

Abstract
Epstein-Barr virus (EBV)-positive Burkitt’s lymphoma cells and EBV-infected B cells elicit humoral factors that inhibit tumor-induced angiogenesis, resulting in tumor necrosis and regression. Of the chemokine factors identified in association with this growth behavior, none have induced complete tumor regression. We have previously identified tissue inhibitors of metalloproteinase (TIMP)-1 in various B cell lymphoma cell lines. Here we show that induction of TIMP-1 expression in an EBV-negative Burkitt’s lymphoma cell line results in a biphasic, in vivo tumor growth pattern in the nude mouse that is essentially identical to EBV-positive Burkitt’s lymphoma cell lines. The initial effect of TIMP-1 is to enhance tumor growth, consistent with the reported anti-apoptotic effect of TIMP-1 on B cell growth. Tumor necrosis and regression then follow the initial period of rapid, increased tumor growth. Only microscopic foci of residual, proliferating tumor cells are observed on biopsy of the tumor site. This latter effect is mediated by TIMP-1 inhibition of an angiogenic response within the developing tumor mass, as demonstrated by immunostaining and microvessel counts. These findings suggest that TIMP-1 is an important mediator of the in vivo growth properties of EBV-positive Burkitt’s lymphoma.
It is well known that Epstein-Barr virus (EBV) infection is associated with several highly aggressive human tumors, including Burkitt’s lymphoma (BL) and nasopharyngeal carcinoma. 1 EBV-positive human BL cells form subcutaneous tumors after inoculation into nude mice and these tumors undergo spontaneous necrosis and complete regression. 2-4 This is in contrast to EBV-negative BL-cells, which demonstrate continued tumor growth without regression. EBV gene products induce humoral factors (eg, CXC chemokines) that result in inhibition of neovascularization and regression of EBV+ BL. 5 However, these effectors fail to induce complete tumor regression suggesting involvement of additional, unidentified factors. 3,4
Matrix metalloproteinases (MMPs) mediate remodeling of the extracellular matrix that is requisite in normal tissue development. Altered MMP activity results in a disruption of extracellular matrix organization that is observed in many developmental abnormalities and disease-related phenotypes. 6-8 The endogenous tissue inhibitors of metalloproteinases (TIMPs) negatively regulate MMP activity associated with a variety of pathological conditions. Reduction or elimination of TIMP gene expression results in enhanced extracellular matrix proteolysis concomitant with progression of many disease states, including tumor progression. 9,10 In contrast, overexpression of TIMPs results in inhibition of tumor cell invasion, tumor growth, metastasis, and angiogenesis. 11-15 Recent transgenic animal models have demonstrated that modulation of TIMP-1 activity in host hepatic tissues can block neoplastic proliferation in the SV40 T-antigen-induced model of hepatocellular carcinoma. 16 TIMP-1 expression in this model inhibits the MMP-dependent processing of insulin-like growth factor binding protein-3, that in turn results in a decrease in the release of insulin-like growth factor II. Other studies have demonstrated that decreased hepatic expression of TIMP-1 can increase host susceptibility to liver metastasis from subcutaneous T cell lymphoma. 17 The results of such studies indicate that TIMPs can indirectly suppress tumor growth in vivo through inhibition of MMP activity. 16,17
However, recent data demonstrate a direct positive correlation between TIMP expression and tumor progression in breast, 18 colorectal, 19 lung, 20 and gastric 21 cancer patients. Although frequently dismissed as a nonspecific host response to the invasive activity of these tumors, several laboratories have demonstrated direct effects of TIMPs on cell growth in vitro. 11,22,23 In human B-cell lymphoma patients TIMP-1 expression directly correlates with the histological grade, ie, high-grade lymphomas have a statistically significant increase in TIMP-1 transcripts compared with low-grade lymphoma tissues as determined by semiquantitative polymerase chain reaction. 24 TIMP-1 expression also correlates with the latency 1,25 of EBV infection in BL cells. Advanced latency (II/III) EBV-positive BL cells express TIMP-1, whereas EBV-negative cells and cells with EBV latency I do not. 26 The aggressive clinical behavior of the EBV-positive BL cells is correlated with altered expression of cell differentiation and activation markers, enhanced resistance to apoptosis, as well as induction of autocrine B-cell growth factors such as interleukin-10. All of these BL tumor markers correlate with TIMP-1 expression in clinical samples. 22,24,26 Furthermore, TIMP-1 expression in an EBV-negative BL cell line results in altered expression of cell differentiation markers, as well as resistance to programmed cell death. 22,26 These findings suggest that enhanced TIMP-1 expression may be causally related to the aggressive phenotypic changes in human B cell tumors associated with EBV infection. These phenotypic changes associated with increased TIMP-1 expression are consistent with the clinically aggressive behavior of BL tumors in human lymphoma patients. However, these in vitro results are in contrast to the reduced tumorigenicity of human EBV-positive BL cells in the nude mouse model observed by many investigators. 3,5 To resolve these divergent effects of TIMP-1 on the growth of EBV-positive (II/III) BL cells, we examined the effect of TIMP-1 expression on the in vivo growth of EBV-negative BL tumors. Our findings are the first in vivo demonstration of divergent effects of TIMP-1 on the same tumor; an early TIMP-1 tumor growth-promoting effect that is followed by a subsequent and dominant anti-angiogenic effect of TIMP-1 resulting in tumor regression.
