

Abstract
Some studies have shown an inverse relationship between microsatellite instability in colon cancer and mutations in p53 and K-ras, whereas others have not. We therefore evaluated these features in a population-based sample of 496 individuals with colon cancer. Microsatellite instability was determined by a panel of 10 tetranucleotide repeats, the Bethesda consensus panel of mono- and dinucleotide repeats, and coding mononucleotide repeats in transforming growth factor-beta receptor type II, hMSH3, BAX, hMSH6, and insulin-like growth factor receptor type II. Mutations in codons 12 and 13 in K-ras were evaluated by sequencing. p53 overexpression (as detected by immunohistochemistry) was used as an indicator of p53 mutation; this was evaluated in 275 of the tumors. K-ras mutations were present in 33.2% of tumors, p53 overexpression in 51.5%, and microsatellite instability (as determined by the Bethesda consensus panel) in 12.5%. K-ras mutations were significantly less common in unstable tumors than stable tumors (11.8% versus 36.9%, P < 0.001). p53 overexpression was significantly less common in unstable tumors than stable tumors (20.0% versus 55.7%, P < 0.001). These inverse relationships between microsatellite instability and ras gene mutations and p53 overexpression were shown to be independent of tumor site in logistic regression analyses. All other measures of instability also showed statistically significant inverse relationships independent of tumor site with alterations in ras and p53, and instability results determined by the panel of 10 tetranucleotide repeats were highly significantly related to those determined by the Bethesda consensus panel. Coding mononucleotide repeat mutations were significantly more common in unstable tumors than stable tumors (85.7% versus 1.0%, P < 0.001). We conclude that there is an inverse relationship between microsatellite instability and mutations in p53 and K-ras, and that the molecular profile of colon cancers with microsatellite instability is characterized by relatively infrequent mutations in K-ras and p53 and relatively frequent mutations in coding mononucleotide repeats.
The relationship between microsatellite instability and mutations in p53 and K-ras in colon cancer is somewhat controversial. Some studies have shown an inverse relationship between instability and mutations in these genes, whereas other studies have not. 1 Possible explanations for these inconsistent results include small studies with insufficient power to show a significant relationship, studies of different populations, and/or different methods for measuring microsatellite instability. In addition, most previous studies did not control for tumor site, a potentially confounding variable because of the high correlation between microsatellite instability and proximal tumor location. 2
The above concerns are addressed in the current study by evaluating microsatellite instability, K-ras, and p53 in a large, population-based sample of colon cancers from the state of Utah. Microsatellite instability is analyzed in several different ways: a panel of 10 tetranucleotide repeats used by us in previous studies, 2-4 the Bethesda consensus panel generated by a National Cancer Institute workshop on microsatellite instability, 5 and mononucleotide repeats within the coding regions of transforming growth factor-β receptor type II (TGFβRII), BAX, hMSH3, hMSH6, and the insulin-like growth factor type II receptor (IGFIIR). 6 We also determine whether relationships between microsatellite instability and alterations in ras and p53 are independent of tumor site (and other variables) in logistic regression analyses.
Materials and Methods
Molecular analysis of colon cancer samples from 496 individuals was performed. These individuals represent the Utah portion of a population-based case-control study of the etiology of colon cancer 7 and includes 154 individuals previously evaluated in a study of microsatellite instability and family history. 2 Study participants were from an eight county area in Utah (Davis, Salt Lake, Utah, Weber, Wasatch, Tooele, Morgan, and Summit counties). Eligibility criteria included diagnosis with first-primary incident colon cancer (ICD-O second edition codes 18.0, 18.2 to 18.9) between October 1, 1991, and September 30, 1994, age between 30 and 79 years at time of diagnosis, and mentally competent to complete the interview. Individuals with adenomatous polyposis coli or inflammatory bowel disease were excluded from the study. Individuals with hereditary nonpolyposis colon cancer were not specifically excluded, but such individuals should comprise only a small fraction of those with colon cancer at the population level; 8 this study sample therefore consists mostly of individuals with sporadic colon cancer. The 496 individuals represent 85.8% (496 of 578) of those diagnosed with colon cancer in the state of Utah between October, 1991, and October, 1994, again underscoring the population-based nature of this study. Colon cancer tissue was microdissected and DNA extracted from formalin-fixed paraffin-embedded tissue blocks as described previously. 9 The respective normal DNA from each individual was extracted from peripheral blood (222 cases) or from paraffin blocks of normal colonic mucosa (274 cases).
Microsatellite Instability
Each tumor was evaluated for microsatellite instability with a panel of 10 tetranucleotide repeats 2 and with the Bethesda consensus panel (mononucleotide repeats BAT-25 and BAT-26 and dinucleotide repeats D5S346, D2S123, and D17S250) generated by the National Cancer Institute workshop on microsatellite instability. 5 The tumors were also evaluated with five coding mononucleotide repeats [(A)10 in TGFBRII, (A)8 in hMSH3, (G)8 in BAX, (G)8 in IGFIIR, and (C)8 in hMSH6]. The primer sequences and polymerase chain reaction (PCR) conditions for the tetranucleotide repeats, coding mononucleotide repeats, and BAT-26 were as described previously. 2,6,10 The primer sequences for the remaining four primer sets of the consensus panel were as described previously. 11 PCR of these primers consisted of 38 cycles of 20 seconds at 95°C, 20 seconds annealing, and 40 seconds at 72°C, followed by a 10-minute extension at 72°C. The initial annealing temperature was 60°C for BAT-25 and D2S123 and 64°C for D17S250 and D52346. This annealing temperature was decreased 1 degree for each of the next seven cycles and was 52°C for the final 30 cycles.
