between COX-2+/+ and COX-2−/− mice (Figure 9, a and b) ▶ . In COX-2+/+ mice, treatment with bleomycin induced a mild fibrotic reaction, which was patchy and consisted of an inflammatory response and moderate thickening of the interstitium (Figure 9, c and e) ▶ . In contrast, in COX-2−/− mice, bleomycin-induced lung injury resulted in an aggressive fibroproliferative response, characterized by increased inflammation, with greater numbers of neutrophils and lymphocytes, and complete loss of alveolar architecture (Figure 9, d and f) ▶ . At higher magnification, increased extracellular matrix protein staining was evident (Figure 9f) ▶ . The increased fibroproliferative response in the lungs of bleomycin-treated COX-2−/− mice compared with COX-2+/+ mice was reinforced by semiquantitative analysis, with mean fibrosis scores of 2.44 ± 0.12 (n = 4) and 1.7 ± 0.14 (n = 6) in COX-2−/− and COX-2+/+ mice, respectively (P < 0.02). In saline-treated animals, mean fibrosis scores were 0.47 ± 0.19 and 0.33 ± 0.21 for COX-2+/+ (n = 7) and COX-2−/− (n = 4) mice, respectively, and were not significantly different.
Figure 9.
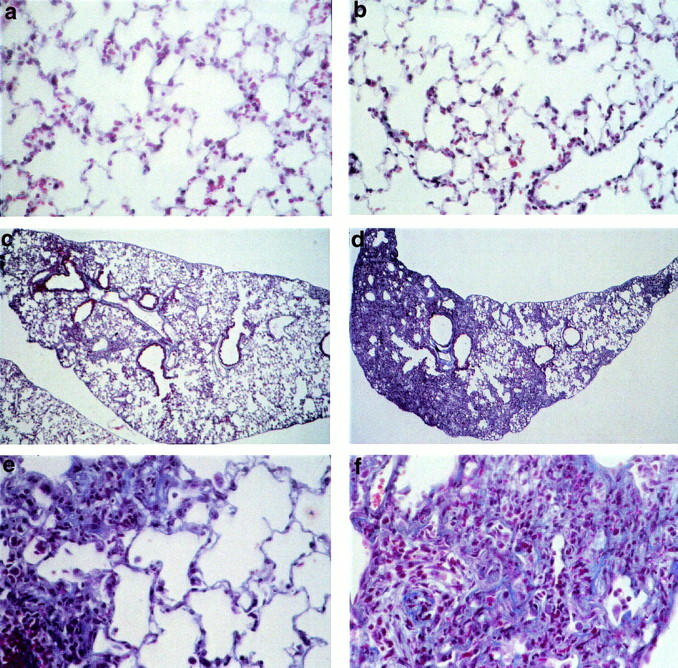
Extracellular matrix protein deposition after bleomycin-induced pulmonary fibrosis in COX-2-deficient mice. COX-2+/+ (a, c, and e) and COX-2−/− (b, d, and f) mice received saline (a and b) or bleomycin sulfate (c–f) via intratracheal instillation. Lungs were harvested after 14 days and sections stained with Masson’s trichrome. In saline-treated animals (a and b) normal lung architecture was preserved. In bleomycin-treated COX-2+/+ mice, mild inflammation and moderate alveolar wall thickening was observed (c and e). In contrast, widespread inflammation and destruction of alveolar structures occurred in COX-2−/− mice instilled with bleomycin (d and f). Original magnifications: ×400 (a, b, e, and f); ×100 (c and d).
Discussion
The pathogenesis of pulmonary fibrosis is thought to be associated with the induction of mediators that promote fibroblast proliferation and extracellular matrix synthesis. However, the reduction of inhibitory mediators of fibroblast function may be equally important. In the present study, we have addressed the hypothesis that a loss of the normal inhibitory mechanisms coupled with an increase in profibrotic mediators plays an important role in the development of pulmonary fibrosis.