Materials and Methods
Cells
BL cell lines were a gift from Dr. Ian Magrath (Pediatric Oncology Branch, National Cancer Institute, Bethesda, MD). The generation of overexpressing TIMP-1 clones (JD38TIMP-1) and vector control clones (JD38LXSN) by retroviral transduction of the parental JD38 BL cells has been previously described. 22 Cell cultures were grown in RPMI 1640 (Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% fetal bovine serum, antibiotics, and l-glutamine. Serum was removed from cultures by washing with phosphate-buffered saline (PBS) before injection into mice.
Mice
Six- to eight-week-old, female, athymic nu/nu mice (National Cancer Institute) were used. Exponential growing cells (0.5 to 0.8 × 10 7 cells in 0.3 ml of PBS per mouse) were injected subcutaneously into the right posterior lateral thoracic wall. Tumor growth was assessed by two-dimensional caliper measurements and recorded as cm 2 every third day for 30 days. These data are presented as tumor area (mean ± SE). Statistical differences between groups were determined using analysis of variance methods.
Histology and Immunostaining
Tumors were removed frozen or fixed in 10% neutral-buffered formalin solution and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin. Immunohistochemistry of tumor sections was performed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) for the staining of murine antigens and the M.O.M. kit (Vector Laboratories) for the staining of human antigens in murine tissue. Murine bone marrow cells were stained with appropriate antibody combinations (Becton Dickinson, San Jose, CA) for identifying cell subsets and analyzed by flow cytometry using a FACScan and Cell Quest analysis software (Becton Dickinson).
Functional TIMP-1 Assay
Supernatants from control JD38-LXSN cells and two independent clones of JD38-TIMP-1 cells were tested for TIMP-1 inhibition of gelatin digestion using reverse zymography as previously reported. 27
Endothelial Cell Migration Assay
Human microvascular endothelial cells–lung (Clonetics, San Diego, CA) were labeled with 5 μmol/L of calcein-AM (Molecular Probes, Eugene, OR) in serum-free media for 2 hours at 37°C and constant rocking. After washing, cells were resuspended in tissue culture media supplemented with 0.1% bovine serum albumin. Fifty thousand cells were added to each well of a Falcon HTS Fluoroblok (3-μm pore) insert. Endothelial basal media (EBM; 200 μl) containing 10% fetal bovine serum, 50 ng/ml bFGF (Collaborative Biomedical Products, Franklin Lakes, NJ) was used as a chemoattractant in the lower wells of both positive controls and cells treated with 15 μl of JD38-LXSN and JD38-TIMP-1 supernatants or 50 nmol/L of rTIMP-1. EBM/0.1% bovine serum albumin was added to the bottom of negative control wells. After 4 hours of incubation at 37°C, the fluorescence of the cells that had migrated through the insert was measured on a plate reader (Perkin Elmer, Emeryville, CA) at 485/530 nm and compared with the fluorescence standard curve of calcein-AM-labeled human microvascular endothelial cells–lung cells at 10, 100, 1,000, 10,000, and 100,000 cells/well.
Results
To study the in vivo growth characteristics of BL, cell lines were inoculated subcutaneously into athymic nude mice. The BL tumor regression was correlated with EBV status and TIMP-1 expression. 26 The results of four representative cell lines are shown in Table 1 ▶ . EBV latency II/III (AG876 and PA682) cell lines expressed TIMP-1 and developed central tumor necrosis before regression. In contrast, EBV-negative tumors (JD38) or tumors with EBV latency I (ST482) failed to express TIMP-1, develop necrosis, or undergo regression. In addition, all EBV latency II/III, TIMP-1-positive BL tumors had shorter lag period before tumors were first detectable with an average time of 4.8 ± 1.3 days compared with 13 ± 5 days for TIMP-1-negative cell lines (P < 0.01). These results indicate that EBV II/III BL tumors grow faster when compared with either EBV I or EBV-negative, TIMP-1-negative BL tumors. These representative results suggest a strong association between TIMP-1 expression, rapid tumor growth, and subsequent tumor necrosis.
Table 1.
TIMP-1 Expression and the Tumorigenicity of Burkitt’s Lymphomas
| BL cell line | ||||
|---|---|---|---|---|
| AG876 | PA686 | JD38 | ST482 | |
| TIMP-1 expression* | ++++ | ++++ | − | − |
| Epstein-Barr virus† | III | III | − | I |
| No. of animals with tumor (% with tumor) | 24 (80%) | 10 (70%) | 17 (57%) | 10 (60%) |
| Necrosis | 19 (79%) | 3 (43%) | 0 | 0 |
To determine whether the rapid growth and necrosis was related to TIMP-1, we expressed TIMP-1 in the EBV-negative Burkitt’s cell line JD38. TIMP-1 expression was mediated by the infection of JD38 cells with LXSN retroviral expression construct expressing human TIMP-1, under control of the viral LTR promoter as previously reported. 22 JD38-LXSN and JD38-TIMP-1 clones were tested for TIMP-1 expression. In vitro secretion of active TIMP-1 was analyzed by reverse zymography of conditioned media samples. Figure 1A ▶ shows TIMP-1 inhibition of gelatinolytic activity by two independent JD38-TIMP-1 cell clones (clones 24 and 20) compared with the TIMP-1-positive control cell line (HT-1080). In contrast, the JD38-LXSN control cells were negative for TIMP-1 expression. However, a low expression of TIMP-2 is produced by control cells that is absent in the cells with up-regulated TIMP-1. We previously reported that JD38 untransfected cells are negative for TIMP-1 expression. 22 Figure 1B ▶ demonstrates TIMP-1 expression in vivo by JD38-TIMP-1 tumors grown subcutaneously in athymic nude mice, and lack of TIMP-1 in JD-38-LXSN tumors. Immunohistochemistry using an antibody against human TIMP-1 was used. In contrast to control JD38-LXSN tumors, intense TIMP-1 staining was detected in the stroma of JD38-TIMP-1 tumors (compare Figure 1B ▶ , top panels). Control cells do not express TIMP-1 in vivo whereas TIMP-1 is readily detected in the cytoplasm and on the surface of JD38-TIMP-1 cells (compare Figure 1B ▶ , bottom panels). However, in JD38-LXSN tumors, TIMP-1 was limited to perivascular staining consistent with basement membrane localization (Figure 1B ▶ , bottom). These findings clearly demonstrate that JD38-TIMP-1 cells produce functional TIMP-1 in vitro and TIMP-1 localization can be demonstrated in tumor cells and stroma.
