

Abstract
Major congenital malformations, including those affecting the cardiovascular system, remain the leading cause of mortality and morbidity in infants of diabetic mothers. Interestingly, targeted mutations of several genes (including VEGF and VEGF receptors) and many teratogenic agents (including excess D-glucose) that give rise to embryonic lethal phenotypes during organogenesis are associated with a failure in the formation and/or maintenance of a functional vitelline circulation. Given the similarities in the pathology of the abnormal vitelline circulation in many of these conditions, we hypothesized that the hyperglycemic insult present in diabetes could cause the resultant abnormalities in the vitelline circulation by affecting VEGF/VEGF receptor signaling pathway(s). In this study we report that hyperglycemic insult results in reduced levels of VEGF-A in the conceptus, which in turn, leads to abnormal VEGF receptor signaling, ultimately resulting in embryonic (vitelline) vasculopathy. These findings and our observation that addition of exogenous rVEGF-A165 within a defined concentration range blunts the hyperglycemia-induced vasculopathy in the conceptus support the concept that VEGF levels can be modulated by glucose levels. In addition, these findings may ultimately lead to novel therapeutic approaches for the treatment of selected congenital cardiovascular abnormalities associated with diabetes.
Offspring of diabetic mothers (both humans and experimental animals) experience a two- to fourfold increase in congenital anomalies. 1,2 Although no particular organ system or tissue seems to be specifically targeted, a variety of cardiovascular anomalies are frequently observed. 3 In addition to the increased incidence of congenital anomalies noted in the live births and stillbirths of diabetic mothers, these mothers experience difficulties in becoming pregnant and exhibit an increased incidence of fetal resorption. 1,4-8 After implantation and before placentation, embryonic growth is dependent on proper development of the yolk sac vasculature and the vitelline circulation. Arrested development or maldevelopment of this vasculature would lead to fetal demise early resulting in the termination of pregnancy; whereas arrest and/or maldevelopment of vasculature(s) at later times, associated with specific organ or tissue development, would contribute to congenital abnormalities in a wide variety of organs and tissues.
It was previously shown that in vitro culture of rat and mouse conceptuses at the primitive streak stage is possible and that conceptuses develop nearly normally for 48 hours during the initial stages of organogenesis. During this period they are nourished via the vitelline circulation, after which further growth and development would require a placental circulation. 9,10 The addition of 20 mmol/L D-glucose (a plasma concentration of glucose often observed in diabetic mothers of humans and of experimental animals) to these cultures results in significant yolk sac and embryonic vasculopathy and abnormal embryonic development. 10 In light of these findings and the data illustrating the crucial roles of VEGF and VEGF receptors (VEGFRs) during early vasculogenesis and angiogenesis, 11-18 we hypothesized that the vasculopathy observed in our cultured conceptuses and in embryos of streptozotocin-induced diabetic pregnant mice 10 was due to abnormalities in the VEGF signaling pathways.
In this report we demonstrate that hyperglycemic insult results in reduced levels of VEGF-A, which in turn, leads to abnormal VEGFR signaling, resulting in embryonic vasculopathy. These findings and our observation that addition of exogenous VEGF-A blunts the hyperglycemia-induced vasculopathy may ultimately lead to novel therapeutic approaches for the prevention and treatment of congenital abnormalities associated with diabetes.
Materials and Methods
Murine Conceptuses
Murine conceptuses 7.5, 8.5, and 9.5 days post-coitus (dpc) were harvested from pregnant CD1 mice (Charles River, Wilmington, MA) as described 19,20 and used for morphological and biochemical studies directly or after defined periods of culture. 10,20,21 VEGF-A-LacZ knock-in mice were generated as described 22 by inserting an internal ribosome entry site (IRES)-LacZ cassette into the 3′UTR (exon 8) of VEGF-A and using a sequence encoding the β-galactose reporter (LacZ cassette) inserted into the noncoding region of exon 8 (3′ end) of the VEGF-A stop codon. An IRES preceded the LacZ coding sequence. This strategy permitted the production of two functional proteins, VEGF-A and LacZ, from a single bicistronic transcript.
In vitro murine conceptuses were cultured as described, in the presence of pooled, heat-inactivated, undiluted rat serum, and oxygenated using a series of gas mixtures with increasing oxygen concentrations as previously described. 9,10 Hyperglycemic culture conditions were achieved by addition of α-D-glucose (Sigma Chemical Co., St. Louis, MO) to a final concentration of 20 mmol/L as described. 10 VEGF/placental growth factor (PlGF) supplementation of control and hyperglycemic cultures was accomplished by addition of 0.2 to 20 pg/ml rVEGF-A165, rVEGF-A120, or rPlGF (Chemicon International, Inc., Temecula, CA; R&D Systems, Inc., Minneapolis, MN). Addition of growth factors or vehicle alone was made either at the start of culture or 3 hours after initiation of culture.
Conceptuses were assessed for the presence or absence of major structural and functional defects including heartbeat, yolk sac circulation, vitelline vessel branching, neural tube closure, and completion of axial rotation. Growth and development were scored by their organ primordia development as described. 23 Heartbeat and blood flow through the vitelline circulation was assessed visually by an Olympus dissecting microscope (Olympus Optical Co., Ltd., Japan) in each group of conceptuses at the termination of the culture period, as recorded and summarized in Table 1 ▶ .
Table 1.