Autocrine induction of PGE2 synthesis in response to TGF-β1 is well documented in lung fibroblasts. 6,12,13,38,39 In this study, we have demonstrated that fibroblasts from fibrotic lungs synthesize less PGE2 both basally and in response to TGF-β1. Similar findings were reported by another group using IL-1β, phorbol myristate, or lipopolysaccharide as stimulants of PGE2 synthesis. 20 This implies that this defect is not confined to TGF-β1 alone but extends to other stimuli. In addition, the defect in PGE2 synthesis may not be specific to the fibroblast cell type, because the lung contains other cells capable of synthesizing PGE2 including macrophages, bronchial epithelial, and smooth muscle cells. 40-42 This premise is further supported by data demonstrating that PGE2 levels are ∼50% lower in bronchoalveolar lavage fluid from patients with pulmonary fibrosis than nonfibrotic controls, 19 despite elevated levels of mediators capable of stimulating PGE2 production. In the present study, we also discovered a number of cell lines derived from nonfibrotic lung, which synthesized low levels of PGE2 basally and in response to TGF-β1. Similar results have been reported previously for human arterial smooth-muscle cell lines stimulated with PDGF and this correlated with a lack of COX-2 expression. 43 Because PGE2 has potent inhibitory effects on fibroblast proliferation and collagen production, this decreased capacity to synthesize PGE2 may affect fibroblast function and contribute to the pathogenesis of pulmonary fibrosis.
We have previously shown that TGF-β1 induces a biphasic response in normal human fetal lung fibroblasts with stimulation of proliferation at low concentrations and inhibition at high concentrations. 6 Autocrine synthesis of PGE2 in response to TGF-β1 was shown to be responsible for the anti-proliferative effects. In the present study, inhibition of PGE2 synthesis with indomethacin abolished the anti-proliferative effect of TGF-β1 in fibroblasts from nonfibrotic lung that were capable of synthesizing PGE2, suggesting that PGE2 also mediates such effects in adult human fibroblasts. The striking lack of TGF-β1-mediated inhibition of proliferation in the fibroblast cell lines from fibrotic lung correlates with their inability to synthesize PGE2. This was also exemplified by the non-PGE2 synthesizing, control cell lines in which TGF-β1 failed to evoke an inhibitory response. In many of these cell lines, increasing the concentration of TGF-β1 produced a further stimulation of proliferation. The mitogenic response to TGF-β1 has previously been shown to be because of autocrine synthesis of PDGF 44,45 and it has recently been shown that PGE2 down-regulates expression of the α subunit of the PDGF receptor. 46 It is therefore possible that the further enhancement in proliferation observed in these cell lines with increasing concentrations of TGF-β1 is because of up-regulation of the PDGF receptor α subunit. Furthermore, in the absence of PGE2 fibroblasts can proliferate in response to leukotriene C4, which is known to be increased in the lungs of patients with idiopathic pulmonary fibrosis. 47,48 Thus in fibrosis, the decreased capacity of fibroblasts to up-regulate PGE2 production in response to profibrotic mediators provides several potential mechanisms for the fibroblast hyperproliferation observed in this disease.
Autocrine synthesis of PGE2 limits TGF-β1-induced procollagen production in human fetal lung fibroblasts. 13 In the current study, fibroblasts from the fibrosis and non-PGE2 synthesizing groups produced approximately twofold and threefold more procollagen, respectively, than group I control fibroblasts in response to TGF-β1. This exaggerated increase in procollagen production coincides with the failure to induce PGE2 synthesis in response to TGF-β1 by these cell lines. Indomethacin did not further stimulate TGF-β1-induced procollagen production in fibroblasts from groups II and III. In group I control fibroblast cell lines, indomethacin showed a tendency to potentiate TGF-β1-induced collagen synthesis by ∼20%, a similar potentiation to that observed previously in fetal lung fibroblasts, 13 although in this study this was not statistically significant. Because the development of pulmonary fibrosis is relatively slow and progressive in most cases, an increase of this magnitude could contribute significantly to the impairment of alveolar function throughout time. However it is unlikely that reduced PGE2 synthesis is the only mechanism involved in the enhanced collagen production by group II and III cell lines in response to TGF-β1.