Figure 1.
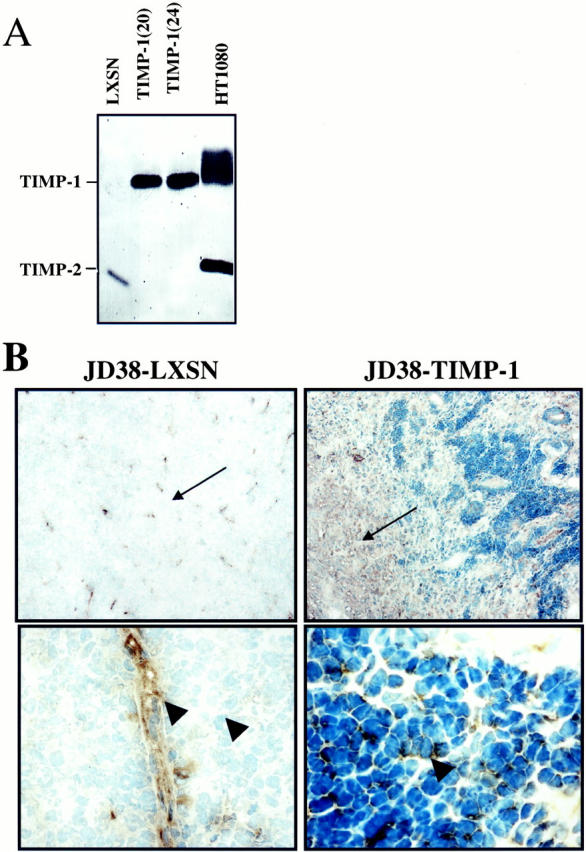
Expression of functional TIMP-1 by JD38 lymphoma cells. A: Reverse zymography showing inhibition of MMP-2-mediated gelatin proteolysis by TIMP-1. Dark areas indicate anti-proteolytic activity at the TIMP-1 band. Control JD38-LXSN cells are TIMP-1-negative, however, low-level expression of TIMP-2 is observed. Two JD38-TIMP-1 clones (clones 20 and 24) show TIMP-1 activity, but no TIMP-2 expression as compared with positive HT1080 control cells. B: Low-power photomicrograph demonstrates expression of TIMP-1 in the stroma of JD38-TIMP-1 tumors, but not in control JD38-LXSN tumors (top, arrows). Expression of TIMP-1 by JD38-TIMP-1 cells whereas control JD38-LXSN are negative (bottom, arrowheads). However, TIMP-1 is detected in control tumors at subendothelial spaces (arrowhead). Original magnifications: ×10 (top), ×40 (bottom).
To demonstrate the effects of TIMP-1 on the growth of BL tumors, control JD38-LXSN and the JD38-TIMP-1 cell clones were again injected subcutaneously into athymic nude mice. Mice were observed and tumor dimensions were measured at 3-day intervals after 1 week. Figure 2A ▶ shows the growth kinetics of mice inoculated subcutaneously with JD38-LXSN control and JD38-TIMP-1 (clone 24) cells. The JD38-TIMP-1 cells formed readily measurable tumors at 7 days after inoculation, whereas control cells required more than 10 days for measurable tumor formation. The growth kinetics of both JD38-TIMP-1 clones in vivo was essentially identical and similar to that of EBV III, TIMP-1+ BL cells listed in Table 1 ▶ . Similar growth patterns for EBV-positive BL tumors have been reported by others. 4 These results indicate that JD38-TIMP-1 tumors show faster initial growth compared with control JD38-LXSN tumors. However, JD38-TIMP-1 tumors show signs of central necrosis before reaching 0.4 cm2. These tumors continued to grow through days 10 to 14 after injection and reached an average size of 0.6 cm 2 before a decrease in tumor volume was detected, as shown in Figure 2B ▶ . In contrast, TIMP-1-negative, JD38-LXSN tumors grew progressively without evidence of necrosis or regression. Four weeks after inoculation, JD38-LXSN tumors reached an average size of 1.4 cm 2 and the mice became moribund. In contrast, JD38-TIMP-1 tumors showed a marked decrease in volume and evidence of scar formation can be observed at the inoculation site (Figure 2B ▶ , bottom). These mice show no evidence of disease. The growth data reach statistical significance (P < 0.01) by day 7 and demonstrate a bimodal effect of TIMP-1 on the kinetics of tumor growth—an initial phase of rapid or enhanced tumor growth followed by necrosis and tumor regression. In summary, TIMP-1 in the absence of EBV induces initial rapid growth in vivo followed by tumor necrosis and regression similar to that shown by EBV-positive, TIMP-1-positive, BL cell tumors (Table 1) ▶ . This effect seems specific for TIMP-1 and is not observed with the TIMP-2 expressing JD38-LXSN cells.
Figure 2.