Effects of VEGF Supplementation on Yolk Sac Vascular Arborization and on the Development of a Functional Vitelline Circulation
| Condition/dose | Normal | Abnormal | ||
|---|---|---|---|---|
| Arborization | Functional circulation | Arborization | Functional circulation | |
| Normoglycemic | ||||
| −VEGF | 120/126 (96%) | 120/126 (96%) | 120/126 (4%) | 120/126 (4%) |
| +VEGF165: | ||||
| 0.2 pg/ml | 9/10 (90%) | 9/10 (90%) | 1/10 (10%) | 1/10 (10%) |
| 2.0 pg/ml | 49/54 (91%) | 49/54 (91%) | 5/54 (9%) | 5/54 (9%) |
| 20.0 pg/ml | 0/20 (0%) | 0/20 (0%) | 20/20 (100%) | 20/20 (100%) |
| 200.0 pg/ml | 0/10 (0%) | 0/10 (0%) | 10/10 (100%) | 10/10 (100%) |
| Hyperglycemic | ||||
| −VEGF | 0/20 (0%) | 0/20 (0%) | 20/20 (100%) | 20/20 (100%) |
| +VEGF165: | ||||
| 0.2 pg/ml | 2/10 (20%) | 2/10 (20%) | 8/10 (80%) | 8/10 (80%) |
| 1.0 pg/ml | 8/22 (36%) | 8/22 (36%) | 14/22 (64%) | 14/22 (64%) |
| 2.0 pg/ml* | 72/80 (90%) | 72/80 (90%) | 8/80 (10%) | 8/80 (10%) |
| 10.0 pg/ml | 48/62 (77%) | 48/62 (77%) | 12/62 (23%) | 12/62 (23%) |
| +VEGF165 @ 3 hours | ||||
| 2.0 pg/ml 10.0 pg/ml | 0/34 (0%) | 0/34 (0%) | 34/34 (100%) | 34/34 (100%) |
| +VEGF 120: | ||||
| 0.2 pg/ml | 8/15 (53%) | 0/15 (0%) | 7/15 (47%) | 15/15 (100%) |
| 10.0 pg/ml | 12/20 (60%) | 5/20 (25%) | 8/20 (40%) | 15/20 (75%) |
| +PGF: | ||||
| 0.2 pg/ml | 0/19 (0%) | 0/19 (0%) | 19/19 (100%) | 19/19 (100%) |
| 10.0 pg/ml | 0/19 (0%) | 0/19 (0%) | 19/19 (100%) | 19/19 (100%) |
*Optimal VEGF-A165 concentration.
β-Galactosidase Staining of Conceptuses
Conceptuses harvested from VEGF-A-LacZ knock-in mice were fixed in 0.2% glutaraldehyde, 2% paraformaldehyde, 2 mmol/L EGTA, and 2 mmol/L MgCl2 in Pipes buffer, pH 6.9. Staining was performed at 37°C in 0.02% X-Gal, 5 mmol/L K3Fe(CN)6, 2 mmol/L MgCl2 in phosphate-buffered saline overnight. Stained conceptuses were washed, postfixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm. Sections were mounted on glass slides and counterstained with Nuclear Fast Red as described. 22
Whole Mount Immunostaining of Conceptuses
Whole mount staining of conceptuses with anti-platelet endothelial cell adhesion molecule-1 (PECAM-1) was performed as described. 10,20 Approximately 20 to 25 randomly selected in vivo grown and cultured conceptuses were evaluated for each experimental group.
Transmission Electron Microscopy (TEM) of Conceptuses
Light level semithin and TEM level thin sections of 8.5 and 9.5 dpc conceptuses were examined using a Zeiss Axiophot light microscope (Carl Zeiss, Oberkochen, Germany) and a Zeiss EM910 electron microscope (Carl Zeiss, Oberkochen, Germany) , respectively, as described elsewhere. 10
Immunoprecipitation and Western Blotting
Preparation of yolk sac and embryo lysates and subsequent immunoprecipitation with anti-VEGFR-2/Flt-1 and Western blotting with anti-VEGF, anti-VEGFR-2/Flt-1 and anti-PY (PY 99) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were performed as described. 10,20,21,24 All blots were scanned into a Macintosh G3 computer (Apple Computer, Brea, CA) using an Arcus II scanner (Agfa-Gevaert, N.V.) and Photoshop 5.0 software (Adobe Systems, Inc. San Jose, CA). All experiments were repeated at least three times using independently prepared lysates.
Results
Hyperglycemia-Induced Embryonic Vasculopathy Is Associated with Decreased VEGF-A Levels
Although it is recognized that hyperglycemia elicits embryonic vasculopathy, the mechanism(s) responsible for this embryopathy remain unclear. 3,5,8,10,25,26 We have reported earlier that hyperglycemic insult results in arrest of vascular development at the primary capillary plexus stage in the yolk sacs of the conceptuses of streptozotocin-induced diabetic mothers and in cultured murine conceptuses. 10 From this observation we hypothesized that perturbations in VEGF-A expression, VEGFR expression, and/or VEGFR signaling may be crucial to the observed vasculopathy.
To assess VEGF-A levels in the murine conceptus, two approaches were taken. In an earlier project a β-galactosidase (LacZ) reporter gene with a preceding IRES was introduced by gene targeting into the 3′ UTR region of VEGF-A. This modification allows the production of both VEGF-A and LacZ from the same bicistronic mRNA created. Therefore the LacZ and VEGF-A expression are transcriptionally coupled. 22 In the first approach, these VEGF-A-LacZ knock-in heterozygous male mice were mated with CD1 mothers. Conceptuses recovered at 7.5 dpc were placed in normoglycemic and hyperglycemic culture as described. 10 Conceptuses were harvested after 24 (Figure 1, a and b) ▶ and 48 hours (Figure 1, d and e) ▶ in culture. Yolk sacs were isolated and 5-μm sections prepared and stained for activity. As illustrated in Figure 1, a, b, d, and e ▶ , we observed a reduced LacZ activity in the hyperglycemia-exposed yolk sacs compared with the normoglycemic yolk sacs, consistent with reduced levels of VEGF-A mRNA after hyperglycemic insult. Specifically, by 8.5 dpc the primitive capillary plexus formed from fused blood islands in the yolk sac. Abundant LacZ expression was observed in the visceral endodermal yolk sac cells, in extraembryonic mesoderm-derived cells and mesothelial cells forming the inner yolk sac layer. Hematopoietic and endothelial cells were unstained (Figure 1a) ▶ . At 9.5 dpc an arborizing vascular network had developed and LacZ/VEGF-A was extensively expressed in the yolk sac endodermal cells and mesenchymal/mesothelial cells (Figure 1d) ▶ . In hyperglycemia-exposed conceptuses, reductions in LacZ/VEGF-A expression were noted (Figure 1, b and e) ▶ , being more pronounced in the visceral endodermal cells compared to mesenchymal/mesothelial cells in the inner layer of the yolk sacs.