Experiments addressing the mechanism by which TGF-β1 induced PGE2 synthesis implicated induction of COX-2. A COX-2 selective inhibitor, NS-398 almost completely inhibited TGF-β1-induced-PGE2 synthesis in all groups. The preferential COX-1 inhibitor, piroxicam had a minimal effect in fibroblasts from groups I and III but it partially inhibited TGF-β1-induced PGE2 synthesis in group II fibroblasts. This may be because of the inhibition of COX-2 by piroxicam because this compound is not totally selective for COX-1 or inhibition of basal COX-1-mediated PGE2 synthesis in this group. Northern analysis showed that in response to TGF-β1, group I control fibroblasts up-regulated steady state COX-2 mRNA levels by at least fourfold whereas effects were minimal for the group III fibrosis and group II control fibroblasts. Although group I cell lines produced more PGE2 basally than cell lines from groups II and III, there did not seem to be any difference in COX-2 mRNA levels between the groups. However, this may reflect the very low levels of basal COX-2 expression that makes accurate quantitation of any small differences between groups difficult. COX-1 mRNA levels, although more abundant, did not increase with TGF-β1 in any of the cell lines studied. Previous studies have suggested that TGF-β1 does not induce steady state levels of COX-2 mRNA in human lung fibroblasts. 38,39 However, these studies were performed throughout 16 to 24 hours whereas we assessed COX-2 mRNA levels after 6 hours stimulation with TGF-β1. COX-2 is an immediate early gene, and levels of mRNA have been shown to return to baseline by ∼8 hours in the murine homologue of COX-2. 49
The failure to up-regulate COX-2 mRNA levels suggests a pretranslational defect in both the group II non-PGE2 synthesizing control cells and the group III fibroblasts derived from fibrotic lung. There are a number of possible explanations for this including a possible promoter-associated defect or decreased mRNA stability. A TGF-β response element has been localized to the COX-2 promoter 50 but this is unlikely to be the source of the defect given that IL-1β also fails to induce COX-2 in fibroblasts from fibrotic lung. 20 In addition, a number of other putative regulatory regions have also been identified in the COX-2 promoter 51 and parts of the 3′-untranslated region of the human COX-2 gene have been shown to affect basal and IL-1β-induced mRNA metabolism. 52 Further studies will be required to determine the mechanisms involved in the dysregulation of COX-2 expression in these cells.
The reason for the limitation in COX-2 induction is unknown. However, there are several potential mechanisms. It may be because of COX-2 polymorphisms that result in the loss of COX-2 inducibility. At present, there is no evidence for the existence of COX-2 polymorphisms in the normal population. Alternatively, the inability to induce COX-2 could reflect an acquired defect that is specific to the lung. For example, viral infection could alter a cell’s ability to induce COX-2. There is evidence to suggest an association between Epstein-Barr virus infection and pulmonary fibrosis. 53,54 Furthermore, in human B cells, the presence of Epstein-Barr virus in either its wild-type or latent form results in a loss of the anti-proliferative effects of TGF-β1. 55 Other DNA tumor viruses such as SV40, adenovirus, and human papilloma virus also confer resistance to TGF-β1-mediated growth inhibition in human keratinocytes. 56 Further studies are required to determine the role of viral infection and COX-2 expression in patients with pulmonary fibrosis.
The decreased capacity to up-regulate COX-2 and PGE2 synthesis in fibroblasts derived from fibrotic lung together with the effects this confers on these cells suggests that this defect may play an important role in the pathogenesis of pulmonary fibrosis. Other studies also support a role for COX-2 in this fibroproliferative pathology. For example, up to 58% of patients with rheumatoid arthritis develop interstitial lung disease. 3 Frequently, these patients are on long-term treatment with nonsteroidal anti-inflammatory drugs and given the data presented here it is possible that persistent pharmacological inhibition of COX-2 may contribute to the development or progression of pulmonary disease in these individuals. In addition, evidence is accumulating that suggests that COX-2 is anti-inflammatory. 57,58 The pathology exhibited by COX-2-deficient mice is also consistent with an anti-fibrotic role for this enzyme. COX-2 null mice, although demonstrating no obvious innate lung pathology, suffer from fibrotic abnormalities of the kidneys, heart, and ovaries, as well as developing peritoneal adhesions. 22,23 In terms of the lung, COX-2-deficient mice sensitized and challenged with ovalbumin develop an increased inflammatory response. 58 Furthermore, we provide evidence for the first time demonstrating that COX-2-deficient mice exhibit enhanced lung injury in response to bleomycin. Histologically, this was characterized by marked inflammation, including increased numbers of neutrophils and lymphocytes, and increased collagen deposition in the lungs of COX-2-deficient mice compared with wild-type mice instilled with bleomycin. Increased COX-2 expression is evident in the lungs of wild-type mice exposed to bleomycin (data not shown) and is consistent with elevated levels of PGE2 after bleomycin-induced pulmonary fibrosis in hamsters. 59 This suggests that PGE2 synthesis via induction of COX-2 is required to promote normal healing and resolution and that COX-2 plays an important role in the regulation of inflammatory and fibroproliferative conditions.