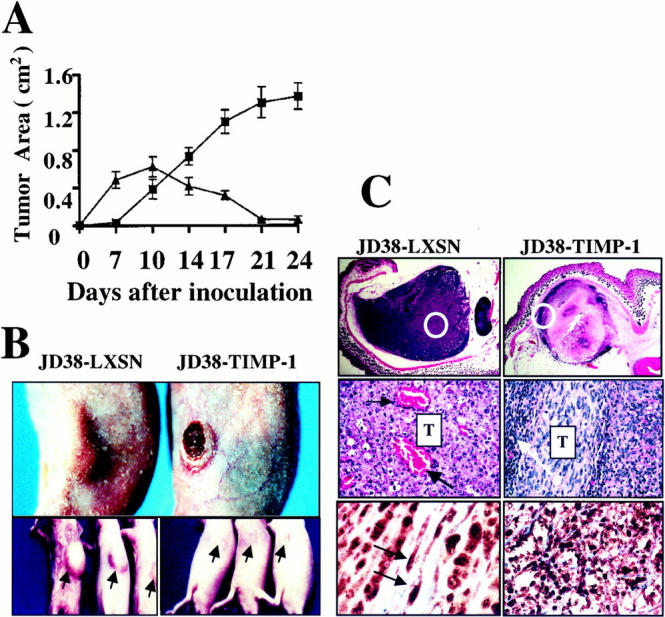
Effects of TIMP-1 up-regulation on growth, tumor necrosis, and histology in the EBV-negative JD38 lymphoma cells. A: Growth kinetics of control JD38-LXSN (filled squares) cells and JD38-TIMP-1 (filled triangles) cells demonstrate a bimodal effect of TIMP-1 on the growth of JD38 BL tumors. The initial rapid phase observed in the growth kinetics is followed by marked tumor regression to no palpable tumor mass. Each data point represents the mean ±SE of tumors in 15 mice. The differences in the mean size of the tumors in the two groups were statistically significant (P < 0.01, analysis of variance) at all time points. B: Change in the gross appearance of JD38-TIMP-1 tumors. At day 14 after tumor cell inoculation, a decrease in the size of JD38-TIMP-1 tumors is evident on gross examination of tumor volumes. In some tumors central necrosis within the tumor mass is evidenced by progression to ulceration (top right). In contrast, control JD38 tumors (top left) appear larger and show no gross evidence of tumor necrosis or ulceration. In addition, complete regression of the tumor mass and scar formation (arrows) are observed 3 weeks after tumor inoculation in the JD38-TIMP-1 tumors (bottom right) compared with continued tumor growth of the control JD38 tumors (bottom left). C: Tumor histology shows that TIMP-1 induces tumor regression that starts around day 10 with central necrosis as shown by JD38-TIMP-1 tumor (top right). In contrast to JD38-TIMP-1 tumors, central areas in control JD38-LXSN tumor are highly vascularized (middle left, black arrows). JD38-TIMP-1 tumors show residual, viable tumor cells in the periphery of the tumor mass, as well as a modest inflammatory infiltrate also at the periphery of the tumor mass. This inflammatory cell response is minimal or absent in control tumors (middle right, white arrow). Although gross examination and measurement of tumor size suggests complete regression of the JD38-TIMP-1 tumors by day 21, we examined the tumor inoculation sites for residual tumor by immunohistochemistry. Immunostaining with an antibody to phosphorylated Rb (pRb, dark nuclear staining) demonstrates that microscopic, residual foci of JD38-TIMP-1 cells (detected by human B cell markers, not shown) stain positive for pRB suggesting that these cells are actively proliferating (bottom right). Control JD38-LXSN tumor cells also show actively proliferating tumor cells by pRb staining, as well as active endothelial cell proliferation (bottom left, arrows). Original magnifications in C: ×5 (top), ×40 (middle), ×20 (bottom right), and ×100 (bottom left).
We then questioned whether the tumor regression was because of an effect of TIMP-1 on tumor cell proliferation or to changes in the tumor microenvironment. To this aim, histological analysis at every time point of the tumor growth was performed. Figure 2C ▶ shows the histology at the onset of necrosis of JD38-TIMP-1 tumors (day 10) as well as the histology of control JD38-LXSN tumors at the same time point. Results demonstrate that JD38-TIMP-1 tumor regression starts with the formation of central necrosis (Figure 2C ▶ , top). Areas of tumor lacking necrosis were seen at the periphery and these areas were found to consist of viable, actively proliferative (pRb-positive) tumor cells (Figure 2C ▶ , middle and bottom). Moreover, few blood vessels were seen in residual areas of viable JD38-TIMP-1 tumor cells. When compared to JD38-TIMP-1 tumors, the histology of control JD38-LXSN tumors demonstrates lack of necrosis as well as an intense neovascularization throughout the tumor mass (Figure 2C ▶ , middle left, arrows). There is also a moderate mononuclear inflammatory cell infiltrate that is confined to the periphery of the centrally necrotic JD38-TIMP-1 tumor mass (Figure 2C ▶ , middle right, arrow). In contrast, a minimal mononuclear inflammatory cell infiltrate is associated with JD38-LXSN tumors (data not shown). These mononuclear inflammatory infiltrates were also observed by others using similar model systems. 3,5 They seem to be a reaction to the central necrosis in the EBV-positive BL tumors and may mediate resolution of the tumor necrosis.
