

Abstract
The presumed precursor lesions of pancreatic ductal adenocarcinoma were recently classified according to their increasing grade of dysplasia and were designated as pancreatic intraepithelial neoplasia (PanIN) 1 through 3. In this study, we tested whether molecular genetic alterations can be correlated with this classification and may help to further categorize the various PanIN grades. We determined the frequencies of allelic loss at chromosomal arms 9p, 17p, and 18q in 81 microdissected duct lesions of various PanIN grades, using a combination of whole genome amplification and microsatellite analysis. In addition we examined the p53 and Dpc4 protein expression patterns by immunohistochemical analysis. In PanIN-1, we did not detect allelic losses. In PanIN-2, allelic losses were found in increasing frequency, and were particularly high in those lesions with moderate-grade dysplasia (low grade, 20, 33, and 17%, loss at 9p, 17p, and 18q, respectively; moderate grade, 46, 77, and 58%). PanIN-3 and invasive carcinomas exhibited abundant losses. Abnormal p53 and Dpc4 protein expression was only rarely identified in PanIN-2 lesions, but occurred frequently in PanIN-3 lesions and invasive carcinomas. The combined genetic and protein expression data support a model in which allelic loss is the first hit in the biallelic inactivation of the p53 and DPC4 tumor suppressor genes. In addition, our data indicate that allelic loss analysis may be useful in separating PanIN-2 lesions with low-grade dysplasia from those PanIN-2 lesions with moderate-grade dysplasia, each potentially representing a distinct progression step toward invasive carcinoma.
Ductal lesions of various morphological types have long been recognized in association with ductal carcinomas of the pancreas and within noncancerous pancreatic tissues. 1-3 Recently, an international expert committee classified these lesions on the basis of histological criteria into three grades of pancreatic intraepithelial neoplasia (PanIN; for detailed information on the PanIN classification see http://pathology.jhu/pancreas_panin). Briefly, PanIN-1 lesions have a flat or papillary mucinous epithelium without cellular atypia, whereas PanIN-2 lesions show increasing signs of cellular atypia and a prevalence of papillary architecture. Finally, PanIN-3 lesions correspond to carcinoma in situ lesions. A test of the reproducibility of the newly adopted PanIN classification among pathologists revealed that the kappa values for observer agreement were fair (0.43, 0.41) for PanIN-1 and PanIN-3 lesions but poor (0.14) for PanIN-2 lesions. 4 These results suggested that further refinement of the PanIN classification is needed, with particular emphasis on a better categorization of the somewhat problematic PanIN-2 lesions.
Genetic data on the various PanIN grades would conceivably provide the necessary means to further refine the histopathological PanIN classification and may also be useful for early detection of high-risk lesions. Mutations in the K-ras oncogene have been identified in PanINs of all grades, 5-9 and are therefore not useful in discriminating PanINs according to their grade and malignant potential. In a small series of lesions, p16 gene mutations were almost exclusively identified in PanIN-3 lesions, 6 whereas loss of p16 protein expression was already found at the PanIN-2 stage. 10 Furthermore, abnormal expression of the p53 protein and lack of Dpc4 protein expression was frequently seen in PanIN-3 lesions but rarely in PanIN-1 or PanIN-2 lesions, 11-15 suggesting that monitoring the expression of p53 and Dpc4 would provide additional criteria to identify PanIN-3 lesions. Two recently published studies reported low to moderate frequencies of allelic losses at 9p, 17p, and 18q in PanIN-1 lesions and moderate to high allelic loss frequencies of these regions for PanIN-3 lesions. 13,15 Although these molecular analyses of PanINs established a first genetic progression model for pancreatic carcinoma, no criteria have yet been generated to further categorize the PanIN-2 lesions, which may represent a very important step in the preinvasive development of pancreatic carcinoma. We therefore attempted to identify such criteria through an extensive analysis of p16, p53, and DPC4 in a series of microdissected PanINs from 22 patients, using a combination of genetic and immunohistochemical techniques.
Materials and Methods
Tissue Sampling and Microdissection
Whipple resection specimens from 21 patients (11 men and 10 women; mean age, 60.1 years) with ductal adenocarcinoma of the pancreas and one male patient with noncancerous pancreatic disease were obtained from the Department of Surgery, University of Kiel, Germany. The specimens were macroscopically examined in the unfixed state, cut along the duct level, and immediately fixed in buffered 4% formalin. An average of three blocks of tumor tissue were excised, embedded in paraffin, cut into 5-μm sections, and stained with hematoxylin and eosin. Ductal lesions were classified according to the recently established PanIN classification. 4 Furthermore, PanIN-2 lesions were subdivided into lesions exhibiting signs of low- and moderate-grade dysplasia. The criteria for low-grade dysplasia were a slight nuclear enlargement (not more than twice that in normal duct epithelium) and a slight increase in nuclear hyperchromasia with only few coarse chromatin granules. Advanced nuclear enlargement and numerous coarse dark chromatin granules were classified as moderate-grade dysplasia.