Both tumoral DNA and normal DNA were PCR amplified with the above primer sets. Microsatellite instability for a given primer set was defined as the appearance of one or more new PCR products either smaller or larger than those produced from normal DNA. Results from the tetranucleotide repeat panel were considered to indicate significant microsatellite instability if three or more of the 10 repeats were unstable. Results were considered to indicate stability if <30% of the repeats were unstable and at least six of the 10 repeats were typed. Results from the consensus panel were considered to indicate significant microsatellite instability if two or more of the five repeats were unstable. Results from the consensus panel were considered to indicate stability if no repeats were unstable and at least four were typed or if one of five repeats were unstable. Using these criteria, 92.1% of tumors were successfully classified as unstable or stable by the tetranucleotide repeats and 90.3% were classified by the consensus panel.
Microsatellite instability was also assessed using one of the consensus panel repeats, BAT-26, by itself. Instability in this mononucleotide repeat has been reported to be highly correlated with generalized dinucleotide repeat instability. 12
Instability in the coding mononucleotide repeats was considered in two ways: instability in any of the five coding repeats, and instability in TGFβRII, the coding repeat most frequently mutated in unstable tumors. 6,13
K-ras Mutations
Codons 12 and 13 of the K-ras gene were evaluated for mutations. Exon 1 of K-ras was amplified as described previously 14 except that primers were tailed with universal primer (UP) and reverse primer (RP) for sequencing. PCR products were sequenced using prism Big Dye terminators and cycle sequencing with Taq FS DNA polymerase. DNA sequence was collected and analyzed on an ABI prism 377 automated DNA sequencer (Applied Biosystems, Foster City, CA).
p53 Expression
Automated immunohistochemical staining for p53 was performed using the D07 mouse monoclonal antibody and the percentage of p53-positive tumor cell nuclei was determined as described previously. 15 This antibody and experimental technique have been shown to be highly specific and predictive for p53 mutations in colon cancer. 16 Immunostained slides were evaluated by one of the authors (JAH) without knowledge of the respective clinical parameters or the results of the other analyses in this study. We defined overexpression of p53 as tumors with 50% or more tumor cell nuclei staining positively with the antibody. 17 Paraffin blocks for this aspect of the study were available on 274 individuals.
Logistic Regression Analysis
Unconditional logistic regression models were fit to estimate the association between microsatellite instability and Ki-ras mutation or p53 overexpression after adjusting for age, sex, and tumor site. In these models, different indicators of microsatellite instability were used to predict a dichotomous dependent variable of wild-type Ki-ras versus mutated Ki-ras or p53-negative (<50% p53 nuclear staining) versus p53 overexpression. These data are reported as the odds ratio and 95% confidence interval for having microsatellite instability but lacking either K-ras mutation or p53 overexpression.
Results
Instability results for the panel of 10 tetranucleotide repeats, the consensus panel, Bat 26 by itself, TGFβRII, and instability with any coding mononucleotide repeat are shown in Table 1 ▶ . Overall instability rates were fairly similar among the various measures, ranging from 10.1 to 13.8%. All measures showed more instability in proximal tumors (19.5 to 23.9%) than distal tumors (1.4 to 4%); these differences were all statistically significant (P < 0.001, chi-square test). A representative example of instability in a tetranucleotide repeat, dinucleotide repeat, noncoding mononucleotide repeat (BAT-26), and a coding mononucleotide repeat (TGFβRII) from the same tumor is shown in Figure 1 ▶ .
Table 1.
Microsatellite Instability (MI) as Determined by Various Measures of Instability
| Instability measure | Overall MI | MI in proximal tumors | MI in distal tumors | P value* |
|---|---|---|---|---|
| 10 tetranucleotides | 13.8% (63/457) | 23.9% (54/226) | 4.0% (8/201) | < 0.001 |
| Consensus panel | 12.5% (56/448) | 22.6% (49/217) | 2.5% (5/202) | < 0.001 |
| BAT-26 | 11.4% (53/466) | 21.2% (47/222) | 2.3% (5/213) | < 0.001 |
| TGFβRII | 10.1% (47/466) | 19.5% (44/226) | 1.4% (3/210) | < 0.001 |
| Any coding mononucleotide | 11.9% (57/481) | 21.4% (49/229) | 3.2% (7/220) | < 0.001 |
*All P values based on chi-square test comparing the percentage of proximal tumors with microsatellite instability versus the percentage of distal tumors with microsatellite instability.
Figure 1.