JD38-TIMP-1 tumors were also analyzed after regression when scar tissue had begun to form and were compared with control tumors of the same age. Examination of tissue excised from the inoculation site in mice 24 days after inoculation with JD38-TIMP-1 tumor cells demonstrated the presence of few residual tumor cells as identified by immunostaining with antibodies against human B-cell markers (data not shown). This small, residual, poorly organized, tumor cell mass was observed in all JD38-TIMP-1 tumors after regression of the bulk of the tumor mass. In contrast, at the same time point (24 days), control JD38-LXSN tumors have continued growth and formed highly vascularized tumors. Proliferation of residual JD38-TIMP-1 cells after tumor regression was confirmed by immunohistochemistry using an antibody against phosphorylated retinoblastoma protein (pRb). The positive nuclear-staining indicates that residual JD38-TIMP-1 cells in the scar tissue are actively proliferating (Figure 2C ▶ , bottom right). Likewise, control JD38-LXSN sections show viable proliferating tumor cells. Interestingly, these control sections also demonstrate nuclear staining of endothelial cells, indicative of active vascular proliferation of the newly established blood vessels (Figure 2C ▶ , bottom left, arrows). These findings also support the conclusion that TIMP-1 does not suppress proliferation of BL cells, either directly or indirectly, but does alter the host response of tumor-induced angiogenesis. The lack of tumor-induced angiogenesis limits tumor size in the JD38-TIMP-1, but does not alter the proliferation of the small residual foci of JD38-TIMP-1 tumor cells that may remain resistant to induction of apoptosis secondary to the effects of TIMP-1.
To determine the role of TIMP-1 in preventing vascularization of BL, tumor sections were analyzed for angiogenesis at day 10 before the onset of necrosis in the JD38-LXSN tumors. Assessment of the angiogenic responses in tumors at later time points was difficult because of the presence of necrosis that interfered with the anti-CD31 antibody staining. Tumor sections were analyzed for the presence of blood vessels by immunohistochemistry using an antibody against the murine endothelial cell antigen CD31. Figure 3A ▶ shows CD31 staining in JD38-LXSN and JD38-TIMP-1 tumors before necrosis. In contrast to control JD38-LXSN tumors, JD38-TIMP-1 tumors demonstrate a decreased frequency of vascular staining by CD31. The insert shows at higher magnification, the absence of blood vessel staining in the central areas of JD38-TIMP-1 tumors before the onset of tumor necrosis. Quantification of tumor angiogenesis was performed by CD31+ vessel counts. These results demonstrate that JD38-TIMP-1 tumors have statistically significantly fewer blood vessels (>50% decrease) compared to the JD38-LXSN control tumors (Figure 3B) ▶ . These findings suggest that up-regulation of TIMP-1 expression in EBV latency II/III BL tumors, inhibits angiogenesis required for sustained tumor growth. The initial rapid growth and subsequent lack of angiogenic response can account for the onset of central necrosis in the TIMP-1-positive BL tumors.
Figure 3.

Inhibition of tumor vascularization and altered bone marrow response to tumors after up-regulation of TIMP-1 expression in EBV-negative JD38 cells. A: CD31 immunostaining of blood vessels at day 10 before the onset of tumor necrosis. In contrast to control tumors, JD38-TIMP-1 tumors demonstrate a reduced number of blood vessels at the center (inset) [original magnifications: ×10, and ×40 (insets)]. B: Number of CD31+ blood vessels of sections shown in A, data are expressed as the mean ±SD of two independent determinations. The difference between the vessel counts was statistically significant (P < 0.05, Student’s t-test). C: Supernatant of JD38-TIMP-1 cells inhibits human microvascular endothelial cell migration as compared with JD38-LXSN control or media-treated cells. Levels of inhibition by JD38-TIMP-1 supernatant are comparable to those of cells treated with 50 nmol/L of human rTIMP-1. Data are mean ±SD of triplicate determinations. Statistical significance (Student’s t-test) was determined by comparison with samples from JD38-LXSN (same as control without conditioned media). D: Bone marrow aspirates 7 days after inoculations of control JD38-LXSN and JD38-TIMP-1 tumor cells. Flow cytometry analysis of dot-plots showing bone-marrow cell distribution according to size (x axis, forward scatter) and cytoplasmic granularity (y axis, side scatter). Arrows indicate those cell populations that changed with TIMP-1. Antibodies against various cell subsets were used. Differences in the expression of NK cell and granulocyte markers were found in the gated area and their relative percentages shown (bottom). Data represent one of two independent determinations.
To further support the in vivo JD-38-TIMP-1 tumor cell action on the host angiogenic response, supernatants from JD38-LXSN and JD38-TIMP-1 tumor cells were tested for their ability to inhibit human microvascular endothelial-cell migration in vitro. Figure 3C ▶ shows inhibition of human microvascular endothelial-cell migration by the supernatant from JD38-TIMP-1 tumor cells. The migration of human microvascular endothelial cells was suppressed by 60% compared with untreated culture media. Conditioned media from TIMP-2 expressing JD38-LXSN cell cultures did not inhibit endothelial cell migration. The percentage of inhibition by the supernatant of JD38-TIMP-1 cells is comparable to the inhibition observed in cells treated with 50 nmol/L of human rTIMP-1 (Figure 3C) ▶ . In previous reports the differential effects of TIMPs on endothelial cell behavior are noted. TIMP-1 has been shown to selectively inhibit endothelial cell migration in vitro, whereas TIMP-2 specifically inhibits endothelial cell proliferation without altering migration. 12,28-30 Taken together, these data indicate that inhibition of vascularization in vivo by TIMP-1 can be mediated, at least in part, by preventing migration of endothelial cells into the tumor mass.