For each lesion an unstained 10-μm parallel section was microdissected, deparaffinized, and overlaid with phosphate-buffered saline. Microdissection was performed using a micromanipulator (Leica, Narishige Micromanipulator, Wetzlar, Germany), as has been described. 8 In a few instances, microdissected cells from the same lesion present in two to three serial sections were pooled to obtain the minimum of ∼100 cells required for genetic analysis.
Whole Genome Amplification by Degenerate Oligonucleotide Primed (DOP)-Polymerase Chain Reaction (PCR)
Approximately 100 cells per lesion were microdissected and transferred to 0.2-ml PCR tubes, containing 40 μl of lysis buffer (10 mmol/L Tris-HCl, pH 8.0, 10 mg/ml Proteinase K, 1% Tween 20), and overlaid with mineral oil. Samples were subsequently incubated at 48°C for 15 hours, followed by 10 minutes at 95°C. The samples were then divided into two 0.2-ml PCR tubes, and whole genome amplification by DOP-PCR 16 was simultaneously performed for each of the two samples.
DOP-PCR was performed in 20 mmol/L Tris-HCl, pH 8.8, 10 mmol/L KCl, 10 mmol/L (NH4)2SO4, 2 mmol/L MgSO4, 1% Triton X-100, 1 mg/ml bovine serum albumin, 3 μmol/L universal primer (5′-CCG ACT CGA GNN NNN NAT GTG G-3′), 200 μmol/L dNTPs, and 3.75 U Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA), in a volume of 50 μl. DNA was amplified using a temperature cycler (PTC-200; MJ Research, Watertown, MA). The initial amplification stage comprised eight cycles at 94°C for 1 minute, 30°C for 1.5 minutes, followed by temperature ramping to 72°C (ramping rate 0.2°C/second), and a final extension at 72°C for 2 minutes. The subsequent cycling stage comprised 40 cycles at 94°C for 1 minute, 62°C for 1 minute, and 72°C for 2 minutes. Per cycle 5 seconds were added to each 72°C extension step, and a final extension step was performed at 72°C for 10 minutes.
Microsatellite PCR
Microsatellite markers D9S319, D9S157, D9S304, D9S171, D17S1832, D17S786, D17S796, TP53-PCR15, D18S877, D18S535, D18S363, D18S46, and D18S474 were selected within 4 cM proximity to the p16 (9p21), p53 (17p13), and DPC4 (18q21) gene loci. PCR primers were selected such that the fragment length was below 250 bp for all markers. Primer sequences are available on request.
Microsatellite-PCR reactions were performed in 96-well microtiter plates, in 20 mmol/L Tris-HCl, pH 8.4, 5 mmol/L KCl, 1.5 mmol/L MgCl2, 100 ng of each primer, 200 μmol/L dNTPs, 60 mmol/L TMAC (Sigma, Taufkirchen, Germany), 1.5% formamide, 2 μl DOP-PCR product as template, and 1.5 units Taq DNA polymerase (Gibco BRL, Karlsruhe, Germany), in a final volume of 15 μl. Reactions were performed in a Hybaid Touchdown temperature cycler (MWG-Biotech, Ebersberg, Germany), for 40 cycles of 94°C for 15 seconds, 55°C for 30 seconds, and 72°C for 30 seconds, and a final extension at 72°C for 5 minutes. PCR products were separated on 6% polyacrylamide, 8 mol/L urea gels and DNA fragments were visualized by silver staining. Gels were independently scored by two investigators (HG and SAH), who were unaware of the histological grade of the lesions. Normal tissues were considered informative if two bands of the expected size were detected, usually separated by at least two bases. Loss of an allele was determined when the corresponding normal tissue was informative and one band (allele) was missing in the neoplastic tissue. In a few ambiguous cases, gels were scanned by an imaging system (Alpha Imager, Biozym, Hessisch Oldendorf, Germany) and analyzed using image analysis software (Gel-Pro Analyzer, Version 3;Media Cybernetics, Spring Field, MD). A reduction in relative intensity of >75% was required for allelic loss.