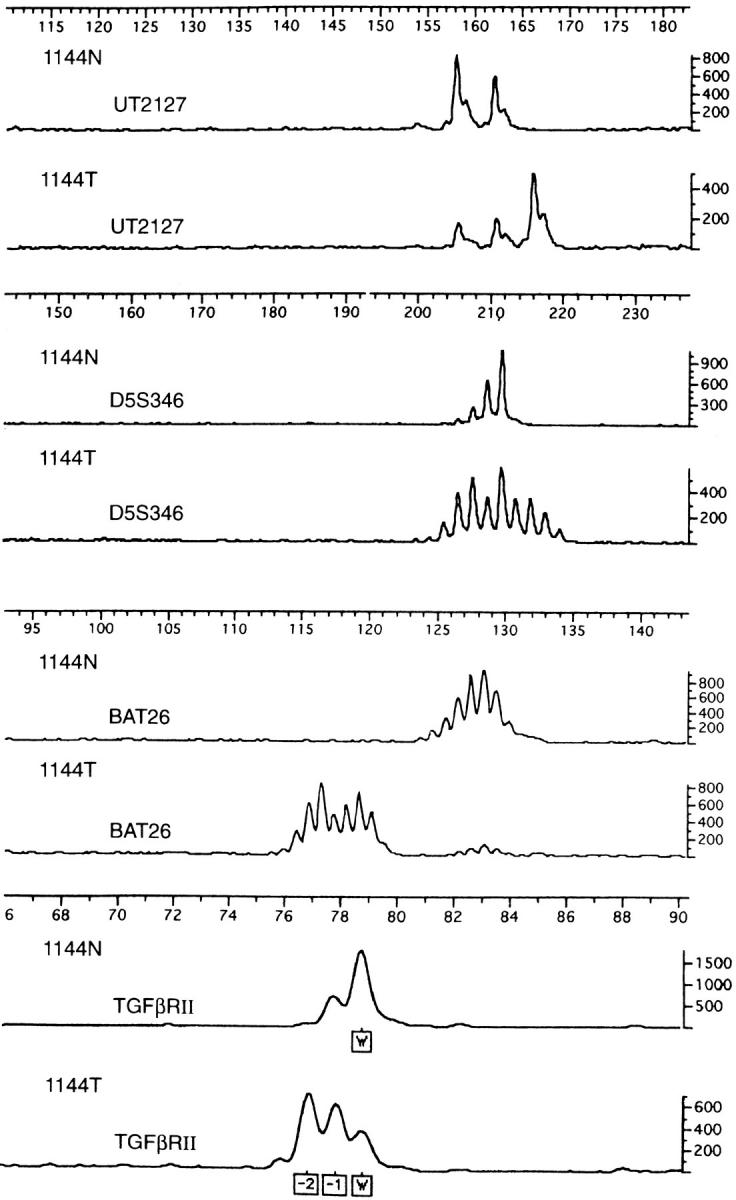
Paired normal (N) and tumor (T) results for tumor 1144 demonstrating microsatellite instability with a tetranucleotide repeat (UT2127), a dinucleotide repeat (D5S346), a noncoding mononucleotide repeat (BAT-26), and a coding mononucleotide repeat (TGFβRII). Size in bp is indicated by the scale above each repeat result; signal amplitude is indicated by the scales on the right. For TGFβRII, W indicates wild type and −1 and −2 indicate 1 and 2 bp deletions.
Codon 12 or 13 K-ras gene mutations were identified in 155 of 467 (33.2%) tumors. The type and frequency of ras gene mutations are detailed in Table 2 ▶ . Ras gene mutations were seen in a higher percentage of proximal (42.5%, 94 of 221) than distal (22.1%, 46 of 208) tumors; this difference was statistically significant (P < 0.001, chi-square test). The relationship between ras gene mutations and microsatellite instability (as determined by the various measures of instability) is summarized in Table 3 ▶ . All measures of instability showed a higher percentage of ras gene mutations in stable tumors (36 to 38.1%) than in unstable tumors (4.7 to 11.8%); these differences were all statistically significant (P < 0.001, chi-square test). A logistic regression analysis revealed that the inverse association of microsatellite instability with ras gene mutations was independent of tumor site, age, and gender. The strength of the inverse association comparing wild-type K-ras to mutant K-ras for the various indicators of instability was similar with odds ratios ranging from 8.3 to 20.0 (Table 4) ▶ , and all were statistically significant (P < 0.01).
Table 2.
Type and Frequency of ras Gene Mutations
| Base pair change* | Amino acid change† | Percentage of ras mutations |
|---|---|---|
| 2G to A | Gly12 to Asp | 34.8 |
| 5G to A | Gly13 to Asp | 23.2 |
| 2G to T | Gly12 to Val | 19.4 |
| 1G to T | Gly12 to Cys | 10.3 |
| 1G to A | Gly12 to Ser | 4.5 |
| 2G to A (H)‡ | Gly12 to Asp | 1.3 |
| 2G to C (H) | Gly12 to Ala | 1.3 |
| 2G to T (H) | Gly12 to Val | 1.3 |
| 1G to C | Gly12 to Arg | 0.6 |
| 2G to C | Gly12 to Ala | 0.6 |
| 1G to A (H) | Gly12 to Ser | 0.6 |
| 1G to T (H) | Gly12 to Cys | 0.6 |
| 5G to A (H) | Gly13 to Asp | 0.6 |
| 1 and 5G to A | Gly12 to Ser | 0.6 |
| Gly13 to Asp |
*1G and 2G are first two bases of codon 12, 5G is second base of codon 13.
†Changed codon (12 or 13) indicated by superscript.
‡H indicates homozygous mutation.
Table 3.
Comparison of K-ras Gene Mutations with Microsatellite Instability
| Instability measure | ras Mutations in stable tumors | ras Mutations in unstable tumors | P value* |
|---|---|---|---|
| 10 tetranucleotides | 38.1% (143/375) | 10.2% (6/59) | < 0.001 |
| Consensus panel | 36.4% (136/374) | 11.8% (6/51) | < 0.001 |
| BAT-26 | 36.8% (144/391) | 8.3% (4/48) | < 0.001 |
| TGFβRII | 36.0% (143/397) | 4.7% (2/43) | < 0.001 |
| Any coding mononucleotide | 36.2% (145/401) | 9.8% (5/51) | < 0.001 |
*All P values based on chi-square test comparing the percentage of stable tumors with ras gene mutations versus the percentage of unstable tumors with ras gene mutations.