These data demonstrate down modulation of the host angiogenic response by TIMP-1-expressing BL tumors. The differences in the reported effects of TIMPs on hematological cells, 31,32 as well as the observed difference in mononuclear cell infiltrates surrounding the JD38-TIMP-1 tumors, prompted us to analyze possible effects of TIMP-1 at distant sites such as the bone marrow. Therefore, bone marrow from mice bearing control JD38-LXSN and JD38-TIMP-1 tumors were removed and analyzed by flow cytometry using a panel of antibodies against various bone-marrow cell populations. Figure 3D ▶ shows the results of this analysis of bone marrow cells obtained 7 days after tumor inoculation. The upper panels depict bone marrow cells according to their size (forward scatter) and their cytoplasm granularity (side scatter). The gated cell population indicates granulocytes and large leukocytes. The arrow indicates an observed decrease in this bone marrow cell population in the mice inoculated with JD38-TIMP-1 BL cells as compared with bone marrow from mice inoculated with JD38-LXSN cells. Bone marrow from mice with JD38-TIMP-1 tumors show both a significant lower percentage of Gr-3-1-positive granulocytic (17.8%) cells and a higher number of NK1.1-positive NK (45.4%) cells, compared with mice bearing JD38-LXSN tumors (Figure 3 ▶ , bottom). This data suggests that in the JD38-LXSN animals an angiogenic stimulus could be driving bone marrow progenitors toward granulocyte differentiation. This observation is consistent with reports suggesting a role for inflammatory cell types in the angiogenesis process. 33-35 Concurrent with the production of angiogenesis effector genes, the expression of genes associated with granulocyte chemotaxis has also been reported in vascularized tumors. 36 Alternatively, TIMP-1 may function, directly or indirectly, to modulate the tumor effects on the bone marrow cell populations. It has recently been demonstrated that TIMP-1 expression in BL cells is responsible for up-regulation of interleukin-10 expression. 24 Resolution of the mechanism of the effects of TIMP-1 expressing BL on bone marrow will require further investigation.
Discussion
Angiogenesis plays an important role in the progression of tumor growth and dissemination. 37 However, most studies in this field are aimed at understanding the angiogenic processes in solid tumors of mainly epithelial origin. 38,39 We have confirmed that, like solid epithelial tumors, vascularization is crucial for the growth of subcutaneous BLs. This model may serve as a useful adjunct in the study of tumor-induced angiogenesis and/or in vivo screening of anti-angiogenic compounds. Using this model we confirm that EBV-positive BL tumors grow rapidly before undergoing necrosis and regression. We found that in vivo, TIMP-1-expressing, EBV-negative BL tumors can mimic this behavior. JD38-TIMP-1 tumors have a faster growth rate than TIMP-1-negative, EBV-negative JD38 control tumors, an effect that is consistent with the growth-promoting effects of TIMP-1 observed in vitro. 26,40 However, in this lymphoma model, TIMP-1 is a potent anti-angiogenic factor that prevents the host angiogenic response required for continued tumor growth. On reaching a critical tumor size, the anti-angiogenic effect of TIMP-1 dominates, resulting in the necrosis and tumor regression. Thus, TIMP-1 interferes with interactions in the tumor-host environment. These observations are consistent with the classical view that angiogenesis is a pivotal requirement for continued tumor growth. 41
The tumor-host environment changes induced by TIMP-1 may also encompass modulation of host inflammatory cells and their functions, in addition to altered endothelial cell migration. Effects such as those observed in the bone marrow of athymic mice with JD38-TIMP-1 tumors are in agreement with the previous reports on the pro-angiogenic function of granulocytes, as mentioned previously. The increase in NK cells in the bone marrow of JD38-TIMP-1 tumor-bearing mice is also consistent with the report on TIMP-1 regulation of B cell factors, such as interleukin-10, that are known to stimulate NK cell production and recruitment. 24 However, examination of tumor tissues suggests that the mononuclear infiltrate at the periphery of JD38-TIMP-1 tumors is a response to tumor necrosis and not a primary mechanism of tumor destruction. Although, our histological analysis of the tumor tissues showed no evidence of an NK-mediated cytotoxic response against the JD38-TIMP-1 tumors, further investigation of this effect is warranted.
We have shown that in EBV-negative JD38 cells, up-regulation of TIMP-1 expression results in similar in vivo growth characteristics as those seen in TIMP-1-positive BL cells with an EBV latency II/III phase of infection. Our study is the first in vivo demonstration of a biphasic effect of TIMP-1 on tumor cell growth. There is an initial growth-promoting effect consistent with the previously reported in vitro growth-promoting 40 and anti-apoptotic effects of TIMP-1. 22,23 However, our data demonstrate that this initial effect is overcome by the ongoing suppression of the host angiogenic response. This anti-angiogenic function eventually becomes dominant during continued tumor growth and results in tumor necrosis and regression. Recent results from one of our laboratories have demonstrated that EBV infection of B cells results in up-regulation of TIMP-1 expression (M Stetler-Stevenson et al, manuscript in preparation). The present model may be a valuable tool for studies on viral mechanism controlling TIMP-1 expression and further implications for the growth and angiogenesis not only of B cell lymphomas, but also in other EBV-associated pathologies. 42,43 In summary, our findings clearly demonstrate that TIMPs have multiple, divergent effects on tumor growth that can be detected in vivo. Further understanding of the role of TIMPs in tumor progression may provide new therapeutic targets for disrupting tumor growth and/or prognostic markers in B cell lymphoma.
Acknowledgments
We thank NCI predoctoral fellows, Ms. Laurel Courtemanch and Ms. Kimberly Proctor, for their excellent technical help; Mrs. Patricia Fetsch for her advice in performing the immunohistochemical staining; Dr. Adrian Senderowicz for providing Rb antibody; and Mr. James Banks for his photographic assistance.