Immunohistochemistry
Immunostaining was performed using the anti-p53 monoclonal antibody DO1 (dilution 1:50; Calbiochem, Oncogene Research Products, Cambridge, MA) and anti-Dpc4 clone A8 (dilution 1:50; Santa Cruz Biotechnology, Santa Cruz, AZ). Antigen retrieval and immunostaining for p53 and Dpc4 were performed as previously described. 17,18 Briefly, antigen retrieval was performed by the pressure cooker method for 3.5 minutes for the p53 antibody and 20 minutes for the Dpc4 antibody. To improve the staining sensitivity a DAB-enhanced detection kit (Ventana Medical System, Tucson, AZ) and an amplification kit (Ventana) were used. All immunostainings were independently assessed by two of the authors who were unaware of the molecular data (JL and VB).
Results
PCR Quality Control
One hundred and thirty-six PanINs derived from 21 cases of pancreatic ductal adenocarcinoma and one patient without neoplastic disease of the pancreas were microdissected from paraffin-embedded tissue specimens. After whole genome amplification, allelic losses at chromosomal regions 9p21 (the location of the p16 gene), 17p13 (p53), and 18q21 (DPC4/SMAD4) were examined by microsatellite analysis using 13 markers that mapped to these regions. To exclude amplification artifacts, we performed two DOP-PCRs per lesion and two microsatellite PCRs for each marker from both DOP-PCR templates, thus generating a data set of four PCRs per marker. A representative example of the microsatellite analysis is shown in Figure 1 ▶ .
Figure 1.
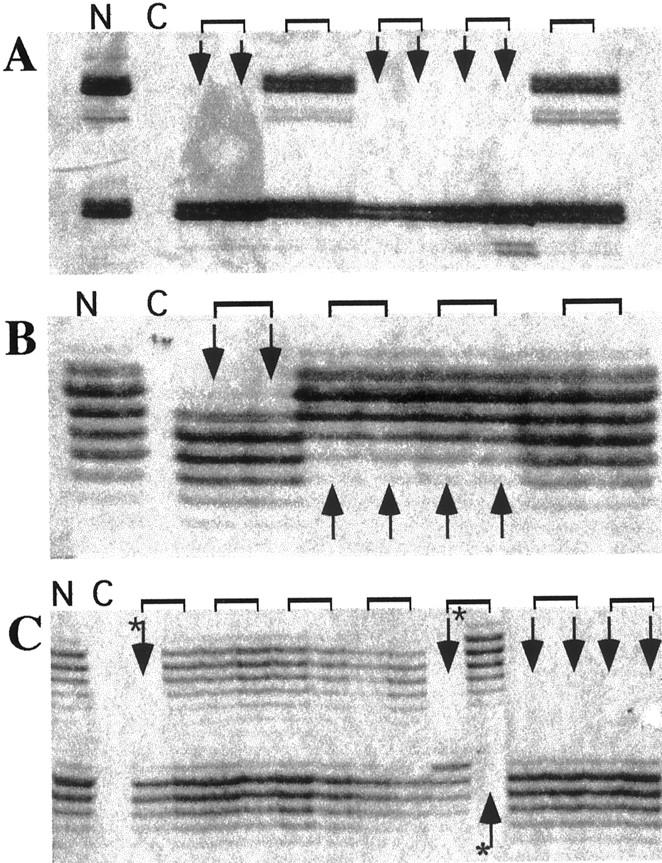
Example of microsatellite analysis of DOP-PCR templates derived from various PanINs. Amplification products were separated on a standard sequencing gel and visualized by silver staining. A: Microsatellite marker D9S304. B: Marker D17S786. C: Marker D18S363. Arrows, samples with allelic loss; brackets, pairs of microsatellite PCRs generated from the same DOP-PCR template; asterisks, examples of discordant PCR amplification patterns. N, normal DNA; C, negative control.
The study included only lesions for which the DOP-PCR yielded material of sufficient quality, as indicated by >75% successful microsatellite PCRs. This criterion was met in 81 lesions. The average heterozygosity rate of our marker panel was 0.53 (ranging from 0.38 to 0.71), and 558 of 1,053 data sets (81 lesions times 13 microsatellite markers) were thus informative. Finally, only data sets with concordant amplification patterns among all four PCRs or among three PCRs with a failed fourth PCR were included in the study. Of the 558 data sets, 367 (66%) met this criterion.
The number of lesions that were excluded from the study either because of insufficient DOP-PCR amplification or because of discordant PCR results for the various PanIN grades revealed no obvious selection bias. PCR was ineffective in 6 of 14 (43%), 7 of 22 (32%), 14 of 28 (50%), 5 of 21 (24%), 7 of 24 (29%), and 16 of 28 (57%) of PanIN-1A, PanIN-1B, PanIN-2 with low-grade dysplasia, PanIN-2 with moderate-grade dysplasia, PanIN-3 lesions, and carcinomas, respectively. Discordant results were observed in 10 of 41 (24%), 11 of 98 (11%), 16 of 86 (19%), 25 of 80 (31%), 21 of 89 (24%), and 10 of 65 (15%) PCRs performed with DNA derived from PanIN-1A, PanIN-1B, PanIN-2 with low-grade dysplasia, PanIN-2 with moderate-grade dysplasia, PanIN-3 lesions, and carcinomas, respectively. Importantly, in 67 data sets the larger allele was lost and in 63 data sets the smaller allele was lost, thus further confirming the reliability of the amplification procedure and the stringency of our inclusion criteria.