Table 4.
Logistic Regression Analyses of Inverse Relationship between Microsatellite Instability and K-ras Mutations and p53 Overexpression
| Ki-ras OR* (95% CI) | p53 OR (95% CI) | |
|---|---|---|
| 10 tetranucleotides | 9.1 (3.4–20.0) | 2.8 (1.2–6.2) |
| Consensus panel | 9.1 (3.3–25.0) | 4.5 (1.8–11.5) |
| Bat-26 | 11.1 (3.7–33.3) | 8.2 (2.6–25.5) |
| TGFβRII | 20.0 (4.6–100) | 9.2 (2.6–33.1) |
| Any coding mononucleotide | 8.3 (3.1–20) | 12.9 (3.6–45.5) |
*OR is odds ratio of the absence of an alteration in K-ras or p53 in tumors with microsatellite instability; CI is confidence interval.
p53 overexpression was identified in 141 of 274 tumors (51.5%). An example of a tumor with p53 overexpression is shown in Figure 2 ▶ . p53 overexpression was present in a higher percentage of distal (60.9%, 70 of 115) than proximal (41.0%, 57 of 139) tumors, this difference was statistically significant (P < 0.002, chi-square test). The relationship between p53 overexpression and microsatellite instability (as determined by the various measures of instability) is summarized in Table 5 ▶ . All measures of instability showed a higher percentage of stable tumors with p53 overexpression (54.3 to 57.2%) than unstable tumors with p53 overexpression (9.1 to 26.3%); these differences were all statistically significant (P < 0.001, chi-square test). A logistic regression analysis revealed that the inverse association of microsatellite instability with p53 overexpression was independent of tumor site, age, and gender. Odds ratios for the inverse association comparing p53 negative (<50% nuclear staining) versus p53 overexpression ranged from 2.8 to 12.9 (Table 4) ▶ , and all were statistically significant (P < 0.05).
Figure 2.
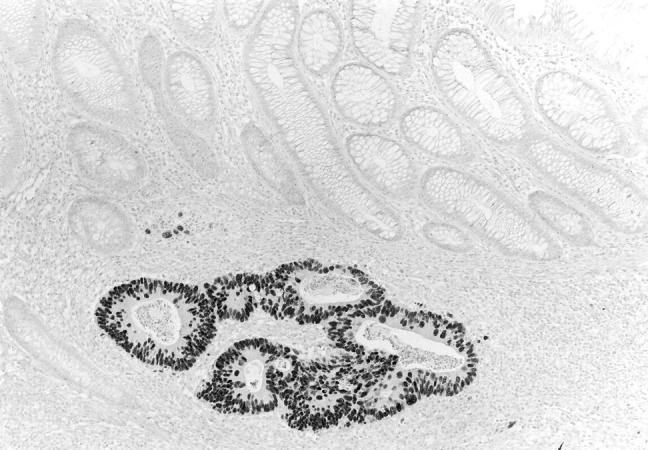
Colon cancer with p53 overexpression. Immunostaining for p53 reveals abundant nuclear staining in the submucosal tumor and no staining in the overlying normal mucosa.
Table 5.
Comparison of p53 Overexpression with Microsatellite Instability
| Instability measure | p53 Overexpression in stable tumors | p53 Overexpression in unstable tumors | P value* |
|---|---|---|---|
| 10 tetranucleotides | 54.3% (120/221) | 26.3% (10/38) | < 0.001 |
| Consensus panel | 55.7% (122/219) | 20.0% (7/35) | < 0.001 |
| BAT-26 | 56.5% (130/230) | 12.1% (4/33) | < 0.001 |
| TGFβRII | 55.7% (132/237) | 11.1% (3/27) | < 0.001 |
| Any coding mononucleotide | 57.2% (135/236) | 9.1% (3/33) | < 0.001 |
*All P values based on chi-square test comparing the percentage of stable tumors with p53 overexpression versus the percentage of unstable tumors with p53 overexpression.
Microsatellite instability in the coding mononucleotide repeats is summarized in Table 6 ▶ ; all observed mutations were frameshifts (addition of one base, deletion of one or two bases). TGFβRII contained the most frequently mutated coding repeat, with length alterations in this poly A repeat in 10.1% (47 of 466) of tumors overall, followed by BAX (6.1%), hMSH3 (5.2%), hMSH6 (2.7%), and IGFIIR (2.3%). As seen in Table 6 ▶ , all coding mononucleotide repeats were more frequently mutated in unstable (as judged by the Bethesda consensus panel) tumors than stable tumors; these differences were all statistically significant (P < 0.001, chi-square test). At least one coding mononucleotide repeat mutation was seen in 85.7% (48 of 56) of unstable tumors but in only 1.0% (4 of 392) of stable tumors; this difference was also statistically significant (P < 0.001, chi-square test).
Table 6.
Type and Frequency of Coding Mononucleotide Repeat Instability
| Coding mononucleotide | Changes in repeat length | Overall mutation frequency | Mutations in unstable tumors* | Mutations in stable tumors* | P value† |
|---|---|---|---|---|---|
| TGFβRII | +1,−1, −2 | 10.1% (47/466) | 74.5% (41/55) | 0.3% (1/383) | < .001 |
| BAX | +1, −1 | 6.1% (23/375) | 39.6% (19/48) | 0.3% (1/309) | < .001 |
| hMSH3 | +1,−1,−2 | 5.2% (21/401) | 39.6% (19/48) | 0.0% (0/329) | < .001 |
| hMSH6 | +1,−1 | 2.7% (13/473) | 16.4% (9/55) | 0.5% (2/390) | < .001 |
| IGFIIR | +1, −1 | 2.3% (9/396) | 19.1% (9/47) | 0.0% (0/327) | < .001 |
*Unstable and stable as defined by the Bethesda consensus panel.