In summary, we have provided functional evidence demonstrating the effects of deficient COX-2 expression in fibroblasts from patients with pulmonary fibrosis. We have also examined the effect of bleomycin-induced lung injury in COX-2-deficient mice. In control fibroblasts, TGF-β1 up-regulates COX-2 expression and PGE2 synthesis that in turn inhibits proliferation and limits collagen production, however, fibroblasts from fibrotic lung lack this anti-proliferative response to TGF-β1 and exhibit enhanced collagen synthesis. Furthermore, we have identified a group of cell lines established from nonfibrotic lung that also fail to induce COX-2 expression and demonstrate loss of TGF-β1-mediated growth inhibition and exhibit exaggerated collagen synthesis. The individuals from whom these cell lines were derived may represent a subset of the normal population who are predisposed to developing pulmonary fibrosis. Finally, we demonstrate that COX-2-deficient mice are more susceptible to bleomycin-induced lung injury, which suggests that augmenting COX-2 or PGE2 levels in the lung may be of therapeutic benefit in patients with pulmonary fibrosis.
Acknowledgments
We thank Dr. Neil Goldsack for collecting lung biopsy specimens and Dr. Tim Hla for donating a human COX-2 plasmid construct.
Footnotes
Address reprint requests to Robin J. McAnulty, Cardiopulmonary Biochemistry and Respiratory Medicine, Royal Free and University College London Medical School, Rayne Institute, 5 University St., London WC1E 6JJ, UK. E-mail: r.mcanulty@ucl.ac.uk.
Supported by the Biotechnology and Biological Sciences Research Council (UK), Aventis Pharmaceuticals (formerly Rhone Poulenc-Rorer), the Wellcome Trust (UK), and the Arthritis Research Campaign (UK).
References
- 1.Turner-Warwick M: In search of a cause of cryptogenic fibrosing alveolitis (CFA): one initiating factor or many? Thorax 1998, 53(Suppl 2):S3-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison NK, Myers AR, Corrin B, Soosay G, Dewar A, Black CM, Du Bois RM, Turner-Warwick M: Structural features of interstitial lung disease in systemic sclerosis. Am Rev Respir Dis 1991, 144:706-713 [DOI] [PubMed] [Google Scholar]
- 3.Gabbay E, Tarala R, Will R, Carroll G, Adler B, Cameron D, Lake FR: Interstitial lung disease in recent onset rheumatoid arthritis. Am J Respir Crit Care Med 1997, 156:528-535 [DOI] [PubMed] [Google Scholar]
- 4.McAnulty RJ, Laurent GJ: Collagen and its regulation in pulmonary fibrosis. Phan SH Thrall RS eds. Pulmonary Fibrosis. 1995, :pp 135-171 Marcel Dekker, New York [Google Scholar]
- 5.Oliver MH, Harrison NK, Bishop JE, Cole PJ, Laurent GJ: A rapid and convenient assay for counting cells cultured in microwell plates: application for assessment of growth factors. J Cell Sci 1989, 92:513-518 [DOI] [PubMed] [Google Scholar]
- 6.McAnulty RJ, Hernandez-Rodriguez NA, Mutsaers SE, Coker RK, Laurent GJ: Indomethacin suppresses the anti-proliferative effects of transforming growth factor-β isoforms on fibroblast cell cultures. Biochem J 1997, 321:639-643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein RH, Polgar P: The effect and interaction of bradykinin and prostaglandins on protein and collagen production by lung fibroblasts. J Biol Chem 1982, 257:8630-8633 [PubMed] [Google Scholar]
- 8.Saltzman LE, Moss J, Berg RA, Hom B, Crystal RG: Modulation of collagen production by fibroblasts: effects of chronic exposure to agonists that increase intracellular cyclic AMP. Biochem J 1982, 204:25-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozaki T, Rennard SI, Crystal RG: Cyclooxygenase metabolites are compartmentalized in the human lower respiratory tract. J Appl Physiol 1987, 62:219-222 [DOI] [PubMed] [Google Scholar]