Allelic Losses in PanINs
The results of the allelic loss study are detailed in Figure 2 ▶ and summarized in Figure 3A ▶ . Of the 81 lesions included in the study, eight were classified as PanIN-1A. None of these early lesions revealed loss of an allele at any of the three loci analyzed. A low frequency of allelic loss was detected for chromosomal regions 17p (2 of 12) and 18q (1 of 14) in PanIN-1B lesions. A low to moderate frequency of allelic loss was found at 9p (2 of 10), 17p (4 of 12), and 18q (2 of 12) in PanIN-2 lesions with low-grade dysplasia. The most significant increase in allelic losses occurred in PanIN-2 lesions with moderate-grade dysplasia (6 of 13 lesions at 9p, 10 of 13 at 17p, and 7 of 12 at 18q), which further increased in the PanIN-3 lesions (13 of 15 at 9p, 6 of 10 at 17p, and 14 of 16 at 18q) and the carcinomas (8 of 8 at 9p, 10 of 11 at 17p, 9 of 11 at 18q). The observed progressive increase in allelic losses in low- to high-grade PanIN lesions support the PanIN classification and the proposed tumor progression model for pancreatic carcinoma. 19
Figure 2.
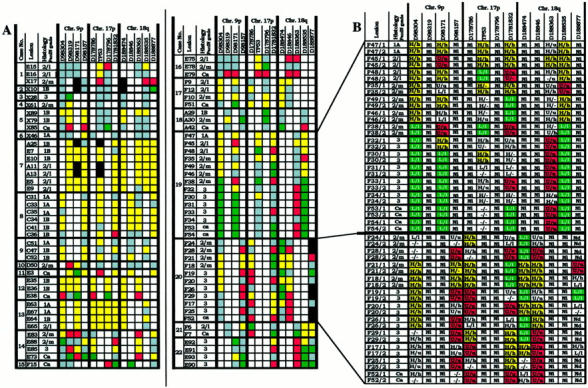
Microsatellite analyses of various grades of PanIN. A: Microdissected lesions were derived from specimens from 21 pancreatic carcinoma patients and from one patient with noncancerous pancreatic disease (case 13). Allelic loss at chromosomal regions 9p, 17p, and 18q, was determined with 13 microsatellite markers. For each microsatellite marker, a data set (squares) of four independent PCRs was generated per lesion. Denotation of the color-coded squares: yellow, heterozygosity; red, allelic loss of the upper allele; green, allelic loss of the lower allele; white, noninformative; gray, excluded data set, black, not done. B: Detailed microsatellite analysis data on two representative cases (cases 19 and 20). Data sets (see A) are represented in data pairs (in uppercase and lowercase) from two microsatellite PCRs that were performed on each of two DOP-PCR templates. 2/L, PanIN-2 lesion with low-grade dysplasia; 2/m, PanIN-2 lesion with moderate-grade dysplasia; U/u, allelic loss of the upper allele (PCR1/PCR2); L/L, allelic loss of the lower allele (PCR1/PCR2); H/h, heterozygosity (PCR1/PCR2); Nd, not done; Ni, noninformative; (−), failed PCR reaction.
Figure 3.