Footnotes
Address reprint requests to William G. Stetler-Stevenson, National Institutes of Health, National Cancer Institute, Laboratory of Pathology, Bldg. 10, Rm. 2A33, 10 Center Dr., MSC #1500, Bethesda, MD 20892-1500. E-mail: sstevenw@mail.nih.gov.
Present address of Theresa A. Bennett: University of New Mexico, Albuquerque, New Mexico.
References
- 1.Cohen JI: Epstein-Barr virus infection. N Engl J Med 2000, 343:481-492 [DOI] [PubMed] [Google Scholar]
- 2.Angiolillo AL, Sgadari C, Sheik N, Reaman GH: Regression of experimental human leukemias and solid tumors induced by Epstein-Barr virus-immortalized B cells. Leukemia Lymphoma 1995, 19:267-276 [DOI] [PubMed] [Google Scholar]
- 3.Tosato G, Sgadari C, Taga K, Jones KD, Pike SE, Rosenberg A, Sechler JMG, Magrath IT, Love LA, Bathia K: Regression of experimental Burkitt’s lymphoma induced by Epstein-Barr virus-immortalized human B Cells. Blood 1994, 83:776-784 [PubMed] [Google Scholar]
- 4.Cherney BW, Sgadari C, Kanegane C, Wang F, Tosato G: Expression of the Epstein-Barr virus protein LPM1 mediates tumor regression in vivo. Blood 1998, 91:2491-2500 [PubMed] [Google Scholar]
- 5.Teruya-Feldstein J, Jaffe ES, Burd PR, Kanegane H, Kingma DW, Wilson WH, Longo DL, Tosato G: The role of Mig, the monokine induced by interferon-gamma, and IP-10, the interferon-gamma-inducible protein-10, in tissue necrosis and vascular damage associated with Epstein-Barr virus-positive lymphoproliferative disease. Blood 1997, 90:4099-4105 [PubMed] [Google Scholar]
- 6.Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H: MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99:81-92 [DOI] [PubMed] [Google Scholar]
- 7.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z: The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 1999, 98:137-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z: MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93:411-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexander CM, Werb Z: Targeted disruption of the tissue inhibitor of metalloproteinases gene increases the invasive behavior of primitive mesenchymal cells derived from embryonic stem cells in vitro. J Cell Biol 1992, 118:727-739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khokha R, Waterhouse P, Yagel S, Lala PK, Overall CM, Norton G, Denhardt DT: Antisense RNA-induced reduction in murine TIMP levels confers oncogenicity on Swiss 3T3 cells. Science 1989, 243:947-950 [DOI] [PubMed] [Google Scholar]
- 11.Takigawa M, Nishida Y, Susuki F, Kishi JI: Induction of angiogenesis in chick yolk-sac membrane by polyamines and its inhibition by tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2). Biochem Biophys Res Commun 1990, 171:1264-1271 [DOI] [PubMed] [Google Scholar]
- 12.Schnaper HW, Grant DS, Stetler-Stevenson WG, Fridman R, D’Orazi G, Murphy AN, Byrd RE, Hoythya M, Fuerst TR, French DL, Quigley JP, Kleimann HK: Type IV collagenase(s) and TIMPs modulate endothelial cell morphogenesis in vitro. J Cell Physiol 1993, 156:235-246 [DOI] [PubMed] [Google Scholar]
- 13.Johnson MD, Kim HRC, Chesler L, Tsaowu G, Bouck N, Polverini PJ: Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. J Cell Physiol 1994, 160:194-202 [DOI] [PubMed] [Google Scholar]
- 14.DeClerck YA, Perez N, Shimada H, Boone TC, Langley KE, Taylor SM: Inhibition of invasion and metastasis in cells transfected with an inhibitor of metalloproteinases. Cancer Res 1992, 52:701-708 [PubMed] [Google Scholar]
- 15.Albini A, Melchiori A, Santi L, Liotta LA, Brown PD, Stetler-Stevenson WG: Tumor cell invasion inhibited by TIMP-2. J Natl Cancer Inst 1991, 83:775-779 [DOI] [PubMed] [Google Scholar]
- 16.Martin DC, Fowlkes JL, Babic B, Khokha R: Insulin-like growth factor II signaling in neoplastic proliferation is blocked by transgenic expression of the metalloproteinase inhibitor TIMP-1. J Cell Biol 1999, 146:881-892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruger A, Fata JE, Khokha R: Altered tumor growth and metastasis of a T-cell lymphoma in timp-1 transgenic mice. Blood 1997, 90:1993-2000 [PubMed] [Google Scholar]
- 18.Remacle A, McCarthy K, Noel A, Maguire T, McDermott E, O’Higgins N, Foidart JM, Duffy MJ: High levels of TIMP-2 correlate with adverse prognosis in breast cancer. Int J Cancer 2000, 89:118-121 [DOI] [PubMed] [Google Scholar]
- 19.Holten-Andersen MN, Stephens RW, Nielsen HJ, Murphy G, Christensen IB, Stetler-Stevenson WG, Brünner N: High preoperative plasma TIMP-1 levels are associated with short survival of patients with colorectal cancer. Clin Cancer Res (2000) 6:4292–4299 [PubMed]
- 20.Ylisirnio S, Hoyhtya M, Turpeenniemi-Hujanen T: Serum matrix metalloproteinases-2, -9 and tissue inhibitors of metalloproteinases-1, -2 in lung cancer—TIMP-1 as a prognostic marker. Anticancer Res 2000, 20:1311-1316 [PubMed] [Google Scholar]