Figure 1.
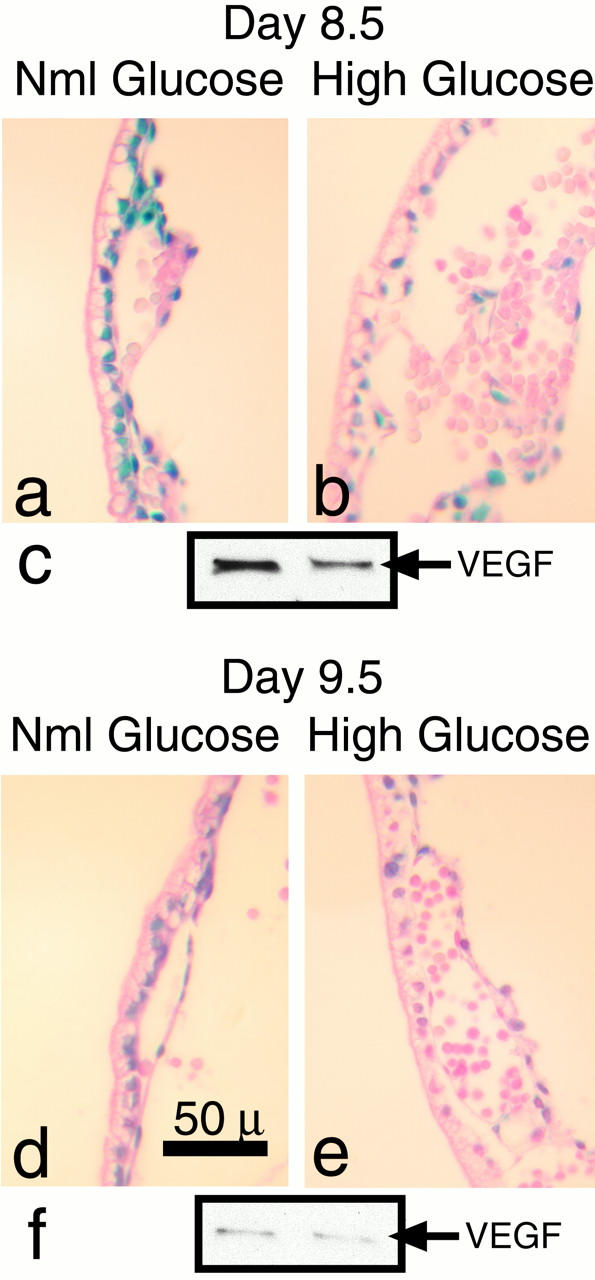
Hyperglycemia causes reduction of VEGF-A mRNA and protein in the murine conceptus. Histochemical analysis of 5-μm sections of yolk sacs from 8.5 (a and b) and 9.5 (d and e) dpc. VEGF-A/LacZ knock-in heterozygous conceptuses. The blue color present in the endodermal, mesodermal and mesothelial cells represents VEGF-A gene induction, whereas the endothelial cells and primitive blood cells are negative. Note that there is decreased intensity of staining in the hyperglycemia-exposed conceptuses (b and e). c and f: Representative Western blots of lysates of 8.5 (c) and 9.5 (f) dpc yolk sacs derived from normoglycemic (left lanes) and hyperglycemia-exposed (right lanes) conceptuses immunoblotted with anti-VEGF. Note the decreased band intensities in the lysates derived from the hyperglycemia-exposed conceptuses.
In the second approach, we performed Western blots on yolk sac lysates at 8.5 and 9.5 dpc. VEGF-A expression was noted throughout all stages of yolk sac vascularization, being barely detectable at 7.5 dpc (data not shown); increased at 8.5 dpc, when the primary capillary plexus has formed but arborization is not yet manifested; and decreased at 9.5 dpc, when there has been completion of branching morphogenesis and establishment of the vitelline circulation. Figure 1, c and f ▶ , illustrates the reduction in VEGF-A protein in the 8.5 and 9.5 dpc hyperglycemic cultures compared to the normoglycemic cultures.
Exogenous VEGF-A Prevents Hyperglycemia-Induced Embryonic Vasculopathy
In light of our findings of reduced levels of VEGF-A in the hyperglycemic cultures, we hypothesized that perhaps treatment of these cultured embryos with exogenous VEGF-A would abrogate or blunt the effects of the hyperglycemic insult.
Because VEGF-A levels are known to be tightly regulated during vasculogenesis, 11,12,27-30 we determined the effects of exogenous VEGF-A165 on yolk sac vascularization. Concentrations of 0.2 to 2.0 pg/ml of VEGF-A165 had no detectable adverse effects on vascular morphology. However, concentrations higher than 20.0 pg/ml elicited abnormalities in yolk sac vascular development, manifested as edematous conceptuses (data not shown). This abnormal yolk sac vasculature was characterized by the presence of an enlarged, tortuous, ectatic vasculature. Our findings are consistent with known VEGF concentration-dependent effects on vascular development and function. 11,12,31,32
When 2 or 10 pg/ml of VEGF-A165 were added to hyperglycemia-exposed cultures at the start of the culture period a marked improvement in the branching morphology of yolk sac vessels was noted (see Table 1 ▶ ). Figure 2 ▶ illustrates the effects of exogenous VEGF-A165 on conceptuses exposed to hyperglycemic culture conditions at 8.5 and 9.5 dpc. At 8.5 dpc the effects of hyperglycemia and VEGF-A165 treatment are not apparent on low power examination of intact conceptuses (Figure 2, a–c) ▶ . However, at 9.5 dpc the effects of hyperglycemic insult are readily observable (Figure 2, d, e, g, and h) ▶ . An arborized yolk sac vasculature, an actively beating heart and blood flow in the vitelline vessel were noted in the normoglycemic conceptuses (Figure 2, d and g) ▶ . In contrast, the yolk sacs of the hyperglycemia-exposed conceptuses displayed an ectatic vascular plexus with no signs of arborization (Figure 2, e and h) ▶ and large, non-fused blood islands toward the ectoplacental cone (Figure 2h) ▶ . The embryos were malformed, with slowly beating hearts and no appreciable blood flow. However, when exogenous VEGF-A165 was added to the cultures (0.2 to 10 pg/ml), the hyperglycemia-exposed conceptuses displayed arborizing yolk sac vascular networks (Figure 2, f and i) ▶ . A striking difference between this group and the hyperglycemia-exposed group was the presence of a faster beating heart and a vigorous blood flow through the yolk sac vasculature.