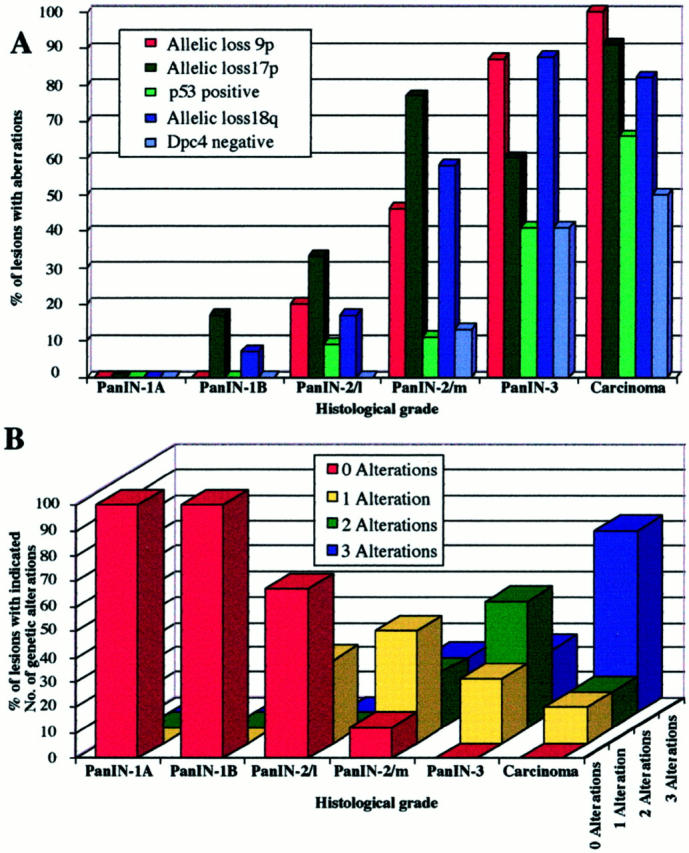
Molecular analyses of various grades of PanIN. A: Frequencies of allelic loss at chromosomal regions 9p, 17p, and 18q together with frequencies of nuclear p53 expression and lack of Dpc4 protein expression. B: Accumulation of genetic alterations. The number of alterations refers to the number of allelic losses at three chromosomal regions (9p, 17p, and 18q) in those lesions that were informative for at least one marker at each of the three loci. PanIN-2/L, PanIN-2 lesion with low-grade dysplasia; PanIN-2/m, PanIN-2 lesion with moderate-grade dysplasia.
The accumulation of multiple genetic alterations in the various PanIN grades was examined in those lesions that were informative for at least one microsatellite marker at each of the three chromosomal regions. The results of this analysis are summarized in Figure 3B ▶ . The nine PanIN-2 lesions with low-grade dysplasia had either no (6 of 9) or one (3 of 9) alteration. The nine PanIN-2 lesions with moderate-grade dysplasia had no (1 of 9), one (4 of 9), two (2 of 9), or three (2 of 9) alterations. The eight PanIN-3 lesions with high-grade dysplasia had one (2 of 8), two (4 of 8), or three (2 of 8) alterations, and the seven carcinomas had one (1 of 7), two (1 of 7), or three (5 of 7) alterations. Again these findings support the PanIN classification and the established tumor progression model. 19
Clonal heterogeneity was assessed in 15 cases for which more than one lesion was available (Figure 2) ▶ . Clonal heterogeneity was postulated when allelic loss data were incompatible, that is, different alleles were lost in lesions from a single case (red and green squares in Figure 2 ▶ ), or earlier lesions revealed allelic loss, whereas later stages were heterozygous (Figure 2 ▶ , yellow squares) at the same locus. Clonal heterogeneity was demonstrated in 6 of these 15 cases (40%; eg, cases 14, 16, 19, 20, 21, and 22; Figure 2 ▶ ). For four of these cases, samples of at least two PanIN-3 and/or invasive carcinomas were available (eg, cases 14, 19, 20, and 22; Figure 2 ▶ ). In all four cases there were PanIN-3 lesions with allelic losses that were incompatible with the allelic loss pattern of the carcinoma or other PanIN-3 lesions (eg, lesions E85 and E73 from case 14; lesions F33 and all other lesions from case 19; lesions F29 and F17 from case 20; and lesions E92 and E93 from case 22). These results indicate that these PanIN-3 lesions cannot simply be classified as “cancerization of the ducts.” 19
p53 and Dpc4 Protein Expression in PanINs
p53 protein expression was analyzed immunohistochemically in 192 PanINs (Figure 3A) ▶ . Thirty-three PanIN-1A and 62 PanIN-1B lesions did not stain for p53. Nuclear accumulation of p53 was found in 2 of 22 (9%) PanIN-2 lesions with low-grade dysplasia, in 4 of 36 (11%) PanIN-2 lesions with moderate-grade dysplasia, and in 16 of 39 (41%) PanIN-3 lesions. Concordant with the literature, 11-13,15,20 14 of 21 (67%) carcinomas showed overexpressed p53, including all carcinomas from patients with p53-positive precursor lesions. Our data suggest that abnormal p53 protein expression patterns essentially do not arise until the PanIN-3 stage of pancreatic tumor development.