- 10.Korn JH: Fibroblast prostaglandin E2 synthesis. Persistence of an abnormal phenotype after short-term exposure to mononuclear cell products. J Clin Invest 1983, 71:1240-1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elias JA: Tumour necrosis factor interacts with interleukin-1 and interferons to inhibit fibroblast proliferation via fibroblast prostaglandin-dependent and independent mechanisms. Am Rev Respir Dis 1988, 138:652-658 [DOI] [PubMed] [Google Scholar]
- 12.Diaz A, Varga J, Jimenez SA: Transforming growth factor-β stimulation of lung fibroblast prostaglandin E2 production. J Biol Chem 1989, 264:11554-11557 [PubMed] [Google Scholar]
- 13.McAnulty RJ, Chambers RC, Laurent GJ: Regulation of fibroblast procollagen production. Transforming growth factor-β1 induces prostaglandin E2 but not procollagen synthesis via a pertussis toxin-sensitive G-protein. Biochem J 1995, 307:63-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yucel-Lindberg T, Ahola H, Nilsson S, Carlstedt-Duke J, Modeer T: Interleukin-1β induces expression of cyclooxygenase-2 mRNA in human gingival fibroblasts. Inflammation 1995, 19:549-560 [DOI] [PubMed] [Google Scholar]
- 15.Mauviel A, Daireaux M, Redini F, Galera P, Loyau G, Pujol J-P: Tumour necrosis factor inhibits collagen and fibronectin synthesis in human dermal fibroblasts. FEBS Lett 1988, 236:47-52 [DOI] [PubMed] [Google Scholar]
- 16.Habenicht AJR, Goerig M, Grulich J, Rothe D, Gronwald R, Loth U, Schettler G, Kommerell B, Ross R: Human platelet-derived growth factor stimulates prostaglandin synthesis by activation and by rapid de novo synthesis of cyclooxygenase. J Clin Invest 1985, 75:1381-1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuroki S, Ohta A, Sueoka N, Katoh O, Yamada H, Yamaguchi M: Determination of various cytokines and type III procollagen aminopeptide levels in bronchoalveolar lavage fluid of the patients with pulmonary fibrosis: inverse correlation between type III procollagen aminopeptide and interferon-γ in progressive patients. Br J Rheumatol 1995, 34:31-36 [DOI] [PubMed] [Google Scholar]
- 18.Ludwicka A, Ohba T, Trojanowska M, Yamakage A, Strange C, Smith EA, Leroy EC, Sutherland S, Silver RM: Elevated levels of platelet-derived growth factor and transforming growth factor-β1 in bronchoalveolar lavage fluid from patients with scleroderma. J Rheumatol 1995, 22:1876-1883 [PubMed] [Google Scholar]
- 19.Borok Z, Gillissen A, Buhl R, Hoyt RF, Hubbard RC, Ozaki T, Rennard SI, Crystal RG: Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am Rev Respir Dis 1991, 144:1080-1084 [DOI] [PubMed] [Google Scholar]
- 20.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Streiter RM, Peters-Golden M: Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express COX-2. J Clin Invest 1995, 95:1861-1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vancheri C, Sortino MA, Tomaselli V, Mastruzzo C, Condorelli F, Bellistri G, Pistorio MP, Canonico PL, Crimi N: Different expression of TNF-α receptors and prostaglandin E2 production in normal and fibrotic lung fibroblasts. Potential implications for the evolution of the inflammatory process. Am J Respir Cell Mol Biol 2000, 22:628-634 [DOI] [PubMed] [Google Scholar]
- 22.Morham SG, Lagenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O: Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 1995, 83:473-482 [DOI] [PubMed] [Google Scholar]
- 23.Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng MB, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM: Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 1995, 378:406-409 [DOI] [PubMed] [Google Scholar]
- 24.Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, duBois RM, Jeffery PK, McAnulty RJ: Transforming growth factors-β1, -β2 and -β3 stimulate fibroblast procollagen in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am J Pathol 1997, 150:981-991 [PMC free article] [PubMed] [Google Scholar]