Figure 2.
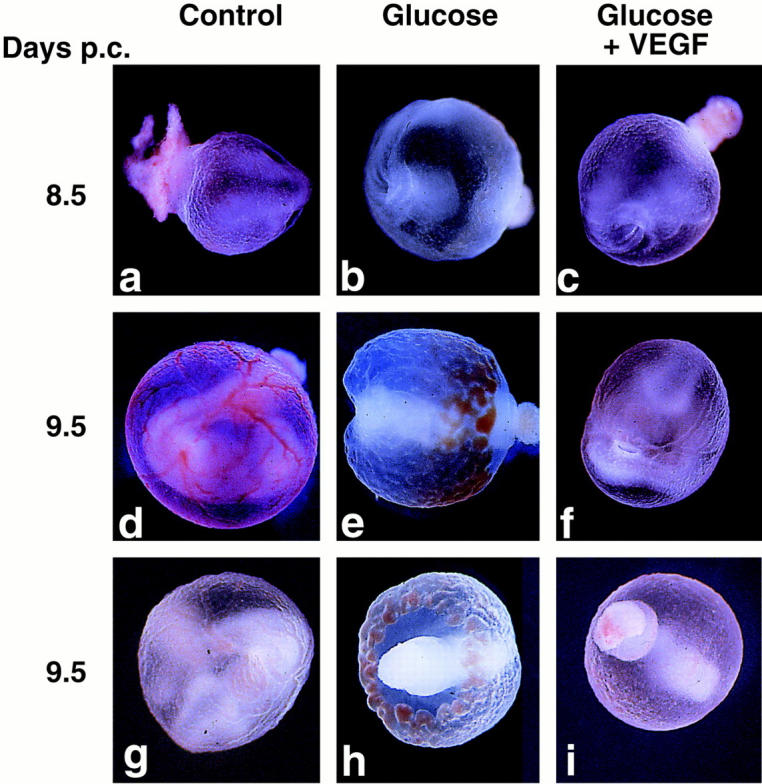
Hyperglycemia-induced vasculopathy is blunted by treatment with exogenous VEGF-A165. Low power representative micrographs of whole conceptuses at 8.5 (a–c) and 9.5 dpc(d–i) under normoglycemic conditions (Control, a, d, and g), hyperglycemic conditions (Glucose, b, e, and h), and hyperglycemic conditions + exogenous VEGF-A165 (Glucose + VEGF-A, c, f, and i). Note the vasculopathy consisting of non-arborizing, ectatic vessels in the hyperglycemic conditions (e and h) and the rescue of a normal arborizing phenotype in the hyperglycemia-exposed conceptuses treated with exogenous VEGF-A165 (f and i).
It is important to note that this significant improvement in yolk sac vascularization and circulation was achieved when exogenous VEGF-A and D-glucose were added to the medium at the start of the culture period and were present for the duration of the culture. To ascertain whether VEGF-A treatment could be preventive, we exposed 7.5 dpc conceptuses to 20 mmol/L D-glucose for an initial 3-hour period (known to elicit yolk sac vasculopathy 10 ) and then transferred the conceptuses into VEGF-A165-supplemented normoglycemic media for the remaining 45 hours of culture. We observed that VEGF-A165 added at this time failed to correct the vasculopathy, indicating that VEGF-A is preventive rather than corrective and its effects are time-dependent.
PECAM-1 staining of whole mounts of yolk sacs harvested from 9.5 dpc normoglycemic, hyperglycemic, and hyperglycemic VEGF-A-treated conceptuses confirmed our results. Specifically, we noted an arrest of yolk sac vascular arborization after hyperglycemic insult (compare Figure 3a to F ▶ igure 3b) and a rescue of yolk sac vascular arborization in hyperglycemia-exposed conceptuses treated with 2 to 10 pg/ml VEGF-A165 (Figure 3c) ▶ . Further, high power and TEM examination of the yolk sac vasculature revealed that addition of exogenous VEGF-A165 to hyperglycemic conceptus cultures prevented the morphological abnormalities of the yolk sac vasculature (Figure 3, d–m) ▶ . Specifically, the loss of intimate endothelial cell-mural cell (pericyte) interactions observed after hyperglycemic insult was prevented with VEGF-A supplementation (compare Figure 3, e, h, and l ▶ with Figure 3, f, i, and m ▶ ). Interestingly, the endodermal cells of the yolk sac also exhibit a morphological appearance similar to control normoglycemic conceptuses when treated with exogenous VEGF-A (compare Figure 3j ▶ with Figure 3, k and m ▶ ), raising the possibility that VEGF-A may exert effects on nonvascular cells during embryonic development. 33,34
Figure 3.