Dpc4 protein expression was analyzed immunohistochemically in 180 PanIN lesions (Figures 3A and 4) ▶ ▶ . Dpc4 protein expression was detected in all 92 PanIN-1 lesions and in all 23 PanIN-2 lesions with low-grade dysplasia, but was not detected in 4 of 31 (13%) PanIN-2 lesions with moderate-grade dysplasia and in 14 of 34 (41%) PanIN-3 lesions. Nine of 18 (50%) carcinomas did not express Dpc4, which is in agreement with reported frequencies. 14,18 In two cases, the carcinoma tissue had retained Dpc4 expression whereas the PanIN-3 lesions were Dpc4-negative. This observation substantiates the clonal heterogeneity that was observed in the allelic loss patterns of these lesions (Figure 2) ▶ . Our results suggest that abnormal Dpc4 protein expression patterns also tend to arise at the PanIN-3 stage.
Figure 4.
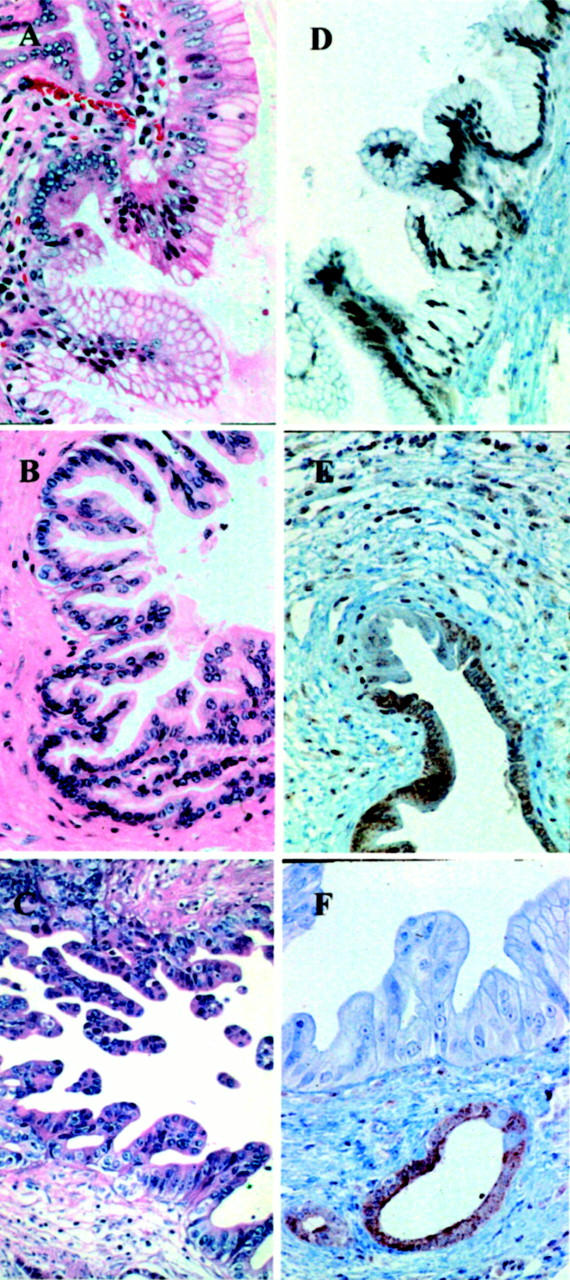
Examples of various grades of duct lesions associated with pancreatic carcinoma. A: Papillary duct lesion, grade PanIN-2 with low-grade dysplasia: slight nuclear enlargement and nuclear crowding. H&E; original magnification, ×250. B: Papillary duct lesion, grade PanIN-2 with moderate-grade dysplasia: moderate nuclear enlargement, nuclear hyperchromasia and loss of polarity. H&E; original magnification, ×250. C: Papillary duct lesion, grade PanIN-3: loss of polarity and structure and increasing branching of the papillae. H&E; original magnification, ×250. D–F: Immunohistochemical staining for Dpc4. D: Duct lesion, mainly PanIN-2 with low-grade dysplasia and partially PanIN-1A, both with positive staining for Dpc4 protein. Original magnification, ×125. E: Duct lesion, grade PanIN-2 with moderate-type dysplasia with nuclear Dpc4 staining, and a few negative nuclei in the adjacent intraductal carcinoma. Original magnification, ×125. F: Duct lesion, mainly grade PanIN-3 without Dpc4 protein expression, but retained Dpc4 expression in adjacent hyperplastic epithelium. Original magnification, ×250.
Discussion
During the Pancreatic Cancer Think Tank meeting held in 1999, a new standardized nomenclature and classification for the proliferative epithelial lesions in pancreatic ducts was introduced. 4 In this classification, PanIN is categorized according to defined histopathological criteria. When tested among experts, however, the reproducibility of the PanIN classification was found to be fair for PanIN-1 and PanIN-3 lesions and poor for PanIN-2 lesions, indicating that additional criteria were needed. 4 Hypothesizing that genetic alterations in various PanINs could provide these criteria, we analyzed allelic losses at chromosomal regions 9p21 (the location of the p16 gene), 17p13 (p53), and 18q21 (DPC4/SMAD4) in a series of PanINs, using genetic and immunohistochemical analyses.