- 21.Yoshikawa T, Saitoh M, Tsuburaya A, Kobayashi O, Sairenji M, Motohashi H, Yanoma S, Noguchi Y: Tissue inhibitor of matrix metalloproteinase-1 in the plasma of patients with gastric carcinoma—a possible marker for serosal invasion and metastasis. Cancer 1999, 86:1929-1935 [DOI] [PubMed] [Google Scholar]
- 22.Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M: In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J Clin Invest 1998, 102:2002-2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li GY, Fridman R, Kim HRC: Tissue inhibitor of metalloproteinases-1 inhibits apoptosis of human breast epithelial cells. Cancer Res 1999, 59:6267-6275 [PubMed] [Google Scholar]
- 24.Guedez L, Mansoor A, Birkdal-Hansen B, Lim MS, Fukushima P, Venzon D, Stetler-Stevenson WG, Stetler-Stevenson M: Tissue inhibitor of metalloproteinase-1 regulation of interleukin-10 in B cell differentiation and lymphomagenesis. Blood 2001, 97:1796-1802 [DOI] [PubMed] [Google Scholar]
- 25.Magrath I: The pathogenesis of Burkitt’s lymphoma. Adv Cancer Res 1990, 55:133-270 [DOI] [PubMed] [Google Scholar]
- 26.Guedez L, Courtemanch L, Stetler-Stevenson M: Tissue inhibitor of metalloproteinase (TIMP-1) induces differentiation of an antiapoptotic phenotype in germinal center B cells. Blood 1998, 92:1342-1349 [PubMed] [Google Scholar]
- 27.Oliver GW, Leferson JD, Stetler-Stevenson WG, Kleiner DE: Quantitative reverse zymography: analysis using gelatinase A and B reverse zymograms. Anal Biochem 1996, 244:161-163 [DOI] [PubMed] [Google Scholar]
- 28.Kraling BM, Wiederschain DG, Boehm T, Rehn M, Mulliken JB, Moses MA: The role of matrix metalloproteinases activity in the maturation of human capillary endothelial cells in vitro. J Cell Sci 1999, 112:1599-1609 [DOI] [PubMed] [Google Scholar]
- 29.Fernandez HA, Kallenbach K, Seghezzi G, Grossi E, Colvin S, Schneider R, Galloway A: Inhibition of endothelial cell migration by gene transfer of tissue inhibitor of metalloproteinases-1. J Surg Res 1999, 82:156-162 [DOI] [PubMed] [Google Scholar]
- 30.Murphy AN, Unsworth EJ, Stetler-Stevenson WG: Tissue inhibitor of metalloproteinases-2 inhibits bFGF-induced human microvascular endothelial cell proliferation. J Cell Physiol 1993, 157:351-358 [DOI] [PubMed] [Google Scholar]
- 31.Guedez L, Lim MS, Stetler-Stevenson WG: The role of metalloproteinases and their inhibitors in hematological disorders. Crit Rev Oncogenesis 1996, 7:205-225 [DOI] [PubMed] [Google Scholar]
- 32.Hayakawa TM: Multiple functions of tissue inhibitors of metalloproteinases (TIMPs): new aspects in hematopoiesis. Platelets 1999, 10:5-6 [DOI] [PubMed] [Google Scholar]
- 33.Coussens L, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D: Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Gene Dev 1999, 13:1382-1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loukinova E, Dong G, Enamorado-Ayala I, Thomas GR, Chen Z, Schreiber H, Van Waes C: Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by host CXC receptor-2 dependent mechanism. Oncogene 2000, 19:3477-3486 [DOI] [PubMed] [Google Scholar]
- 35.Mueller MD, Lebovic DI, Garrett E, Taylor RN: Neutrophil infiltrating the endometrium express vascular endothelial growth factor: potential role in endometrial angiogenesis. Fertil Steril 2000, 74:107-112 [DOI] [PubMed] [Google Scholar]
- 36.McQuibban G, Gong JH, Tam EM, McCulloch CAG, Clark-Lewis I, Overall CM: Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000, 289:1200-1205 [DOI] [PubMed] [Google Scholar]
- 37.Hanahan D, Weinberg RA: The hallmarks of cancer. Cell 2000, 100:57-70 [DOI] [PubMed] [Google Scholar]
- 38.Thorgeirsson UP, Yoshiji H, Sinha CC, Gomez DE: Breast cancer: tumor neovasculature and the effect of tissue inhibitor of metalloproteinases-1 (TIMP-1) on angiogenesis. In Vivo 1996, 10:137-144 [PubMed] [Google Scholar]
- 39.Twardowski P, Gradishar WJ: Clinical trails of antiangiogenic agents. Curr Opin Oncol 1997, 9:584-589 [DOI] [PubMed] [Google Scholar]
- 40.Yamashita K, Suzuki M, Iwata H, Teruhiko K, Hamaguchi M, Shinagawa A, Noguchi T, Hayakawa T: Tyrosine phosphorylation is crucial for growth signaling by tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2). FEBS Lett 1996, 396:103-107 [DOI] [PubMed] [Google Scholar]
- 41.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]
- 42.Jaffe E, Chan J, Frizzera G, Mori S, Feller S, Feller A, Ho F: Report of the workshop on nasal and related extranodal angiocentric T/natural killer cell lymphomas. Definitions, differential diagnosis, and epidemiology. Am J Surg Pathol 1996, 20:103-107 [DOI] [PubMed] [Google Scholar]
- 43.Raabtraub N, Flynn K, Lanier A, Pagano J: The differentiated form of nasopharyngeal carcinomas contains Epstein-Barr virus DNA. Int J Cancer 1987, 39:25-29 [DOI] [PubMed] [Google Scholar]