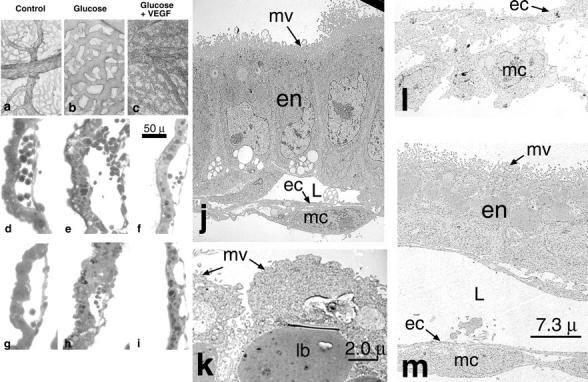
Hyperglycemia-induced disorganization of endothelial cells and adjacent mural cells is rescued by treatment with exogenous VEGF-A165. Low power representative micrograph of PECAM-1 stained 9.5 dpc yolk sacs harvested from normoglycemic (a), hyperglycemic (b), and hyperglycemic + exogenous VEGF-A165 cultures (c). Note the lack of arborization and ectatic vessels in the hyperglycemia-exposed conceptuses (b) and the rescue of an arborizing phenotype in the in the hyperglycemia-exposed conceptuses treated with exogenous VEGF-A165 (c). d–i: High power micrographs illustrating the perturbation of the intimate endothelial cell-mural cell interactions following hyperglycemic insult at 8.5 (compare normoglycemic, d, with e) and 9.5 dpc (compare normoglycemic, g, with h). f and i: Illustrations of the rescue of intimate endothelial cell-mural cell interactions after addition of exogenous rVEGF-A165 to the cultures (compare d with f and g with i). Scale bar, 50 μm (d–i). j–m: Representative transmission electron micrographs of control (j), hyperglycemic (k and l), and hyperglycemic 9.5 dpc conceptuses treated with VEGF-A165 (m). j: Illustratation of a yolk sac comprised of polarized endodermal cells (en) with apical microvilli (mv) and a microvascular lumen (L), lined by flattened endothelial cells (ec) intimately invested by mesothelial cells (mc). In contrast, k illustrates a hyperglycemic yolk sac comprised of endodermal cells with blunted microvilli and large lysosomal bodies (lb). l: Illustratation of the yolk sac vasculature, which is comprised of plump endothelial cells (ec) and mesothelial cells (mc) that have lost their intimate associations with the endothelium. m: A hyperglycemic yolk sac treated with exogenous rVEGF-A165. Its endodermal cells (en) are polarized with apical microvilli (mv). Its vasculature is again noted to be comprised of flattened endothelial cells (ec) intimately invested by mesothelial cells (mc). Scale bars, 7.3 μm (j, l, and m) and 2.0 μm (k).
Hyperglycemia-Induced Vasculopathy Correlates with a Reduction in VEGFR-2/Flk-1 Activation and Complex Formation
In light of our findings of reduced VEGF-A expression after hyperglycemic insult and partial rescue of yolk sac vascular development by addition of exogenous VEGF-A, we reasoned that decreased VEGF-A165 levels could alter VEGFR-2 activation and subsequent interaction with a variety of signaling and adapter moieties which associate with the VEGFR-2 after VEGF engagement. 35-37
We conducted experiments to investigate the patterns of VEGFR-2/Flk-1 expression under normoglycemic and hyperglycemic culture conditions. Western blot analysis of VEGFR-2 levels revealed that although expression levels changed over time (7.5 to 9.5 dpc) in culture, hyperglycemia did not appear to alter expression levels (Figure 4A) ▶ . Immunoprecipitation of VEGFR-2 followed by Western blotting using anti-VEGFR-2/Flk-1 antibodies confirmed this (Figure 4B) ▶ .
Figure 4.

VEGFR-2/Flk-1 signaling is blunted in hyperglycemic conditions, but partially restored by treatment with exogenous VEGF-A165. A: Representative Western blot of 7.5, 8.5, and 9.5 dpc yolk sac lysates derived from conceptuses cultured in normoglycemic (NG), hyperglycemic (HG), and hyperglycemic conditions plus exogenous VEGF-A165 (HG + VEGF) probed with antibodies directed against VEGFR-2. B: Representative immunoprecipitation of VEGFR-2 followed by a Western blot with anti-VEGFR-2/Flk-1 antibody of 7.5, 8.5, and 9.5 dpc yolk sac lysates derived from conceptuses cultured in normoglycemic (NG), hyperglycemic (HG), and hyperglycemic conditions plus exogenous VEGF-A165 (HG + VEGF). Note that although there appears to be differing expression levels of VEGFR-2 over the time period studied, no changes were observed after hyperglycemic insult or VEGF-A165 addition. C: Representative immunoprecipitation of VEGFR-2 followed by a Western blot with anti-phosphotyrosine (PY) antibodies of 7.5, 8.5, and 9.5 dpc yolk sac lysates derived from conceptuses cultured in normoglycemic (NG), hyperglycemic (HG) and hyperglycemic conditions plus exogenous VEGF-A165 (HG + VEGF). Note that hyperglycemic insult significantly reduces VEGFR-2/Flk-1 tyrosine phosphorylation and the complement of phosphoproteins coprecipitating with VEGFR-2 at all three time points and that addition of exogenous VEGF-A165 to the hyperglycemic cultures partially restores VEGFR-2/Flk-1 tyrosine phosphorylation and the complement of coprecipitating phosphoproteins.
Investigation of the signaling complex formed following engagement of VEGFR-2/Flk-1 was performed by immunoprecipitation of VEGFR-2 followed by Western blot analysis using an anti-phosphotyrosine antibody (Figure 4C) ▶ . This pull-down study revealed differential tyrosine phosphorylation of VEGFR-2/Flk-1 and several phosphoproteins associated with VEGFR-2/Flk-1 37 at 7.5, 8.5, and 9.5 dpc (see lanes 7.5 NG, 8.5 NG, and 9.5 NG). Hyperglycemic insult was noted to diminish dramatically the tyrosine phosphorylation of VEGFR-2/Flk-1 itself and the association of these phosphoproteins with VEGFR-2 (see lanes 7.5 HG, 8.5 HG, and 9.5 HG and compare them to the corresponding NG lanes).
Exogenous VEGF-A Treatment Partially Prevents the Effects of Hyperglycemia on VEGFR-2 Tyrosine Phosphorylation and Complex Formation
After supplementation of hyperglycemic conceptus cultures with exogenous VEGF-A165, partial restoration of the tyrosine phosphorylation of the VEGFR-2 and the associated phosphoproteins was noted at 8.5 and 9.5 dpc (see lanes 8.5 HG + VEGF-A and 9.5 HG + VEGF-A and compare them with lanes 8.5 NG and HG and 9.5 NG and HG).