†All P values based on chi-square test comparing the percentage of stable tumors with the respective mononucleotide repeat mutation versus the percentage of unstable tumors with that mutation.
Table 7 ▶ shows a comparison of the panel of 10 tetranucleotide repeats with the other measures of instability. Microsatellite instability as determined by the 10 tetranucleotide repeats was significantly related to microsatellite instability as determined by the consensus panel, BAT-26, TGFβRII, or instability in any coding mononucleotide repeat (P < 0.001, chi-square test). Table 8 ▶ shows a comparison of the consensus panel with BAT-26 by itself. There are very few tumors in which either BAT-26 or the consensus panel alone is unstable, and there is a significant relationship between these two measures of microsatellite instability (P < 0.001, chi-square test).
Table 7.
Comparison of Panel of 10 Tetranucleotide Repeats with other Measure of Instability
| Panel of 10 tetranucleotide repeats* | |||
|---|---|---|---|
| Stable | Unstable | P value | |
| Consensus panel | |||
| Stable | 369 | 2 | |
| Unstable | 5 | 51 | < 0.001 |
| BAT-26 | |||
| Stable | 383 | 9 | |
| Unstable | 4 | 49 | < 0.001 |
| TGFβRII | |||
| Stable | 382 | 16 | |
| Unstable | 3 | 44 | < 0.001 |
| Any coding mononucleotide | |||
| Stable | 384 | 13 | |
| Unstable | 8 | 49 | < 0.001 |
*P values are based on chi-square tests comparing microsatellite instability as determined by the panel of 10 tetranucleotide repeats with microsatellite instability determined by the other measures of instability.
Table 8.
Comparison of BAT-26 (by Itself) with the Consensus Panel
| Consensus panel | BAT-26 | ||
|---|---|---|---|
| Stable | Unstable | P value* | |
| Stable | 385 | 1 | |
| Unstable | 5 | 51 | <0.001 |
*P value based on a chi-square test comparing microsatellite instability as determined by BAT-26 by itself versus instability determined by the consensus panel.
Discussion
This study shows highly statistically significant inverse relationships between microsatellite instability and K-ras gene mutations and p53 overexpression in colon cancers. K-ras mutations were identified in 33.2% of tumors. This is consistent with previous studies that, with rare exceptions, 18 have identified K-ras mutations in ∼30 to 40% of colon cancers. 19-28 Overexpression of p53 has been used by many studies as an indicator of p53 mutational status. Although some 29,30 have questioned the validity of this practice, others 16 have shown that the antibody, experimental technique, and high threshold for positivity used by us in this study lead to immunohistochemical results that do correlate well with p53 mutational status, at least in colorectal tumors. We therefore conclude that our results also suggest an inverse relationship between microsatellite instability and p53 mutations. It should be noted, however, that a lack of concordance between p53 mutations and overexpression would not invalidate our highly statistically significant results with overexpression, and that, regardless of the underlying mechanism, overexpression may still be useful in identifying different pathways to colon cancer.
As this (Table 1) ▶ and other studies 2 have shown, microsatellite instability is also highly correlated with tumor site, as it is much more commonly seen in proximal tumors than distal tumors. It could be argued, then, that the relative lack of ras gene mutations and p53 overexpression in unstable tumors could have been because of the proximal site of these tumors rather than their instability. This is less of a concern with ras gene mutations, as in our study such mutations were actually more common in proximal tumors. p53 overexpression in our study was more common in distal tumors, however, and a previous study 31 suggested that microsatellite instability was not an independent predictor of p53 mutational status if tumor location was considered. Our logistic regression analyses, however, indicate that the inverse relationships between microsatellite instability and ras gene mutations and p53 overexpression are independent of tumor site (Table 4) ▶ .
The inverse relationship between microsatellite instability and ras mutations and p53 overexpression was also independent of the type of microsatellite used for instability analysis. The inverse relationship was seen with a panel of 10 tetranucleotide repeats, the Bethesda consensus panel (a mixture of dinucleotide and mononucleotide repeats), the mononucleotide BAT-26 by itself, the coding mononucleotide in TGFβRII, and with instability in any of five coding mononucleotide repeats (Tables 3 and 5) ▶ ▶ . The relative lack of ras gene mutations and p53 overexpression in unstable tumors thus seems to be a general characteristic of such tumors and is not limited to a subset with instability in a certain type of microsatellite. The inverse relationship with K-ras and p53 alterations was not seen in tumors with low levels (<30%) of instability (as defined by the Bethesda consensus panel, data not shown), consistent with a previous study of ours linking cigarette smoking to only high levels of microsatellite instability. 32
Although some previous studies have shown an inverse relationship between microsatellite instability and ras and p53 mutations, others have not. 1 This discrepancy is probably not because of the use of different types of microsatellites for instability analysis in the various studies, as we have shown (Tables 3 and 5) ▶ ▶ that the inverse relationship can be seen with mononucleotide (coding and noncoding), dinucleotide, and tetranucleotide repeats. It is possible that different populations of individuals were studied, and, indeed, the situation may be different for tumors from individuals with hereditary nonpolyposis colon cancer. 33 Our population-based study would be predicted to consist mostly of individuals with sporadic tumors, especially since previous estimates of hereditary nonpolyposis colon cancer at the population level were inflated by the inclusion of founder mutations peculiar to Finland. 8,34,35 Indeed, subsequent germline analysis of individuals with unstable tumors from the current study have identified only two with hereditary nonpolyposis colon cancer (data not shown).