We found a progressive increase in allelic losses at all three chromosomal regions in PanINs from low to high grade, with the highest rate of allelic loss identified in carcinomas (Figure 3A) ▶ . The majority (75%) of PanIN-3 lesions revealed losses at two or three of the chromosomal regions tested, whereas 67% of PanIN-2 lesions with moderate dysplasia had losses at one or two of the three regions (Figure 3B) ▶ . These data are in agreement with earlier molecular studies showing that the successive accumulation of genetic changes paralleled the severity of ductal dysplasia. 6,13,15 Our data also support the previously suggested precursor nature of the ductal lesions and the proposed tumor progression model for pancreatic neoplasia. 14,19
Recently, two independent reports also described allelic losses at 9p, 17p, and 18q in various grades of PanINs. 13,15 The allelic loss frequencies that we identified at these three regions in PanIN-1 and PanIN-3 lesions are in good agreement with those reported by Yamano and colleagues. 15 PanIN-2 lesions were not included in their study. 15 The allelic loss frequencies reported by Heinmöller and colleagues, 13 however, are discordant with ours and those of Yamano and colleagues, 15 in that they found a significant rate of allelic loss at chromosomes 9p and 18q (32 and 35%, respectively) and a somewhat lower rate at 17p (15%) in PanIN-1 lesions. In addition, their frequencies of loss at 9p and 18q did not differ significantly among PanIN-1B, -2, and -3 lesions. Finally, they did not find an accumulation of clonal genetic changes with increasing PanIN grades. 13 The discrepancy in the allelic losses reported by Heinmöller and colleagues 13 and those reported here and by Yamano and colleagues 15 may very well be explained by differences in the histological classification systems applied to the investigated lesions.
As expected, in our study abnormal p53 and Dpc4 protein expression patterns were most prevalent in the invasive carcinomas (67 and 50% of carcinomas, respectively). 12,14,18,20-22 Importantly, abnormal expression of both proteins seemed to arise mainly at the PanIN-3 stage (each near 40% of lesions, Figure 3A ▶ ). Our genetic analyses, however, had revealed that the majority of allelic losses occurred at the PanIN-2 stage, with 88% of moderate-grade PanIN-2 lesions harboring allelic losses (Figures 2 and 3B) ▶ ▶ . The combined genetic and immunohistochemical data thus support a model for pancreatic carcinogenesis in which allelic loss precedes the mutational event in the biallelic inactivation of the p53 and DPC4 tumor suppressor genes. Moreover, our data suggest that allelic loss analysis may be useful in separating PanIN-2 lesions with low-grade dysplasia from those PanIN-2 lesions with moderate-grade dysplasia, each potentially representing a distinct progression step toward invasive carcinoma.
A major challenge for the near future lies in the development of early diagnosis strategies to identify those patients at the precarcinoma in situ stage with a high risk for developing pancreatic carcinoma. Our identification of allelic loss as the first hit in the inactivation of the p53 and DPC4 genes together with the observed marked increase in allelic losses at the transition between PanIN-2 lesions with low-grade dysplasia and those with moderate-grade dysplasia (15 to 35% and 45 to 80% losses for the three chromosomal regions, respectively; Figure 3A ▶ ) could form the basis for designing studies to test the positive predictive value of allelic losses for the development of pancreatic carcinoma in risk patients. In this context, recent advances in the application of fluorescence in situ hybridization technology suggest that the analysis of pancreatic juice for allelic losses as a means for early diagnosis may indeed become feasible. 23
Acknowledgments
We thank Mieke Schutte and Michael Goggins for helpful discussions and comments on the manuscript.
Footnotes
Address reprint requests to Stephan Hahn, Department of Internal Medicine, University of Bochum, In der Schornau 23-25, 44892 Bochum, Germany. E-mail: stephan.hahn@ruhr-uni-bochum.de.
Supported by grants from the Deutsche Krebshilfe (to S. A. H., I. S.-W., W. S., J. L., and G. K.), from the European Community Biomed 2 Project, BMH4-CT98-3085 (to S. A. H., I. S.-W., W. S.), from the BMBF (01GB9708; to W. S., I. S. W.), and from the Ruhr-Universität-Bochum, FORUM (to S. A. H., I. S.-W.).