Rescue of the Embryonic Vasculature by VEGF Is Specific for VEGF-A165 and Time-Dependent
In light of our findings of robust prevention of hyperglycemia-induced yolk sac vasculopathy with VEGF-A165 supplementation (86% restitution in arborization and in functional circulation) compared with a complete (100%) arrest of arborization and lack of a functional vitelline circulation caused by hyperglycemic insult, 10 we examined the protective capabilities of other VEGF-A isoforms and family members. The freely diffusible murine form of VEGF-A120 used in the identical concentration range elicited moderate, partial improvement of yolk sac vessel branching (60% restitution in arborization), but only a modest improvement in functional circulation (25%) in the vitelline vasculature. Parallel studies using PlGF at the identical concentration range resulted in no detectable rescue in arborization or functional circulation of the embryonic vasculature. In addition, when VEGF-A165 was added to the cultures 3 hours after initiation of hyperglycemia, it failed to improve either arborization (0%) or functional circulation (0%; Table 1 ▶ ).
Discussion
Congenital defects known to be associated with maternal diabetes mellitus are numerous and varied. Several theories have been postulated regarding potential mechanisms by which maternal diabetes might induce dysmorphogenesis; however, the pathogenesis remains unknown. 1-10,38
Similarities of the vasculopathies observed after hyperglycemic insult at the primitive streak stage of development 10 and that noted in mice containing targeted disruption of either VEGF or its receptors 11-18,27-29 suggested the possibility that the arrest of yolk sac vascular development at the primary capillary plexus stage noted after hyperglycemic insult could be due to modulation of VEGF/VEGFR signaling pathways. Indeed, our findings of reduced VEGF-A expression in 8.5 and 9.5 dpc conceptuses cultured in 20 mmol/L D-glucose support this hypothesis. Furthermore, prevention of yolk sac vasculopathy by supplementation of the hyperglycemic cultures with exogenous rVEGF-A165 supports this concept, as do our findings that the observed VEGF-mediated prevention was mediated by a specific VEGF isoform, VEGF-A165.
That the observed hyperglycemia-induced vasculopathy was related to altered VEGFR engagement and subsequent complex formation was supported by our findings of attenuations of VEGFR-2 (Flk-1) tyrosine phosphorylation and coprecipitation with several phosphoproteins known to be associated with VEGFR-2 37 after engagement. That reduced VEGF-A levels were responsible for this blunting of VEGFR tyrosine phosphorylation and signaling complex formation was confirmed by restitution of VEGFR-2 tyrosine phosphorylation levels and increased coprecipitation of tyrosine-phosphorylated signaling complex components in cultures supplemented with exogenous rVEGF-A165. Our examination of VEGFR-1 (Flt-1) revealed low constitutive expression that made analysis of its tyrosine phosphorylation profiles unreliable (data not shown). Further, exogenous addition of its high affinity ligand, PlGF-1, did not improve the phenotype of the hyperglycemic cultured conceptuses and was noted to elicit yolk sac vasculopathy in normoglycemic cultured conceptuses (data not shown). Additionally, previous studies have shown that although VEGFR-1 (Flt-1) deficiency results in embryonic death, reconstitution with a truncated VEGFR-1 (Flt-1) devoid of its cytoplasmic domain rescues the phenotype. 39 These data, along with our current findings, are consistent with the notion that VEGFR-1 (Flt-1) may function as a cell surface reservoir or presenter of VEGF family ligands, rather than as a direct signaling moiety in certain circumstances. The roles played by the other VEGF-A receptors, VEGFR-3 15 and neuropilin-1, 16 in this model are unknown and beyond the scope of this report. Future studies focusing on these receptors may shed more light on the mechanisms involved in hyperglycemia-induced yolk sac and embryonic vasculopathy.
The exact mechanisms by which altered VEGF/VEGFR signaling cause yolk sac vasculopathy are unknown. Other investigators have demonstrated changes in PECAM-1 phosphorylation state after VEGF treatment of endothelial cells. 40 In prior studies using this conceptus culture model and conceptuses harvested from diabetic mice we have noted changes in PECAM-1 tyrosine phosphorylation, leading to persistent PECAM-1/Src homology 2 domain-containing protein tyrosine phosphatase (SHP-2) association; increased expression and activation of protein kinase C (PKCε), and increased serine/threonine phosphorylation leading to decreased PECAM-1/γ-catenin association. 10,21 Because these changes in PECAM-1 phosphorylation state have been associated with modulation of cell proliferation, cell migration, gene expression, and cytoskeletal organization, 21,24,41-43 it is possible that these alterations in PECAM-1 phosphorylation state are, in part, responsible for the resulting vasculopathy. Figure 5 ▶ is our working model, illustrating the effects of hyperglycemia-induced reduced VEGF-A expression on VEGFR tyrosine phosphorylation, adapter and signaling molecule association, and tyrosine phosphorylation and subsequent downstream signaling, involving changes in PECAM-1 phosphorylation state and binding of adapter, signaling, and cytoskeletal molecules. It remains to be determined whether these observed changes in PECAM-1 phosphorylation state and protein associations are causative and/or related to the arrest in vasculogenesis and angiogenesis observed after hyperglycemic insult during vasculogenesis and angiogenesis in the murine conceptus.
Figure 5.

Current working model of the effects of hyperglycemia on vascular development of the murine yolk sac. Reduction of VEGF-A expression by endodermal, mesodermal and endothelial cells in the yolk sac leads to reduced VEGFR-2 activation and subsequent interactions with several adapter and signaling molecules. This, in turn, leads to altered downstream signaling cascades which result in arrest of vascular development. Our prior studies demonstrating increased PECAM-1 tyrosine phosphorylation and SHP-2 binding as well as increased PKCε activity, increased PECAM-1 serine/threonine phosphorylation, and decreased γ-catenin binding after hyperglycemic insult 10,21 are consistent with this working model.