The most likely explanation for the failure of some previous studies to identify relationships between instability and alterations in ras and p53 is that many of the studies were of relatively small numbers of tumors and thus lacked sufficient power to demonstrate a statistically significant inverse relationship. Indeed, many of these studies did show relatively less ras and p53 mutations in unstable tumors, but the difference did not always reach statistical significance. Statistically significant results were seen in two studies of microsatellite instability and ras gene mutations 31,36 and in six studies of instability and p53 alterations. 1,31,36-39 Some of the studies with significant and nonsignificant results dealt with the possibly confounding variable of tumor site by considering only proximal tumors. 1,40-42 The only previous study to use a multivariate analysis found that the inverse relationship between microsatellite instability and ras gene mutations was independent of tumor site, but that the inverse relationship between instability and p53 mutations was not. 31 Our study represents the largest number of tumors analyzed in these ways to date and is the first to demonstrate statistically significant inverse relationships between microsatellite instability and alterations in both ras and p53 that are independent of tumor site in a logistic regression analysis.
In agreement with other studies, 6,13 TGFβRII contained the most frequently mutated coding repeat, and instability in all five coding repeats was significantly more common in unstable tumors than in stable tumors (Table 6) ▶ . A mutation in at least one coding repeat was significantly more common in unstable tumors than stable tumors (85.7% versus 1.0%). The molecular profile of colon cancers with microsatellite instability is therefore characterized by relatively infrequent ras and p53 mutations and relatively frequent mutations in coding mononucleotide repeats.
The various measures of microsatellite instability showed very similar results in our study (Tables 1, 3, 4, and 5) ▶ ▶ ▶ and were highly correlated with one another (Table 7 and 8) ▶ ▶ . A previous study 11 suggested that tetranucleotide repeat instability may not be a good indicator of generalized instability, but our panel of 10 tetranucleotide repeats was highly correlated with the Bethesda consensus panel of mononucleotide and dinucleotide repeats as well as with BAT-26, a mononucleotide repeat that is highly correlated with generalized dinucleotide repeat instability. 12 Our current study does not indicate which is the best panel of microsatellites for instability analysis. The choice of such a panel may depend on several factors, including cost, time, and the purpose of a study. For example, if a fast and relatively inexpensive study of microsatellite instability alone is desired, it is hard to argue against using BAT-26 (as long as it is compared to results with germline DNA) 10,43 by itself, as some investigators may decide that information gained (if any) by using the other four microsatellites in the Bethesda panel does not justify the added expense and time. If the purpose of a study is to evaluate loss of heterozygosity as well as microsatellite instability, then a panel of repeats from the chromosomal location(s) of interest may be more appropriate.
In conclusion, we observed significant inverse relationships between microsatellite instability and alterations in K-ras and p53. These inverse relationships were independent of tumor site and the type of microsatellite (mono-, di-, or tetranucleotide repeat) used for instability analysis. In addition, coding mononucleotide repeat mutations were significantly more common in unstable tumors than stable tumors. The molecular profile of colon cancers with microsatellite instability is therefore characterized by relatively infrequent mutations in K-ras and p53 and relatively frequent mutations in coding mononucleotide repeats. These different profiles of stable and unstable tumors most likely reflect different molecular pathways to sporadic colon cancer: the microsatellite stable (but chromosomally unstable) 44 pathway, probably initiated by APC mutations, 45 and the microsatellite instability pathway, in which early β-catenin mutations are sometimes seen but in which the initiating event in most tumors is unknown. 46 These different molecular pathways and/or the specific genetic changes we report may in turn reflect different carcinogenic influences, such as diet or tobacco 32 and alcohol use. Future studies that stratify colon cancers on the basis of these genetic changes may identify factors that contribute to one pathway or the other, relationships that might be obscured if the genetic heterogeneity of colon cancer is not taken into account.
Acknowledgments
We thank the Genomics Core Facility and the Sequencing Core Facility of the University of Utah Health Sciences Center for their assistance in genotyping and sequencing; Leslie Palmer for her assistance in collection and processing of tumor blocks; and Khe-Ni Ma for her assistance in data analysis.
Footnotes
Address reprint requests to Wade S. Samowitz, M.D., Dept. of Pathology, University of Utah Health Sciences Center, 50 North Medical Dr., Salt Lake City, Utah 84132. E-mail: wsamowitz@msscc.med.utah.edu.