J. Lüttges and H. Galehdari both made equal contributions to this work.
References
- 1.Cubilla AL, Fitzgerald PJ: Morphological lesions associated with human primary invasive carcinoma nonendocrine pancreas cancer. Cancer Res 1976, 36:2690-2698 [PubMed] [Google Scholar]
- 2.Kozuka S, Sassa R, Takai T, Masamoto K, Nagasawa S, Saga S, Hasegawa K, Takeuchi M: Relation of pancreatic duct hyperplasia to carcinoma. Cancer 1979, 43:1418-1428 [DOI] [PubMed] [Google Scholar]
- 3.Klöppel G, Bommer G, Rückert K, Seifert G: Intraductal proliferation in the pancreas and its relationship to human and experimental carcinogenesis. Virchows Arch Pathol Anat 1980, 387:221-233 [DOI] [PubMed] [Google Scholar]
- 4.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra D, Klöppel G, Longnecker DL, Lüttges J, Offerhaus GJA: Pancreatic intraepithelial neoplasia (PanIN): a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol (in press) [DOI] [PubMed]
- 5.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R: Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 1996, 110:227-231 [DOI] [PubMed] [Google Scholar]
- 6.Moskaluk CA, Hruban RH, Kern SE: p16 and K-ras mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997, 57:2140-2143 [PubMed] [Google Scholar]
- 7.Sugio K, Molberg K, Albores SJ, Virmani AK, Kishimoto Y, Gazdar AF: K-ras mutations and allelic loss at 5q and 18q in the development of human pancreatic cancers. Int J Pancreatol 1997, 21:205-217 [DOI] [PubMed] [Google Scholar]
- 8.Lüttges J, Möllmann B, Menke MAOH, Clemens A, Klimpfinger M, Sipos B, Klöppel G: Duct changes and K-ras mutations in the disease-free pancreas: analysis of type, age relation and spatial distribution. Virchows Arch 1999, 435:461-468 [DOI] [PubMed] [Google Scholar]
- 9.Lüttges J, Schlehe B, Menke MAOH, Vogel I, Henne-Bruns D, Klöppel G: The K-ras mutation pattern in pancreatic ductal adenocarcinoma is usually identical to that in associated normal, hyperplastic and metaplastic duct epithelium. Cancer 1999, 85:1703-1710 [PubMed] [Google Scholar]
- 10.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH: Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998, 58:4740-4744 [PubMed] [Google Scholar]
- 11.Boschman CR, Stryker S, Reddy J, Rao MS: Expression of p53 protein in precursor lesions and adenocarcinoma of human pancreas. Am J Pathol 1994, 145:1291-1295 [PMC free article] [PubMed] [Google Scholar]
- 12.DiGiuseppe JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allison DC, Cameron JL, Offerhaus GJ: Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994, 101:684-688 [DOI] [PubMed] [Google Scholar]
- 13.Heinmöller E, Dietmaier W, Zirngibl H, Heinmöller P, Scaringe W, Jauch KW, Hofstädter F, Rüschoff J: Molecular analysis of microdissected tumors and preneoplastic intraductal lesions in pancreatic carcinoma. Am J Pathol 2000, 157:83-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilentz RE, Iacobuzio DC, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE, Hruban RH: Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000, 60:2002-2006 [PubMed] [Google Scholar]
- 15.Yamano M, Fujii H, Takagaki T, Kadowaki N, Watanabe H, Shirai T: Genetic progression and divergence in pancreatic carcinoma. Am J Pathol 2000, 156:2123-2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung VG, Nelson SF: Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci USA 1996, 93:14676-14679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lüttges J, Diederichs A, Menke MAOH, Vogel I, Kremer B, Klöppel G: Ductal lesions in patients with chorionic pancreatitis show K-ras mutations in a frequency similar to that in the normal pancreas and lack nuclear immunoreactivity for p53. Cancer 2000, 88:2495-2504 [DOI] [PubMed] [Google Scholar]
- 18.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000, 156:37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hruban RH, Wilentz RE, Kern SE: Genetic progression in the pancreatic ducts. Am J Pathol 2000, 156:1821-1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalthoff H, Schmiegel W, Roeder C, Kasche D, Schmidt A, Lauer G, Thiele HG, Honold G, Pantel K, Riethmüller G: p53 and K-RAS alterations in pancreatic epithelial cell lesions. Oncogene 1993, 8:289-298 [PubMed] [Google Scholar]
- 21.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa LT, Yeo CJ, Kern SE: p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54:3025-3033 [PubMed] [Google Scholar]
- 22.Hahn SA, Schutte M, Hoque ATM, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350-353 [DOI] [PubMed] [Google Scholar]
- 23.Fukushige S, Furukawa T, Satoh K, Sunamura M, Kobari M, Koizumi M, Horii A: Loss of chromosome 18q is an early event in pancreatic ductal tumorigenesis. Cancer Res 1998, 58:4222-4226 [PubMed] [Google Scholar]