In addition to its well-documented effects on vascular endothelial development, maintenance, remodeling, proliferation, differentiation, and permeability, VEGF also has been shown to affect non-endothelial cell behavior. Selected epithelial and mesenchymal cell populations have been documented to respond to VEGF isoforms, 33,34,44 indicating that this growth factor/receptor family is likely to affect the development of a wide range of cells, tissues, and organs during development. In our model we noted striking changes in the endodermal cells of the yolk sac with supplementation of the hyperglycemic cultures with exogenous rVEGF-A165. Specifically, the endodermal cells exhibited reformation of their apical microvilli and re-established their polarity and intimate interactions with the endothelium of the yolk sac vasculature on rVEGF-A165 supplementation. In addition, rVEGF-A165 supplementation also correlated with the re-establishment of intimate endothelial cell-mural cell interactions. 45,46 It remains to be determined whether these observed changes in non-endothelial cellular behaviors are directly caused by addition of rVEGF-A165, or are the result of changes in the endothelial cells elicited by rVEGF-A165 supplementation.
Interestingly, diabetes in adults also results in vasculopathy; however, it has been associated with increased VEGF expression. 25,26 The elevated VEGF expression is thought to be a major mediator of retinopathy, nephropathy, and neuropathy, but also may play a beneficial role in collateralization in diabetic cardiovascular disease. This increased level of VEGF expression is in apparent contradiction with our findings, however; in postnatal diabetes hyperglycemia targets mature organs, tissues, and vasculature, whereas in our experiments developing organs, tissues, and vascular beds are exposed and therefore may respond differently to a similar insult.
The in vitro murine whole conceptus culture used in our studies is a useful model to study in situ vasculogenesis in the yolk sac. It permits evaluation of the effects of a single factor (eg, excess glucose, VEGF) on conceptuses, which cannot be achieved in vivo. Furthermore, the anatomically intact relationships among all three germ layers allow signaling (physiological or pathological) among different cell types necessary to form a vessel, which cannot be observed in other in vitro approaches.
Our observations also raise the possibility of future use of targeted delivery of selected VEGF isoforms to prevent specific congenital abnormalities. This approach, however, awaits the further development of specific delivery and exquisitely controlled expression systems.
Footnotes
Address reprint requests to Drs. Joseph A. Madri/Emese Pinter, Departments of Pathology and Pediatrics, Yale University School of Medicine, P.O. Box 208023, New Haven, CT 06520. E-mail: joseph.madri@yale.edu or emese.pinter@yale.edu.
Supported in part by a grant from the Charles H. Hood Foundation (to E. P.) and by U.S. Public Health Service grants R37-HL28373 and PO1-KD-38979 (to J. A. M.).
References
- 1.Eriksson UJ, Hakan-Borg LA, Forsberg H, Styrud J: Diabetic embryopathy: studies with animal and in vitro models. Diabetes 1991, 40:94-98 [DOI] [PubMed] [Google Scholar]
- 2.Kitzmiller JL, Buchanan TA, Kjos S, Combs AC, Ratner RE: Preconception care of diabetes, congenital malformations, and spontaneous abortions. Diabetes Care 1996, 19:514-541 [DOI] [PubMed] [Google Scholar]
- 3.Ferencz C, Rubin JD, McCarter RJ, Clark EB: Maternal diabetes and cardiovascular malformations: predominance of double outlet right ventricular and truncus arteriosus. Teratology 1990, 41:319-326 [DOI] [PubMed] [Google Scholar]
- 4.Schwarz R, Teramo KA: Effects of diabetic pregnancy on the fetus and newborn. Semin Perinatol 2000, 24:120-135 [DOI] [PubMed] [Google Scholar]
- 5.Lee AT, Plump A, DeSimone C, Cerami A, Bucala R: A role for DNA mutations in diabetes-associated teratogenesis in transgenic embryos. Diabetes 1995, 44:20-24 [DOI] [PubMed] [Google Scholar]
- 6.Baker L, Piddlington R: Diabetic embryopathy: a selective review of recent trends. J Diabetes Comp 1993, 7:404-412 [DOI] [PubMed] [Google Scholar]
- 7.Pampfer S, Vanderheyden I, McCracken JE, Vesela J, DeHertogh R: Increased cell death in rat blastocysts exposed to maternal diabetes in utero and to high glucose or tumor necrosis factor-α in vitro. Development 1997, 124:4827-4836 [DOI] [PubMed] [Google Scholar]
- 8.Ellington SK: Effects of excess glucose on mammalian post-implantation embryos. Int J Dev Biol 1997, 41:299-306 [PubMed] [Google Scholar]
- 9.Pinter E, Reece EA, Leranth CZ, Sanyal MK, Hobbins JC, Mahoney MJ, Naftolin F: Yolk sac failure in embryopathy due to hyperglycemia: ultrastructural analysis of yolk sac differentiation associated with embryopathy in rat conceptuses under hyperglycemic conditions. Teratology 1986, 33:73-84 [DOI] [PubMed] [Google Scholar]
- 10.Pinter E, Mahooti S, Wang V, Imhof BA, Madri JA: Hyperglycemia-induced vasculopathy in the murine vitelline vasculature: correlation with PECAM-1/CD31 tyrosine phosphorylation state. Am J Pathol 1999, 154:1367-1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrara N, Carvermoore K, Chen H, Dowd M, Lu L, Oshera KS, Powellbraxton L, Hillan KJ, Moore MW: Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380:439-442 [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Powling J, Moons L, Collen D, Risau W, Nagy A: Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380:435-439 [DOI] [PubMed] [Google Scholar]
- 13.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC: Failure of blood island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 14.Shalaby F, Ho J, Stanford WL, Fisher KD, Schuh AC, Schwartz L, Bernstein A, Rossant J: A requirement for Flk-1 in primitive and definitive hematopoiesis and vasculogenesis. Cell 1997, 89:981-990 [DOI] [PubMed] [Google Scholar]