Supported by grants CA48998 and CA61757 from the National Cancer Institute. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
References
- 1.Fujiwara T, Stolker JM, Watanabe T, Rashid A, Longo P, Eshleman J, Booker S, Lynch HT, Jass JR, Green JS, Kim H, Jen J, Vogelstein B, Hamilton SR: Accumulated clonal genetic alterations in familial and sporadic colorectal carcinomas with widespread instability in microsatellite sequences. Am J Pathol 1998, 153:1063-1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samowitz WS, Slattery ML, Kerber RA: Microsatellite instability in human colonic cancer is not a useful clinical indicator of familial colorectal cancer. Gastroenterology 1995, 109:1765-1771 [DOI] [PubMed] [Google Scholar]
- 3.Samowitz WS, Slattery ML: Microsatellite instability in colorectal adenomas. Gastroenterology 1997, 112:1515-1519 [DOI] [PubMed] [Google Scholar]
- 4.Samowitz WS, Slattery ML: Transforming growth factor β receptor type 2 mutations and microsatellite instability in sporadic colorectal adenomas and carcinomas. Am J Pathol 1997, 151:33-35 [PMC free article] [PubMed] [Google Scholar]
- 5.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani N, Srivastava S: A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 6.Samowitz WS, Slattery ML: Regional reproducibility of microsatellite instability in sporadic colorectal cancer. Genes Chromosom Cancer 1999, 26:106-114 [PubMed] [Google Scholar]
- 7.Slattery ML, Potter JD, Caan BJ, Edwards SL, Coates A, Ma K-N, Berry TD: Energy balance and colon cancer: beyond physical activity. Cancer Res 1997, 57:75-80 [PubMed] [Google Scholar]
- 8.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick RB, Kaariainen H, Eskelinen M, Jarvinen H, Mecklin J-P, de la Chapelle A: Incidence of hereditary nonpolyposis colorectal cancer and feasibility of molecular screening for the disease. N Engl J Med 1998, 338:1481-1487 [DOI] [PubMed] [Google Scholar]
- 9.Spirio LN, Samowitz WS, Robertson J, Robertson M, Burt RW, Leppert MF, White R: Alleles of APC modulate the frequency and classes of mutations that lead to colon polyps. Nat Genet 1998, 20:385-388 [DOI] [PubMed] [Google Scholar]
- 10.Samowitz WS, Slattery ML, Potter JD, Leppert MF: BAT-26 and BAT-40 instability in colorectal adenomas and carcinomas and germline polymorphisms. Am J Pathol 1999, 154:1637-1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Rüschoff J: Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997, 57:4749-4756 [PubMed] [Google Scholar]
- 12.Hoang J-M., Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R: BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res 1997, 57:300-303 [PubMed] [Google Scholar]
- 13.Yamamoto H, Sawai H, Perucho M: Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res 1997, 57:4420-4426 [PubMed] [Google Scholar]
- 14.Sidransky D, Tokino T, Hamilton SR, Kinzler KW, Levin B, Frost P, Vogelstein B: Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science 1992, 256:102-105 [DOI] [PubMed] [Google Scholar]
- 15.Lynch BJ, Komaromy-Hiller G, Bronstein IB, Holden JA: Expression of DNA topoisomerase I, DNA topoisomerase II-alpha, and p53 in metastatic malignant melanoma. Hum Pathol 1998, 29:1240-1245 [DOI] [PubMed] [Google Scholar]
- 16.Baas IO, Mulder JR, Offerhaus GJ, Vogelstein B, Hamilton SR: An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994, 172:5-12 [DOI] [PubMed] [Google Scholar]
- 17.Rashid A, Zahurak M, Goodman SN, Hamilton SR: Genetic epidemiology of mutated K-ras proto-oncogene, altered suppressor genes, and microsatellite instability in colorectal adenomas. Gut 1999, 44:826-833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kern SE, Fearon ER, Tersmette KWF, Enterline JP, Leppert M, Nakamura Y, White R, Vogelstein B, Hamilton SR: Allelic loss in colorectal carcinoma. JAMA 1989, 261:3099-3103 [DOI] [PubMed] [Google Scholar]
- 19.Andreyev HJN, Norman AR, Cunningham D, Oates JR, Clarke PA: Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst 1998, 90:675-684 [DOI] [PubMed] [Google Scholar]
- 20.Laurent-Puig P, Olschwang S, Delattre O, Remvikos Y, Asselain B, Melot T, Validire P, Muleris M, Girodet J, Salmon RJ, Thomas G: Survival and acquired genetic alterations in colorectal cancer. Gastroenterology 1992, 102:1136-1141 [PubMed] [Google Scholar]
- 21.Finkelstein SD, Sayegh R, Bakker A, Swalsky P: Determination of tumor aggressiveness in colorectal cancer by K-ras-2 analysis. Arch Surg 1993, 128:526-532 [DOI] [PubMed] [Google Scholar]
- 22.Dix BR, Robbins P, Soong R, Jenner D, House AK, Iacopetta BJ, : the General Surgeons at Sir Charles Gairdner Hospital: The common molecular genetic alterations in Duke’s B and C colorectal carcinomas are not short-term prognostic indicators of survival. Int J Cancer 1994, 59:747-751 [DOI] [PubMed] [Google Scholar]
- 23.Breivik J, Meling GI, Spurkland A, Rognum TO, Gaudernak G: K-ras mutation in colorectal cancer: relations to patient age, sex and tumour location. Br J Cancer 1994, 69:367-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersen SN, Løvig T, Breivik E, Gaudernack G, Meling GI, Rognum TO: K-ras mutations and prognosis in large bowel carcinomas. Scand J Gastroenterol 1997, 32:62-69 [DOI] [PubMed] [Google Scholar]