- 25.Khalil N, Bereznay O, Sporn M, Greenberg AH: Macrophage production of transforming growth factor β and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 1989, 170:727-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Broekelmann TJ, Limper AH, Colby TV, McDonald JA: Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci USA 1991, 88:6642-6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westergren-Thorsson G, Hernnas J, Sarnstrand B, Oldberg A, Heinegard D, Malmstrom A: Altered expression of small proteoglycans, collagen and transforming growth factor-beta 1 in developing bleomycin-induced pulmonary fibrosis in rats. J Clin Invest 1993, 92:632-637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS: Transforming growth factor rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA 1986, 83:4167-4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J: Adenovector-mediated gene transfer of active transforming-growth factor-β1 induces prolonged severe fibrosis in rat lung. J Clin Invest 1997, 100:768-776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giri SN, Hyde DM, Hollinger MA: Effect of antibody to transforming growth factor-beta on bleomycin-induced accumulation of lung collagen in mice. Thorax 1993, 48:959-966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giri SN, Hyde DM, Braun RK, Gaarde W, Harper JR, Pierschbacher MD: Antifibrotic effect of decorin in a bleomycin hamster model of lung fibrosis. Biochem Pharmacol 1997, 54:1205-1216 [DOI] [PubMed] [Google Scholar]
- 32.Munger JS, Huang X, Kawakatsu H, Griffiths MJD, Dalton SL, Wu J, Pittet J-P, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D: The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96:319-328 [DOI] [PubMed] [Google Scholar]
- 33.Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I: Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest 1999, 104:5-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jordana M, Schulman J, McSharry C, Irving LB, Newhouse MT, Jordana G, Gauldie J: Heterogeneous proliferative characteristics of human adult lung fibroblasts lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis 1988, 137:579-584 [DOI] [PubMed] [Google Scholar]
- 35.Wong E, Bayly C, Waterman HL, Riendeau D, Mancini JA: Conversion of prostaglandin G/H synthase-1 into an enzyme sensitive to PGHS-2-selective inhibitors by a double His513? Arg and lle523? Val mutation. J Biol Chem 1997, 272:9280-9286 [DOI] [PubMed] [Google Scholar]
- 36.Frolich JC: A classification of NSAIDs according to the relative inhibition of cyclooxygenase isoenzymes. Trends Pharmacol Sci 1997, 18:30-34 [DOI] [PubMed] [Google Scholar]
- 37.Aschroft T, Simpson JM, Timbrell V: Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 1988, 41:467-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diaz A, Chepenik KP, Korn JH, Reginato AM, Jimenez SA: Differential regulation of cyclooxygenases 1 and 2 by interleukin-1β, tumour necrosis factor-α and transforming growth factor-β1 in human lung fibroblasts. Exp Cell Res 1998, 241:222-229 [DOI] [PubMed] [Google Scholar]
- 39.Jackson BA, Goldstein RH, Roy R, Cozzani M, Taylor L, Polgar P: Effects of transforming growth factor β and interleulin-1β on expression of cyclooxygenase 1 and 2 and phospholipase A2 mRNA in lung fibroblasts and endothelial cells in culture. Biochem Biophys Res Commun 1993, 197:1465-1474 [DOI] [PubMed] [Google Scholar]
- 40.Hempel SL, Monick MM, Hunninghake GW: Lipopolysaccharide induces prostaglandin H synthase-2 protein and mRNA in human alveolar macrophages and blood monocytes. J Clin Invest 1994, 93:391-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asano K, Lilly CM, Drazen JM: Prostaglandin G/H synthase-2 is the constitutive and dominant isoform in cultured human lung epithelial cells. Am J Physiol 1996, 271:L126-L131 [DOI] [PubMed] [Google Scholar]