- 15.Dumont DJ, Jussila L, Taipale J, Lyboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K: Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 1998, 282:946-949 [DOI] [PubMed] [Google Scholar]
- 16.Soker S, Takashima S, Mial J, Neufeld G, Kalagsbrun M: Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92:735-745 [DOI] [PubMed] [Google Scholar]
- 17.Fong GH, Rossant J, Gertsenstein M, Breitman ML: Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 18.Breier G, Clauss M, Risau W: Coordinate expression of vascular endothelial growth factor receptor-1 (flt-1) and its ligan suggests a paracrine regulation of murine vascular development. Dev Dyn 1995, 204:228-239 [DOI] [PubMed] [Google Scholar]
- 19.New DAT: Whole-embryo culture and the study of mammalian embryos during organogenesis. Biol Rev 1976, 53:81-122 [DOI] [PubMed] [Google Scholar]
- 20.Pinter E, Barreuther M, Lu TT, Imhof BA, Madri JA: Platelet-endothelial cell adhesion molecule-1 (PECAM-1/CD31) tyrosine phosphorylation state changes during vasculogenesis in the murine conceptus. Am J Pathol 1997, 150:1523-1530 [PMC free article] [PubMed] [Google Scholar]
- 21.Ilan N, Cheung L, Pinter E, Madri JA: Platelet-endothelial cell adhesion molecule-1 (CD31), a scaffolding molecule for selected catenin family members whose binding is mediated by different tyrosine and serine/threonine phosphorylation. J Biol Chem 2000, 275:21435-21443 [DOI] [PubMed] [Google Scholar]
- 22.Miguerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A: Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol 1999, 212:307-322 [DOI] [PubMed] [Google Scholar]
- 23.Kaufman MH: Atlas of Mouse Development. 1990:pp 5-85 Academic Press London
- 24.Ilan N, Mahooti S, Rimm DL, Madri JA: PECAM-1 (CD31) functions as a reservoir for and a modulator of tyrosine-phosphorylated beta-catenin. J Cell Sci 1998, 112:3005-3014 [DOI] [PubMed] [Google Scholar]
- 25.Natrajan R, Bai W, Lanting L, Gonzales N, Nadler J: Effect of high glucose on vascular endothelial factor expression in vascular smooth muscle cells. Am J Physiol 1997, 273:H2224-H2231 [DOI] [PubMed] [Google Scholar]
- 26.Duh E, Aiello LP: Perspectives in diabetes: vascular endothelial growth factor and diabetes. Diabetes 1999, 48:1899-1906 [DOI] [PubMed] [Google Scholar]
- 27.Ferrara N, Davis-Smith T: The biology of vascular endothelial growth factor. Endocrinol Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 28.Ferrara N: Endothelial growth factor: molecular and biological aspects. Curr Topics Microbiol Immunol 1999, 237:1-30 [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P, Collen D: Vascular development and disorders: molecular analysis and pathogenetic insights. Kidney Int 1998, 53:1519-1549 [DOI] [PubMed] [Google Scholar]
- 30.Miquerol L, Langille BL, Nagy A: Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 2000, 127:3941-3946 [DOI] [PubMed] [Google Scholar]
- 31.Drake CJ, Little CD: Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc Natl Acad Sci USA 1995, 92:7657-7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drake CJ, Larue A, Ferrara N, Little CD: VEGF regulates cell behavior during vasculogenesis. Dev Biol 2000, 224:178-188 [DOI] [PubMed] [Google Scholar]
- 33.Oberg C, Waltenberger J, Claesson-Welsh L, Welsh M: Expression of protein tyrosine kinases in islet cells: possible role of the Flk-1 receptor for beta-cell maturation from duct cells. Growth Factors 1994, 10:115-126 [DOI] [PubMed] [Google Scholar]
- 34.Rooman I, Schuit F, Bouwens L: Effect of vascular endothelial growth factor on growth and differentiation of pancreatic ductal epithelium. Lab Invest 1997, 76:225-232 [PubMed] [Google Scholar]
- 35.Hidaka M, Stanford WL, Bernstein A: Conditional requirement for Flk-1 receptor in the in vitro generation of early hematopoietic cells. Proc Natl Acad Sci USA 1999, 96:7370-7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuh AC, Faloon P, Hu QL, Bhiamani M, Choi K: In vitro hematopoietic and endothelial potential of flk−/− embryonic stem cells and embryos. Proc Natl Acad Sci USA 1999, 96:2159-2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo D, Jia Q, Song H-V, Warren RS, Donner DB: Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. J Biol Chem 1995, 270:6729-6733 [DOI] [PubMed] [Google Scholar]
- 38.Reece EA, Homko CJ, Wu YK: Multifactorial basis of the syndrome of diabetic embryopathy. Teratology 1996, 54:171-182 [DOI] [PubMed] [Google Scholar]
- 39.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M: Ftl-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci USA 1998, 95:9349-9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W: Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998, 111:1853-1865 [DOI] [PubMed] [Google Scholar]
- 41.Lu TT, Yan LG, Madri JA: Integrin engagement mediates tyrosine dephosphorylation on platelet-endothelial cell adhesion molecule 1. Proc Natl Acad Sci USA 1996, 93:11808-11813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim CS, Wang T, Madri JA: Platelet endothelial cell adhesion molecule-1 expression modulates endothelial cell migration in vitro. Lab Invest 1998, 78:583-590 [PubMed] [Google Scholar]
- 43.Schimmenti LA, Yan HC, Madri JA, Albelda SM: Platelet endothelial cell adhesion molecule, PECAM-1 modulates cell migration. J Cell Physiol 1992, 153:417-428 [DOI] [PubMed] [Google Scholar]
- 44.Oberg-Welsh C, Sandler S, Andersson A, Welsh M: Effects of vascular endothelial growth factor on pancreatic duct cell replication and the insulin production of fetal islet-like cell clusters in vitro. Mol Cell Endocrinol 1997, 126:125-132 [DOI] [PubMed] [Google Scholar]
- 45.Benjamin LE, Hemo I, Keshet E: A plasticity window for blood vessel remodeling is defined by pericyte coverage of preformed endothelial netowrk and is regulated by PDGF-B and VEGF. Development 1998, 125:1591-1598 [DOI] [PubMed] [Google Scholar]
- 46.Hirschi KK, D’Amore PA: Pericytes in the microvasculature. Cardiovasc Res 1996, 32:687-698 [PubMed] [Google Scholar]