- 25.Wadler S, Bajaj R, Neuberg D, Agarwal V, Haynes H, Benson AB: Prognostic implications of c-Ki-ras 2 mutations in patients with advanced colorectal cancer treated with 5-fluorouracil and interferon: a study of the Eastern Cooperative Oncology Group (EST2292). Cancer J Sci Am 1997, 3:284-288 [PubMed] [Google Scholar]
- 26.Hardingham JE, Butler WJ, Roder D, Dobrovic A, Dymock RB, Sage RE, Roberts-Thomson IC: Somatic mutations, acetylator status, and prognosis in colorectal cancer. Gut 1998, 42:669-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerottini JP, Caplin S, Saraga E, Givel JC, Benhattar J: The type of K-ras mutation determines prognosis in colorectal cancer. Am J Surg 1998, 175:198-202 [DOI] [PubMed] [Google Scholar]
- 28.Ahnen DJ, Feigl P, Quan G, Fenoglio-Preiser C, Lovato LC, Bunn PA, Stemmerman G, Wells JD, MacDonald JS, Meyskens FL: Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res 1998, 58:1149-1158 [PubMed] [Google Scholar]
- 29.Tolbert DM, Noffsinger AE, Miller MA, DeVoe GW, Stemmermann GN, Macdonald JS, Fenoglio-Preiser CM: p53: immunoreactivity and single-strand conformational polymorphism analysis often fail to predict p53 mutational status. Mod Pathol 1999, 12:54-60 [PubMed] [Google Scholar]
- 30.Dix B, Robbins P, Carrello S, House A, Iacopetta B: Comparison of p53 gene mutation and protein overexpression in colorectal carcinomas. J Pathol 1998, 184:390-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breivik J, Lothe RA, Meling GI, Rognum TO, Borresen-Dale A, Gaudernack G: Different genetic pathways to proximal and distal colorectal cancer influenced by sex-related factors. Int J Cancer 1997, 74:664-669 [DOI] [PubMed] [Google Scholar]
- 32.Slattery ML, Curtin K, Anderson K, Ma KN, Ballard L, Edwards S, Schaffer D, Potter J, Leppert M, Samowitz WS: Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J Natl Cancer Inst 2000, 92:1831-1836 [DOI] [PubMed] [Google Scholar]
- 33.Craanen ME, Blok P, Offerhaus GJ, Tytgat GN: Recent developments in hereditary nonpolyposis colon cancer. Scand J Gastroenterol 1996, 218:92-97 [DOI] [PubMed] [Google Scholar]
- 34.Salovaara R, Loukola A, Kristo P, Kaariainen H, Ahtola H, Eskelinen M, Harkonen H, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Jarvinen H, Mecklin J-P, Aaltonen LA, de la Chapelle A: Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000, 18:2193-2200 [DOI] [PubMed] [Google Scholar]
- 35.Nystrom-Lahti M, Kristo P, Nicolaides NC, Chang S-Y, Aaltonen LA, Moisio AL, Jarvinen HJ, Mecklin J-P, Kinzler KW, Vogelstein B, de la Chapelle A, Peltomaki P: Founding mutations and Alu-mediated recombination in hereditary colon cancer. Nat Med 1995, 1:1203-1206 [DOI] [PubMed] [Google Scholar]
- 36.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M: Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363:558-561 [DOI] [PubMed] [Google Scholar]
- 37.Cottu PW, Muzeau F, Estreicher A, Fléjou J, Iggo R, Thomas G, Hamelin R: Inverse correlation between RER+ status and p53 mutation in colorectal cancer cell lines. Oncogene 1996, 13:2727-2730 [PubMed] [Google Scholar]
- 38.Rüschoff J, Dietmaier W, Lüttges J, Seitz G, Bocker T, Zirngibl H, Schlegel J, Schackert HK, Jauch KW, Hofstaedter F: Poorly differentiated colonic adenocarcinoma, medullary type. Am J Pathol 1997, 150:1815-1825 [PMC free article] [PubMed] [Google Scholar]
- 39.Salahshor S, Kressner U, Pahlman L, Glimelius B, Lindmark G, Lindblom A: Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosom Cancer 1999, 26:247-252 [PubMed] [Google Scholar]
- 40.Forster S, Sattler H, Hack M, Romanakis K, Rohde V, Seitz G, Wullich B: Microsatellite instability in sporadic carcinomas of the proximal colon: association with diploid DNA content, negative protein expression of p53, and distinct histomorphologic features. Surg J 1998, 123:13-18 [PubMed] [Google Scholar]
- 41.Kim H, Jen J, Vogelstein B, Hamilton SR: Clinical and pathologic characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994, 145:148-156 [PMC free article] [PubMed] [Google Scholar]
- 42.Iacopetta BJ, Welch J, Soong R, House AK, Zhou XP, Hamelin R: Mutation of the transforming growth factor-beta type II receptor gene in right-sided colorectal cancer: relationship to clinicopathological features and genetic alterations. J Pathol 1998, 184:390-395 [DOI] [PubMed] [Google Scholar]
- 43.Pyatt R, Chadwick RB, Johnson CK, Adebamowo C, de la Chapelle A, Prior TW: Polymorphic variation at the BAT-25 and BAT-26 loci in individuals of African origin: implications for microsatellite instability testing. Am J Pathol 1999, 155:349-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lengauer C, Kinzler KW, Vogelstein B: Genetic instability in colorectal cancers. Nature 1997, 386:623-627 [DOI] [PubMed] [Google Scholar]
- 45.Olschwang S, Hamelin R, Laurent-Puig P, Thuille B, De Rycke Y, Li Y-J, Muzeau F, Girodet J, Salmon R-J, Thomas G: Alternative genetic pathways in colorectal carcinogenesis. Proc Natl Acad Sci USA 1997, 94:12122-12127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML: β-catenin mutations are more frequent in small colorectal adenomas than larger adenomas and invasive carcinomas. Cancer Res 1999, 59:1442-1444 [PubMed] [Google Scholar]