- 42.Pang L, Knox AJ: Effect of interleukin-1β, tumour necrosis factor-α and interferon-γ on the induction of cyclo-oxygenase-2 in cultured human airway smooth muscle cells. Br J Pharmacol 1997, 121:579-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bornfeldt KE, Campbell JS, Koyama H, Argast GM, Leslie CC, Raines EW, Krebs EG, Ross R: The mitogen-activated protein kinase pathway can mediate growth inhibition and proliferation in smooth muscle cells. Dependence on the availability of downstream targets. J Clin Invest 1997, 100:875-885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leof EB, Proper JA, Goustin AS, Shipley GD, DiCorleto PE, Moses HL: Induction of c-sis mRNA and activity similar to platelet-derived growth factor by transforming growth factor β: a proposed model for indirect mitogenesis involving autocrine activity. Proc Natl Acad Sci USA 1986, 83:2453-2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R: TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 1990, 63:515-524 [DOI] [PubMed] [Google Scholar]
- 46.Boyle JE, Lindroos PN, Rice AB, Zhang L, Zeldin DC, Bonner JC: Prostaglandin-E2 counteracts interleukin-1β-stimulated upregulation of platelet-derived growth factor α-receptor on rat pulmonary myofibroblasts. Am J Respir Cell Mol Biol 1999, 20:433-440 [DOI] [PubMed] [Google Scholar]
- 47.Baud L, Perez J, Denis M, Ardaillou R: Modulation of fibroblast proliferation by sulfidopeptide leukotrienes: effect of indomethacin. J Immunol 1987, 138:1190-1195 [PubMed] [Google Scholar]
- 48.Wilborn J, Bailie M, Coffey M, Burdick M, Strieter R, Golden-Peters M: Constitutive activation of 5-lipoxygenase in the lungs of patients with idiopathic pulmonary fibrosis. J Clin Invest 1996, 97:1827-1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschmann HR: TIS10, a phorbol ester tumour promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem 1991, 266:12866-12872 [PubMed] [Google Scholar]
- 50.Yang X, Hou F, Taylor L, Polgar P: Characterization of human cyclooxygenase 2 gene promoter localization of a TGF-β response element. Biochim Biophys Acta 1997, 1350:287-292 [DOI] [PubMed] [Google Scholar]
- 51.Tazawa R, Xu XM, Wu KK, Wang LH: Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun 1994, 203:190-199 [DOI] [PubMed] [Google Scholar]
- 52.Gou Q, Liu CH, Ben-Av P, Hla T: Dissociation of basal and cytokine-induced transcript stabilization of the human cyclooxygenase-2 mRNA by mutagenesis of the 3′-untranslated region. Biochem Biophys Res Commun 1998, 242:508-512 [DOI] [PubMed] [Google Scholar]
- 53.Egan JJ, Stewart JP, Hasleton PS, Arrand JR, Carroll KB, Woodcock AA: Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax 1995, 50:1234-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, Woodcock AA: The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1999, 159:1336-1341 [DOI] [PubMed] [Google Scholar]
- 55.Arvanitakis L, Yaseen N, Sharma S: Latent membrane protein-1 induces cyclin D2 expression, pRB hyperphosphorylation, and loss of TGF-β1-mediated growth inhibition in EBV-positive B cells. J Immunol 1995, 155:1047-1056 [PubMed] [Google Scholar]
- 56.Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, Pittelkow MR, Munger K, Howley PM, Moses HL: TGF-β1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990, 61:777-785 [DOI] [PubMed] [Google Scholar]
- 57.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA: Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 1999, 5:698-701 [DOI] [PubMed] [Google Scholar]
- 58.Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, Tiano HF, Lee CA, Lagenbach R, Roggli VL, Zeldin DC: Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest 1999, 104:721-732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandler DB, Giri SN: Changes in plasma concentrations of prostaglandins and plasma angiotensin-converting enzyme during bleomycin-induced lung fibrosis in hamsters. Am Rev Respir Dis 1983, 128:71-76 [DOI] [PubMed] [Google Scholar]